

Abstract
Context
Human NR5A1/SF-1 mutations cause 46,XY disorder of sex development (DSD) with broad phenotypic variability, and rarely cause adrenal insufficiency although SF-1 is an important transcription factor for many genes involved in steroidogenesis. In addition, the Sf-1 knockout mouse develops obesity with age. Obesity might be mediated through Sf-1 regulating activity of brain-derived neurotrophic factor (BDNF), an important regulator of energy balance in the ventromedial hypothalamus.
Objective
To characterize novel SF-1 gene variants in 4 families, clinical, genetic and functional studies were performed with respect to steroidogenesis and energy balance.
Patients
5 patients with 46,XY DSD were found to harbor NR5A1/SF-1 mutations including 2 novel variations. One patient harboring a novel mutation also suffered from adrenal insufficiency.
Methods
SF-1 mutations were studied in cell systems (HEK293, JEG3) for impact on transcription of genes involved in steroidogenesis (CYP11A1, CYP17A1, HSD3B2) and in energy balance (BDNF). BDNF regulation by SF-1 was studied by promoter assays (JEG3).
Results
Two novel NR5A1/SF-1 mutations (Glu7Stop, His408Profs*159) were confirmed. Glu7Stop is the 4th reported SF-1 mutation causing DSD and adrenal insufficiency. In vitro studies revealed that transcription of the BDNF gene is regulated by SF-1, and that mutant SF-1 decreased BDNF promoter activation (similar to steroid enzyme promoters). However, clinical data from 16 subjects carrying SF-1 mutations showed normal birth weight and BMI.
Conclusions
Glu7Stop and His408Profs*159 are novel SF-1 mutations identified in patients with 46,XY DSD and adrenal insufficiency (Glu7Stop). In vitro, SF-1 mutations affect not only steroidogenesis but also transcription of BDNF which is involved in energy balance. However, in contrast to mice, consequences on weight were not found in humans with SF-1 mutations.
Introduction
The nuclear receptor steroidogenic factor 1 (SF-1/NR5A1) is a master regulator of adrenal and gonadal development, including sexual determination and differentiation, as well as steroidogenesis and reproduction [1], [2]. SF-1 also plays a pivotal role in the development of the ventromedial hypothalamic nucleus (VMH) and for functions of the pituitary gland [3], [4]. In addition, expression of SF-1 has been identified in the spleen, skin and in small amounts in the placenta [5], [6], [7]. SF-1 was first identified in 1992 for its function as a transcriptional regulator of steroidogenic genes and was named accordingly [8]. Later, the Sf-1 KO mice were reported with a severe phenotype including lack of adrenal glands and a complete sex reversal of 46,XY animals [9]. Finally in 1999, the first human being with adrenal insufficiency and 46,XY disorder of sexual development (DSD) harboring a heterozygote SF-1 mutation was described [10]. Meanwhile, numerous SF-1 mutations have been identified [2], yet the exact function of SF-1 explaining the broad phenotype associated with SF-1 mutations remains elusive [11], [12].
SF-1 is encoded by the NR5A1 gene which is located on the long arm of chromosome 9 (9q33). The gene consists of 7 exons but only 6 exons are coding. SF-1 has 461 amino acids and comprises a DNA binding domain with two zinc fingers, an accessory DNA binding domain, a hinge region and a ligand binding domain [1]. The structure of SF-1 is greatly conserved among animal species [13]. To date more than 70 human NR5A1 mutations have been described (Human Gene Mutations Database, www.hgmd.cf.ac.uk). Most of these mutations are found in a heterozygote state [11], [14] and only a few in a homozygote [15], or compound heterozygote state [16]. So far, no correlation between phenotype and genotype, and also no clear pattern of heredity has been seen as both sporadic and familiar presentations exist [11]. Furthermore, possibility of dominant negative effect of heterozygote NR5A1 mutations has been debated without convincing results [11].
The clinical presentation of SF-1 deficiency is very variable. The first human individual with a heterozygote Gly35Glu SF-1 mutation had 46,XY DSD and adrenal insufficiency [10]. So far, only two additional SF-1 mutations causing adrenal insufficiency have been reported [15], [17]. The heterozygote Arg255Leu mutation was found in a girl with symptoms of adrenal insufficiency and normal ovarian differentiation and function [17], and the homozygote Arg92Gln mutation was present in a boy with adrenal failure and 46,XY DSD [15]. By contrast, NR5A1 mutations are frequently found in patients with 46,XY DSD with apparently normal function of the adrenal cortex [11], [14], [18]. Similarly, some NR5A1 mutations were found in 46,XX females with premature ovarian failure or ovarian insufficiency with normal adrenal function [11], [19].
SF-1 deficiency also affects the central regulation of reproduction and energy balance [20]. The pituitary Sf-1 KO mouse model showed that SF-1 is an essential regulator of gonadotropin (LH, FSH) expression [4], [21], [22]. These mice present with hypogonadotropic hypogonadism reflected by sexual immaturity, low weight of gonads and sterility [4]. Apart from the pituitary gland, SF-1 is also required for the development, organization and function of the ventromedial hypothalamus (VMH) [3], [23]. Mouse models have shown that a loss of SF-1 stimulation leads to disorganization of the VMH, thereby impairing its function related to anxiety, thermoregulation, sexual behavior and energy balance [24]. Selective deletion of Sf-1 in the VMH in mice prenatally resulted in late onset obesity [25], while the same deletion postnatally led to diet induced obesity and deregulated thermogenesis [25]. However, these possible effects of SF-1 deficiency have not yet been studied in humans harboring SF-1 mutations.
In this context, the brain-derived neurotrophic factor (BDNF) is an important regulator of energy balance [26]. It is a highly conserved neurotrophin which is thought to be a SF-1 target gene [27]. BDNF is expressed in several appetite-regulating centers in the hypothalamus and the hindbrain in both mouse and human [28]. Depletion of Bdnf or its receptor (TrkB) in mice results in excessive feeding, weight gain and features accompanied by the metabolic syndrome [26], [29]. Abnormal locomotor activity and late-onset obesity was also observed in a Bdnf heterozygote knockout mouse model or when Bdnf was inactivated in the central nervous system [29], [30]. In addition, reduced expression of BDNF was described in association with obesity in the leptin receptor deficient mouse [31], the Alzheimer disease mouse [32] and the Sf-1 KO mouse [3]. In humans, two reports show a relationship between BDNF (locus 11p14) and obesity [33], [34]. Patients with WAGR syndrome (Wilm’s tumor, aniridia, genitourinary anomalies and mental retardation, OMIM 194072) with a heterozygous 11p14 deletion including the BDNF gene suffer all from childhood onset obesity, while WAGR syndrome patients without genetic anomalies including the BDNF gene have normal prevalence of obesity [33]. Additionally, a girl with obesity and impaired cognitive function who has only one functional copy of the BDNF gene has been described [34].
We hypothesize that human SF-1 mutations may affect metabolism and that this effect could be mediated in part through BDNF. Therefore, in this study we characterize novel NR5A1 mutations, one being associated with the rare phenotype of adrenal insufficiency and 46,XY DSD. We describe the effect of these SF-1 mutations in vitro on transcription of genes involved in steroidogenesis and on the BDNF gene which is important for central regulation of food intake. Finally, we describe some weight related parameters in our small cohort of NR5A1 patients.
Patients and Methods
Patients and ethical approval
Five patients of Czech Republic and Spanish origin with unsolved 46,XY DSD were studied. Main characteristics of patients and families are shown in Table 1, family trees are depicted in Figure 1A and biochemical data are available as Table S1. All studied subjects and/or their legal guardians gave written informed consent for the hormonal and molecular studies, which were approved by the respective ethical committees of the involved centers: Ethics commissions of Vall d’Hebron Research Institute and CIBERER, Barcelona, Spain, and University Hospital Bern, Switzerland.
Table 1. Clinical and genetic characteristics of 5 patients harboring mutations in the NR5A1 gene.
| Patient | Origin,YOB | Karyotype | Assignedsex | Genital anatomyat birth | Gonadalfunction (age) | Adrenalfunction (age) | NR5A1 genemutation | Familyhistory |
| 1 | Czech, 2010 | 46,XY | Male | Perineal hypospadia.Palpable testicles. | At 12 months: BaselineT low, FSH slightlyelevated. | Adrenal insufficiency(high ACTH, low cortisol,high PRA, normal aldosterone). | Compound heterozygote:c.19G>T, p.Glu7Stop;c.887C>T, p.Thr296Met | F = c.887C>T, p.Thr296Met |
| M = WT | ||||||||
| 2 | Spanish, 2013 | 46,XY | Male to femaleat 4 months | Clitoris with redundant skin.Palpable glans but no corpora cavernosa.Posterior labial fusion.Gonads palpable in genital folds (<1 ml).No Müllerian ducts. | Abnormal at 3.5 months:Baseline T low, FSH high;Very low T responseto hCG stimulation. | Normal at baselineaged 3.5 months. | Heterozygote: c.1222_1223insC, p.His408Profs*159 | M = WT |
| F = “carrier” (hypospadias) | ||||||||
| ICSI product from both parents’ gametes. | ||||||||
| 3 | Spanish, 2011 | 46,XY | Male | Scrotal hypospadias.Penis length 2 cm.Bilateral scrotal gonads.No Müllerian ducts. | Abnormal: T slightly decreased,but normal precursor responseto hCG stimulation. AMHlow at age 17–20 days. | Normal at baselineaged 17 days. | Heterozygote:c.937C>T,p.Arg313Cys | F = WT |
| M = WT | ||||||||
| 4 | Spanish, 2010 | 46,XY | Male | Scrotal hypospadias. Penis length 1.5 cm.Bilateral scrotal gonads (1 ml).No Müllerian ducts. | T high at birth. AMH low at 2 months. | ND | Heterozygote:c.937C>T,p.Arg313Cys | Bichorial twin of patient 5. |
| 1st twin | F = carrier (operated hypospadias; testis volume L 6 ml/R 12 ml). | |||||||
| M = unknown | ||||||||
| ICSI product from donor ova and father’s sperm. | ||||||||
| 5 | Spanish, 2010 | 46,XY | Male | Scrotal hypospadias.Penis length 1.5 cm.Unilateral cryptorchidism,scrotal testis 0.5 ml. | T high at birth.AMH low at 2 months. | ND | Heterozygote:c.937C>T,p.Arg313Cys | Bichorial twin of patient 4. |
| 2nd twin |
YOB: year of birth; F: father; M: mother; WT: wild type; L: left; R: right; ND – not determined.
Figure 1. Genetic information on 5 subjects carrying NR5A1 mutations.

A. Family trees of 4 families and 7 affected individuals (5 patients and 2 parents) are shown. Scheme of the NR5A1 gene showing the mutations identified in the reported patients (above the scheme) and all reported SF-1 mutations causing adrenal insufficiency (below the scheme). Electropherograms of novel mutations are also depicted. The NR5A1 gene is composed of coding (black) and non-coding sequences (gray). Exons are indicated by numbers.
Case reports (Table 1 and 2, Figure 1, Table S1)
Table 2. Weight related parameters of subjects carrying (heterozygote) NR5A1 mutations.
| Subject | Karyotype | NR5A1 mutation(s) | Gestationalage at birth | Birth weight | Current age | Actual weight | BMI | |||||
| (weeks) | (g) | (SD) | (y, m) | (kg) | (SD) | (kg/m2) | (SD) | |||||
| Present study ( Figure 1 ) | ||||||||||||
| F1, II.1 | 46,XY | Glu7Stop;Thr296Met | 31 | 1430 | −0.83 | 3 y 1 m | 16.30 | 0.59 | 17.80 | 1.39 | ||
| F2, II.1 | 46,XY | His408Profs*159 | 40 | 3200 | −0.76 | - | - | - | - | - | ||
| F3, II.1 | 46,XY | Arg313Cys | 38 | 2630 | −1.77 | - | - | - | - | - | ||
| F4, II.1 | 46,XY | Arg313Cys | 36 | 2470 | −0.75 | 1 y 10 m | 12.70 | 0.30 | 15.30 | −0.26 | ||
| F4, II.2 | 46,XY | Arg313Cys | 36 | 1390 | −4.87 | 1 y 10 m | 13.00 | 0.57 | 15.40 | −0.47 | ||
| F2, I.1_Fa | 46,XY | His408Profs*159 | - | - | - | 32 y | 80.00 | - | 27.00 | 0.50 | ||
| F4, I.1_Fa | 46,XY | Arg313Cys | - | - | - | 30 y | 90.00 | - | 26.60 | 0.40 | ||
| Patients from Camats et al. ( Table 1 , Ref. 11) | ||||||||||||
| 1 | 46,XY | Val20Leu | 41 | 2970 | −1.90 | 5 y | 21.80 | 0.80 | 15.30 | 0 | ||
| 2 | 46,XY | His24Tyr | 40 | 3600 | 0 | 17 y | 85.00 | - | 25.70 | 1.33 | ||
| 5 | 46,XY | Gly90Arg | 40 | 3100 | −1.28 | 14 y 6 m | 51.40 | 0 | 19.70 | 0.04 | ||
| 6 | 46,XY | Pro130ArgfsX165 | - | - | - | 1 y | 8.90 | −1.15 | 15.90 | −0.69 | ||
| 7 | 46,XY | Gln206ThrfsX20 | 41 | 3820 | 1.21 | 5 y | 26.30 | 2.90 | 21.70 | 2.81 | ||
| 8 | 46,XY | Leu231_Leu233dup | 40 | 3280 | −0.67 | 5.5 y | 28.00 | 2.85 | 21.40 | 2.45 | ||
| 9 | 46,XX | Pro235Leu | - | - | - | 19 y | 58.00 | - | 24.00 | 0.60 | ||
| 1_Fa | 46,XY | Val20Leu | - | - | - | 32 y | 71.00 | - | 24.00 | 0.84 | ||
| 5_Mo | 46,XX | Gly90Arg | - | - | - | 35 y | 63.00 | - | 23.40 | 0.76 | ||
F: family; Fa: father; Mo: mother.
Patient 1 from Czech Republic, was delivered at 31 weeks gestation because of HELLP (hemolysis, elevated liver enzymes, low platelets) syndrome of the mother. Birth weight was 1430 g (5–10th percentile) and length was 38 cm (5–10th percentile). Physical exam revealed perineal hypospadias but no other anomalies. Karyotype was 46,XY. Neonatal period was unremarkable. However, at the age of three months, he was admitted for adrenal failure (hyponatremia, hyperkalemia, episode of hypoglycemia, dehydration) in the course of an acute, viral respiratory infection. Baseline levels of ACTH and plasma renin activity were elevated. Cortisol response to ACTH stimulation was low confirming adrenal insufficiency.
Patient 2 from Spain (with parents from Argentina of European descents) was investigated during fetal development because of a discordant genital sex by ultrasound (female) compared to the genetic sex (46,XY). He was conceived by ICSI from both parents gametes. Owing to the father‘s history of hypospadias, the NR5A1 gene was analysed in the father showing an heterozygous mutation. The same mutation was detected in fetal material. Although there was no consanguinity, the mother was also analysed to predict possible compound heterozygocity or homozygocity. The patient was delivered at 40 weeks gestation with a normal weight and length. External genitalia showed a clitoris-like genital tubercle, posterior labial fusion, no visible urethral meatus nor vaginal opening. Small (<1 ml) gonads were palpable in the genital folds. Male sex was assigned. The neonatal period was uneventful and endocrine evaluation was not performed until the age of 3.5 months when baseline ACTH, cortisol and 17-hydroxy-progesterone (17OHP) were normal, while baseline testosterone (T) was low and FSH high for age; T response to hCG stimulation was low. Therefore, at 4 months of age, gender was reassigned to female due to the severely feminized external genitalia.
Patient 3 from Spain was delivered at 38 weeks gestation with normal weight and length; he was evaluated at 14 days due to ambiguous genitalia (scrotal hypospadias, penis length 2 cm and gonads palpable in the scrotal folds). Karyotype was 46,XY. Baseline cortisol and aldosterone were normal as were the measured steroid precursors. Baseline FSH was slightly elevated, AMH and inhibin B were low. T response to hCG stimulation was low while the T/DHT ratio was normal.
Patients 4 and 5 from Spain were delivered as bichorionic twins at 36 weeks gestation with the 1st twin showing normal weight and lenght while the 2nd was small for gestational age (SGA). They were obtained by ICSI from the father’s sperm and a donated ova to avoid the mother’s genetic disease (epidermiolysis bullosa). Both babies presented with ambiguous genitalia: scrotal hypospadias with a penis length of 1.5 cm, both gonads palpable (1 ml) in the scrotal folds (1st twin) and unilateral cryptorchidism and one palpable gonad (0.5 ml in the 2nd twin). The father had been operated for hypospadias during childhood. His testes biopsies at 9 years of age revealed diminished seminiferous tubule diameter and fertility index in the left testis while these parameters were normal in the right testis). Neonatal period was uneventful in both twins. At birth (2 days), baseline T, precursors and gonadotropins were normal for age and AMH was low at 2 months.
Genetic analyses
Genomic DNA was isolated from peripheral blood leukocytes. All exons and part of adjacent introns of the NR5A1 gene were amplified and sequenced as previously described [11]. Obtained sequences were analysed against the NR5A1/SF-1 GenBank entries NT_008470.19 (genomic DNA), NM_004959.4 (mRNA) and NP_004950.2 (protein).
In vitro functional studies
Human embryonic kidney cells (HEK293) and human placental choriocarcinoma cells (JEG3) were used for functional studies. HEK293 cells were cultured in DMEM supplemented with 10% fetal calf serum, 1% penicillin/streptomycin and 1% sodium pyruvate (Gibco, Paisley, UK). JEG3 cells were cultured in MEM supplemented with 10% fetal calf serum, 1% penicillin/streptomycin and 1% L-glutamine (Gibco).
Promoter luciferase reporter vectors for steroidogenic enzymes HSD3B2, CYP11A1, CYP17A1 (-227CYP17A1_Δluc, -152CYP11A1_pGL3, -301HSD3B2_pGL3) and corresponding empty control vectors (Δ_luc, pGL3), as well as cDNA for wild-type (WT) SF-1/NR5A1 were available from previous work [35], [36]. Luciferase reporter vectors for human promoters I, IV, V and VII of the BDNF gene (pGL4.15_hBDNFpI, pGL4.15_hBDNFpIV, pGL4.15_hBDNFpV, pGL4.15_hBDNFpVII) were kindly provided by Dr. P. Pruunsild and Prof. T. Timmusk (Tallinn University of Technology, Tallinn, Estonia). Mutant NR5A1 expression vectors (c.19T, c.887T, c.937T, c.1222_1223insC) and SF-1 cis element mutant pGL4.15_hBDNFpI (c.-876_873CTTT) were generated by PCR-based site-directed mutagenesis using specific primers (available upon request) following the QuickChange protocol by Stratagene (Agilent Technologies Inc., Santa Clara, CA, USA).
For promoter activity studies, cells were cultured in 24-well plates and transiently transfected with WT or mutant NR5A1 and WT or mutant promoter luciferase reporter constructs using Lipofectamine 2000 (Invitrogen, AG, Basel, Switzerland) for 48 hours, and the Dual-Luciferase Reporter (DLR) Assay System (Promega AG, Wallisellen, Switzerland) was used for readout as previously described [11]. Specific Firefly luciferase readings were normalized against Renilla control readings and expressed as relative light units (RLU). Experiments were repeated at least 3 times in duplicates.
Statistical analysis
Data are shown as mean±SEM of at least three independent experiments. Statistical analysis was examined by t-test with Microsoft Office Excel (Windows 2003, Microsoft Inc.). Significance was set at *p<0.05, **p<0.01.
Results
Genetic analysis
We identified 2 novel and 2 known NR5A1 sequence variations in 5 patients newly diagnosed with SF-1 deficiency (Figure 1). Patient 1 (family 1), a boy with adrenal insufficiency and 46,XY DSD harbored compound heterozygote novel c.19G>T and c.887C>T variations in the NR5A1 gene coding for Glu7Stop in the DNA binding domain and Thr296Met in the ligand binding domain. Glu7Stop was not found in either parents, thus qualifying as a de novo mutation. By contrast, heterozygocity for Thr296Met was detected in the healthy father (Figure 1). Patient 2 (family 2) was heterozygote for a novel insertion c.1222_1223insC which is predicted to result in a frameshift elongating the SF-1 protein by 159 amino acids (His408Profs*159). The mutation was inherited from an affected carrier father (Figure 1). In patients 3 to 5 from two families (3 and 4), a heterozygote c.937C>T mutation in exon 5 was found changing Arg313Cys in the ligand-binding domain (Table 1, Figure 1). In family 3, the mutation was de novo (patient 3), whereas in family 4 (patients 4 and 5), the mutation was inherited in two dizygotic twins from an affected carrier father (Table 1, Figure 1). This mutation was recently reported in a patient manifesting with distal hypospadias and a bifid scrotum containing testes [37].
In vitro studies of the impact of SF-1 variants on steroidogenesis and energy balance
Impact of identified SF-1 mutations on steroidogenesis was studied in non-steroidogenic HEK293 cells by assessing their transcriptional activity on the promoters of the HSD3B2, CYP11A1 and CYP17A1 genes (Figure 2). The Glu7Stop mutation revealed a complete lack of transcriptional activation for all promoter constructs. The Thr296Met variant showed similar transactivation activity as WT SF-1 indicating that it is not a disease-causing variant but rather a polymorphism (SNP). Interestingly, the His408Profs*159 and Arg313Cys mutations showed normal activity when tested on the HSD3B2 promoter construct, but their transactivation activity was significantly decreased when tested on the CYP11A1 and CYP17A1 promoters.
Figure 2. Promoter reporter studies for reported NR5A1 mutations.
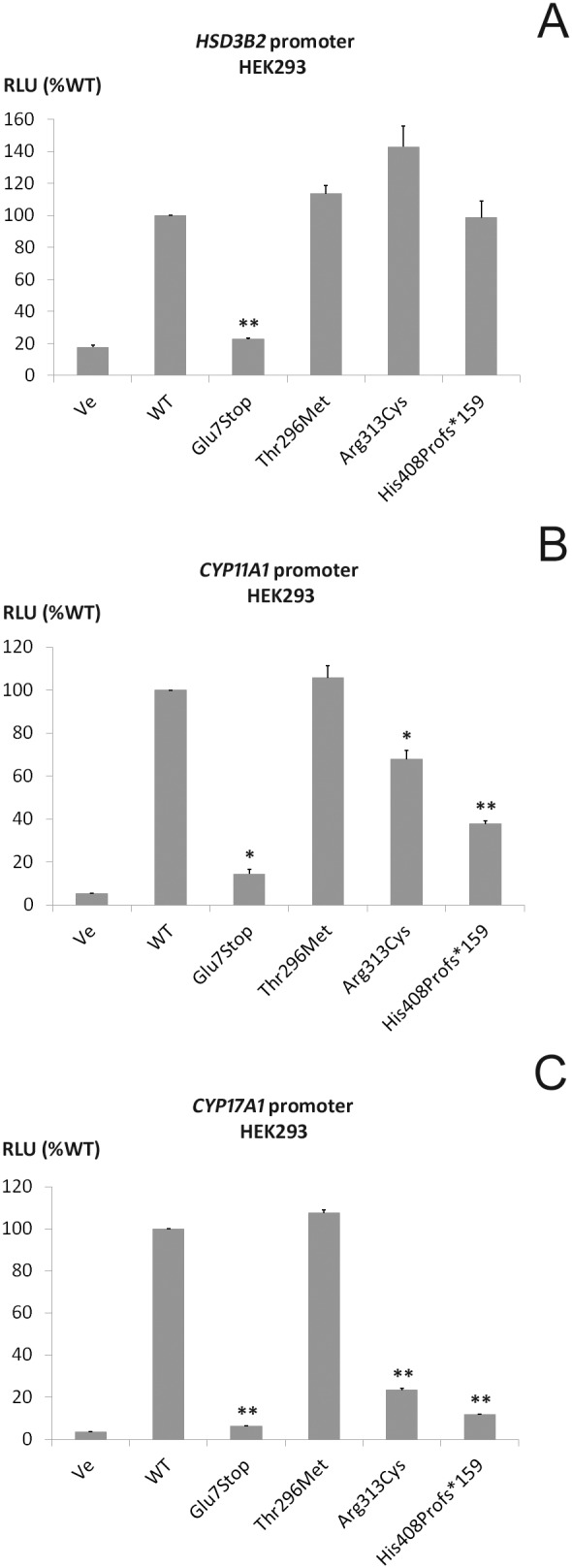
. Human embryonic kidney HEK293 cells were transiently transfected with wild-type (WT) or mutant SF-1 and promoter luciferase reporter constructs of the genes for steroidogenic enzymes HSD3B2 (A), CYP11A1 (B), CYP17A1 (C). Luciferase activity was measured with the Promega Dual Luciferase assay system. Results are expressed as percentage of WT SF-1 activity. Independent experiments were performed in duplicate at least 3 times. Error bars represent the mean and SEM. *p<0.05; **p<0.01.
To study potential involvement of SF-1 on central energy balance, the transcriptional regulation of SF-1 on the promoters of the BDNF gene was assessed in JEG3 cells. According to literature, there are many alternative BDNF variants (17 BDNF and 12 antisense BDNF variants) in human due to use of different promoters [28]. For initial experiments, we used four different human BDNF promoter constructs, namely hBDNF I, IV, V and VII. Promoter constructs I and IV were chosen as mouse Bdnf transcripts I and IV are primarily expressed in the brain (area of VMH) and their promoters are reported to be regulated by Sf-1 [27]. Similarly, promoters V and VII were assessed for reported expression in the human brain [28]. In our JEG3 cell system, among those promoters, we were only able to transactivate the hBDNF promoter I by WT SF-1 (data not shown). Thus, further studies involving SF-1 and human BDNF were performed with this hBDNF promoter I (Figure 3). In this promoter, we found a putative SF-1 cis-element at c.-874 to -867 (CAAGGACA). To confirm that this cis-element in the hBDNF promoter is regulated by SF-1, we constructed a promoter with a mutant SF-1 site and assessed its activity by co-transfection with WT SF-1. Upon co-transfection with WT SF-1, the mutant BDNF promoter lost activity when compared with the WT promoter (Figure 3A). In this system, SF-1 mutants Glu7Stop, Arg313Cys and His408Profs*159 showed only weak transactivation activity compared to WT SF-1 confirming a possible effect of SF-1 on BDNF and thus energy balance. By contrast, the Thr296Met sequence variation had similar transactivation power on the hBNDF promoter I as seen with WT SF-1 (Figure 3B).
Figure 3. Promoter reporter studies for SF-1 regulating the human BDNF promoter I and comparative studies of specific NR5A1 mutations on BDNF promoter activity.

A consensus SF-1 transcription binding site was identified in the wild-type (WT) human BDNF promoter I. This cis-element was mutated to assess the role of SF-1 on BDNF transcription. Human placental JEG3 cells were transiently transfected with the WT or the SF-1 element mutant (Mt) BDNF promoter reporter with or without SF-1, and promoter activity was assessed by the Promega Dual Luciferase assay (A). After showing that SF-1 regulates the WT BDNF promoter I specifically (A), the ability of specific SF-1 mutations to trans-activate the BDNF promoter I was investigated (B). Results are expressed as percentage of WT SF-1 activity. Independent experiments were performed in duplicate 3 times. Error bars represent the mean and SEM. *p<0.05; **p<0.01.
Weight related parameters of patients harboring SF-1 mutations
To address the question whether SF-1 deficiency may have metabolic consequences, we collected clinical data from our cohort of patients with SF-1 mutations. We were able to obtain data from 16 subjects with heterozygote SF-1 mutations including patients and their (affected) relatives (Table 2). Birth weight in singletons was normal (n = 8; median −0.83 SD, range −1.9 to 1.21). BMI of subjects currently being 1–17 years of age was also normal (n = 9; median 0.04 SD, range −0.69 to 2.81), as was BMI of 5 adults (median 0.6 SD, range 0.4 to 0.84). Thus in our small cohort of rather young patients with heterozygote SF-1 mutations overweight or obesity seems not an issue.
Discussion
In this study, two novel SF-1 mutations (Glu7Stop, His408Profs*159) were identified in 5 patients with SF-1 deficiency, all manifesting with 46,XY DSD and one (Glu7Stop) presenting with adrenal insufficiency that is rarely associated with SF-1 mutations. The disease causing impact of the identified mutations was confirmed by functional studies in cell models assessing transcriptional activity of wild-type and mutant SF-1 on genes involved in steroidogenesis (CYP11A1, CYP17A1, HSD3B2) and central energy balance (BDNF).
To date, only three SF-1 mutations have been implicated with adrenal insufficiency (heterozygote Gly35Glu and Arg255Leu, homozygote Arg92Gln) [10], [15], [17]. With patient 1, we add a novel SF-1 mutation (Glu7Stop) to this series. Our in vitro studies suggest that Glu7Stop is a loss of function SF-1 mutation. By contrast, the second SF-1 sequence variation (Thr296Met) identified in patient 1 is rather a simple polymorphism. First, it does not differ in functional assays when compared to WT SF-1. Second, this variant (rs201151141) has also been detected at an allelic frequency of 0.001 in a cohort of 662 normal subjects studied for a large-scale genome sequencing project (dbSNP database, http://www.ncbi.nlm.nih.gov/projects/SNP/snp).
The novel His408Profs*159 mutation identified in the SF-1 gene in patient 2 codes for a longer protein of 567 amino acids compared to WT SF-1 (461 aa). This mutant had an impact on both the steroidogenic promoters and the BDNF promoter (Figure 2). Interestingly, this heterozygous mutation was first detected in the father when investigated because of his history of childhood hypospadias and infertility. He nevertheless fathered a child through ICSI but the fetus’s genetic and phenotypic sex was discordant. At birth the 46,XY child’s genital phenotype was almost completely feminized and hormonal work-up at 4 months of age revealed low androgens prompting female sex assignment although the mutation was the same as in the father. This illustrates again for a novel NR5A1 mutation the wide phenotypic spectrum within the same family.
The Arg313Cys mutation found in patients 3–5 in our study was recently reported in a patient with hypospadias [37]. Similar to our functional assays, Arg313Cys showed reduced transactivation on the promoters of the AMH and CYP11A1 genes [37]. Although Arg313 is located in the highly conserved helix 5 of the ligand-binding domain, this same position has also been described for an Arg313His change (c.838G>A) in males with hypospadias [38], [39]. Thus, position Arg313 of SF-1 may be a hot spot for mutations. Interestingly, this mutation appeared de novo in our family 4, while in family 5, the mutation was transmitted by an affected father.
In theory, SF-1 mutations could also have metabolic consequences for affected patients. But so far, no clinical data existed on this topic. Observations from the Sf-1 KO mice models suggest that loss of SF-1 is associated with impaired energy balance and low temperature expenditure leading to late-onset type of obesity [25]. Deletion of BDNF gene was described in obese patients with WAGR syndrome [33]. Among many other factors regulating appetite, SF-1 together with BDNF are expressed in the VMH of mice. Tran et al. described a significantly decreased expression of the Bdnf gene in Sf-1 +/− KO mice [27]. They also identified two promoter variants of the murine Bdnf gene which were specifically used in the VMH and their activity was related to SF-1 dosage [27]. In the presented study, we tried to establish the role of SF-1 in the regulation of human BDNF, an important player for central energy balance and thus obesity [40]. In fact, our experiments now show that the human BDNF promoter I is regulated by SF-1, and that SF-1 mutations have impaired transactivation activity on this promoter similar to impaired effect on promoters regulating genes of steroidogenesis. These results indicate that SF-1 might be a co-regulator of energy balance and that mutations in SF-1 may therefore also lead to metabolic consequences (e.g. obesity) in humans.
In addition to the in vitro studies, we were also able to collect some clinical data related to energy balance in 16 patients with heterozygote SF-1 mutations (Table 2). However, in our small cohort of 9 patients aged 1–17 years and 5 adults, we did not find weights and BMIs consistent with overweight or obesity. Therefore, presented clinical data in humans are not in line with data found in mice [25], [26], although in vitro data in human and mouse are similar [25], [27]. These negative results might be ‘true’ negative results or may be explained by the following shortcomings of our presented study. First, small number of studied patients, which are in addition too young to observe metabolic consequences. Second, all patients are heterozygote for their SF-1 mutation and carry one wild-type SF-1 allele while most studied mice were Sf-1 complete KO. Further collaborative studies are therefore needed to gather clinical data of more patients and follow-up on a bigger cohort longitudinally.
Supporting Information
Biochemical data of patients included in this study. Values outside the age- and sex-specific reference range are given in bold. 1PRA: plasma renin activity; 2AMH: anti-Müllerian hormone; 3Synacthen (250 µg/1.73 m2 BSA); 4hCG: 600 IU/48 h×6; 5hCG: 1000 IU/24 h×3.
(DOC)
Data Availability
The authors confirm that all data underlying the findings are fully available without restriction. All relevant data are within the paper and its Supporting Information files.
Funding Statement
This work was supported by grants of the Swiss National Science Foundation (320030-146127) to CEF, the Instituto de Salud Carlos III, Madrid, Spain CIBERER U-712 to MFC and the AGAUR (University and Research Management and Evaluation Agency), Barcelona, Spain (2009SGR31) to LA, and by ESPE (European Society of Pediatric Endocrinology) Research Fellowship grants to JM and NC (sponsored by Novo Nordisk A/S). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Hoivik EA, Lewis AE, Aumo L, Bakke M (2010) Molecular aspects of steroidogenic factor 1 (SF-1). Mol Cell Endocrinol 315: 27–39. [DOI] [PubMed] [Google Scholar]
- 2. Ferraz-de-Souza B, Lin L, Achermann JC (2011) Steroidogenic factor-1 (SF-1, NR5A1) and human disease. Mol Cell Endocrinol 336: 198–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Tran PV, Lee MB, Marin O, Xu B, Jones KR, et al. (2003) Requirement of the orphan nuclear receptor SF-1 in terminal differentiation of ventromedial hypothalamic neurons. Mol Cell Neurosci 22: 441–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Zhao L, Bakke M, Krimkevich Y, Cushman LJ, Parlow AF, et al. (2001) Steroidogenic factor 1 (SF1) is essential for pituitary gonadotrope function. Development 128: 147–154. [DOI] [PubMed] [Google Scholar]
- 5. Morohashi K, Tsuboi-Asai H, Matsushita S, Suda M, Nakashima M, et al. (1999) Structural and functional abnormalities in the spleen of an mFtz-F1 gene-disrupted mouse. Blood 93: 1586–1594. [PubMed] [Google Scholar]
- 6. Patel MV, McKay IA, Burrin JM (2001) Transcriptional regulators of steroidogenesis, DAX-1 and SF-1, are expressed in human skin. J Invest Dermatol 117: 1559–1565. [DOI] [PubMed] [Google Scholar]
- 7. Ramayya MS, Zhou J, Kino T, Segars JH, Bondy CA, et al. (1997) Steroidogenic factor 1 messenger ribonucleic acid expression in steroidogenic and nonsteroidogenic human tissues: Northern blot and in situ hybridization studies. J Clin Endocrinol Metab 82: 1799–1806. [DOI] [PubMed] [Google Scholar]
- 8. Lala DS, Rice DA, Parker KL (1992) Steroidogenic factor I, a key regulator of steroidogenic enzyme expression, is the mouse homolog of fushi tarazu-factor I. Mol Endocrinol. 6: 1249–1258. [DOI] [PubMed] [Google Scholar]
- 9. Sadovsky Y, Crawford PA, Woodson KG, Polish JA, Clements MA, et al. (1995) Mice deficient in the orphan receptor steroidogenic factor 1 lack adrenal glands and gonads but express P450 side-chain-cleavage enzyme in the placenta and have normal embryonic serum levels of corticosteroids. Proc Natl Acad Sci U S A 92: 10939–10943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Achermann JC, Ito M, Hindmarsh PC, Jameson JL (1999) A mutation in the gene encoding steroidogenic factor-1 causes XY sex reversal and adrenal failure in humans. Nat Genet 22: 125–126. [DOI] [PubMed] [Google Scholar]
- 11. Camats N, Pandey AV, Fernandez-Cancio M, Andaluz P, Janner M, et al. (2012) Ten novel mutations in the NR5A1 gene cause disordered sex development in 46,XY and ovarian insufficiency in 46,XX individuals. J Clin Endocrinol Metab 97: E1294–1306. [DOI] [PubMed] [Google Scholar]
- 12. Sarafoglou K, Ahmed SF (2012) Disorders of sex development: challenges for the future. J Clin Endocrinol Metab 97: 2292–2294. [DOI] [PubMed] [Google Scholar]
- 13. Taketo M, Parker KL, Howard TA, Tsukiyama T, Wong M, et al. (1995) Homologs of Drosophila Fushi-Tarazu factor 1 map to mouse chromosome 2 and human chromosome 9q33. Genomics 25: 565–567. [DOI] [PubMed] [Google Scholar]
- 14. Kohler B, Lin L, Ferraz-de-Souza B, Wieacker P, Heidemann P, et al. (2008) Five novel mutations in steroidogenic factor 1 (SF1, NR5A1) in 46,XY patients with severe underandrogenization but without adrenal insufficiency. Hum Mutat 29: 59–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Achermann JC, Ozisik G, Ito M, Orun UA, Harmanci K, et al. (2002) Gonadal determination and adrenal development are regulated by the orphan nuclear receptor steroidogenic factor-1, in a dose-dependent manner. J Clin Endocrinol Metab 87: 1829–1833. [DOI] [PubMed] [Google Scholar]
- 16. Bashamboo A, Ferraz-de-Souza B, Lourenco D, Lin L, Sebire NJ, et al. (2010) Human male infertility associated with mutations in NR5A1 encoding steroidogenic factor 1. Am J Hum Genet 87: 505–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Biason-Lauber A, Schoenle EJ (2000) Apparently normal ovarian differentiation in a prepubertal girl with transcriptionally inactive steroidogenic factor 1 (NR5A1/SF-1) and adrenocortical insufficiency. Am J Hum Genet 67: 1563–1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lin L, Achermann JC (2008) Steroidogenic factor-1 (SF-1, Ad4BP, NR5A1) and disorders of testis development. Sex Dev 2: 200–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lourenco D, Brauner R, Lin L, De Perdigo A, Weryha G, et al. (2009) Mutations in NR5A1 associated with ovarian insufficiency. N Engl J Med 360: 1200–1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Majdic G, Young M, Gomez-Sanchez E, Anderson P, Szczepaniak LS, et al. (2002) Knockout mice lacking steroidogenic factor 1 are a novel genetic model of hypothalamic obesity. Endocrinology 143: 607–614. [DOI] [PubMed] [Google Scholar]
- 21. Jacobs SB, Coss D, McGillivray SM, Mellon PL (2003) Nuclear factor Y and steroidogenic factor 1 physically and functionally interact to contribute to cell-specific expression of the mouse Follicle-stimulating hormone-beta gene. Mol Endocrinol 17: 1470–1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Halvorson LM, Kaiser UB, Chin WW (1996) Stimulation of luteinizing hormone beta gene promoter activity by the orphan nuclear receptor, steroidogenic factor-1. J Biol Chem 271: 6645–6650. [DOI] [PubMed] [Google Scholar]
- 23. Ikeda Y, Luo X, Abbud R, Nilson JH, Parker KL (1995) The nuclear receptor steroidogenic factor 1 is essential for the formation of the ventromedial hypothalamic nucleus. Mol Endocrinol 9: 478–486. [DOI] [PubMed] [Google Scholar]
- 24. Schimmer BP, White PC (2010) Minireview: steroidogenic factor 1: its roles in differentiation, development, and disease. Mol Endocrinol 24: 1322–1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kim KW, Zhao L, Donato J Jr, Kohno D, Xu Y, et al. (2011) Steroidogenic factor 1 directs programs regulating diet-induced thermogenesis and leptin action in the ventral medial hypothalamic nucleus. Proc Natl Acad Sci U S A 108: 10673–10678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Unger TJ, Calderon GA, Bradley LC, Sena-Esteves M, Rios M (2007) Selective deletion of Bdnf in the ventromedial and dorsomedial hypothalamus of adult mice results in hyperphagic behavior and obesity. J Neurosci 27: 14265–14274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tran PV, Akana SF, Malkovska I, Dallman MF, Parada LF, et al. (2006) Diminished hypothalamic bdnf expression and impaired VMH function are associated with reduced SF-1 gene dosage. J Comp Neurol 498: 637–648. [DOI] [PubMed] [Google Scholar]
- 28. Pruunsild P, Kazantseva A, Aid T, Palm K, Timmusk T (2007) Dissecting the human BDNF locus: bidirectional transcription, complex splicing, and multiple promoters. Genomics 90: 397–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kernie SG, Liebl DJ, Parada LF (2000) BDNF regulates eating behavior and locomotor activity in mice. EMBO J 19: 1290–1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rios M, Fan G, Fekete C, Kelly J, Bates B, et al. (2001) Conditional deletion of brain-derived neurotrophic factor in the postnatal brain leads to obesity and hyperactivity. Mol Endocrinol 15: 1748–1757. [DOI] [PubMed] [Google Scholar]
- 31. Stranahan AM, Arumugam TV, Mattson MP (2011) Lowering corticosterone levels reinstates hippocampal brain-derived neurotropic factor and Trkb expression without influencing deficits in hypothalamic brain-derived neurotropic factor expression in leptin receptor-deficient mice. Neuroendocrinology 93: 58–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kohjima M, Sun Y, Chan L (2010) Increased food intake leads to obesity and insulin resistance in the tg2576 Alzheimer’s disease mouse model. Endocrinology 151: 1532–1540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Han JC, Liu QR, Jones M, Levinn RL, Menzie CM, et al. (2008) Brain-derived neurotrophic factor and obesity in the WAGR syndrome. N Engl J Med 359: 918–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gray J, Yeo GS, Cox JJ, Morton J, Adlam AL, et al. (2006) Hyperphagia, severe obesity, impaired cognitive function, and hyperactivity associated with functional loss of one copy of the brain-derived neurotrophic factor (BDNF) gene. Diabetes 55: 3366–3371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Huang N, Miller WL (2000) Cloning of factors related to HIV-inducible LBP proteins that regulate steroidogenic factor-1-independent human placental transcription of the cholesterol side-chain cleavage enzyme, P450scc. J Biol Chem 275: 2852–2858. [DOI] [PubMed] [Google Scholar]
- 36. Fluck CE, Miller WL (2004) GATA-4 and GATA-6 modulate tissue-specific transcription of the human gene for P450c17 by direct interaction with Sp1. Mol Endocrinol 18: 1144–1157. [DOI] [PubMed] [Google Scholar]
- 37. Allali S, Muller JB, Brauner R, Lourenco D, Boudjenah R, et al. (2011) Mutation analysis of NR5A1 encoding steroidogenic factor 1 in 77 patients with 46, XY disorders of sex development (DSD) including hypospadias. PLoS One 6: e24117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ciaccio M, Costanzo M, Guercio G, De Dona V, Marino R, et al. (2012) Preserved fertility in a patient with a 46,XY disorder of sex development due to a new heterozygous mutation in the NR5A1/SF-1 gene: evidence of 46,XY and 46,XX gonadal dysgenesis phenotype variability in multiple members of an affected kindred. Horm Res Paediatr 78: 119–126. [DOI] [PubMed] [Google Scholar]
- 39. Adamovic T, Chen Y, Thai HT, Zhang X, Markljung E, et al. (2012) The p.G146A and p.P125P polymorphisms in the steroidogenic factor-1 (SF-1) gene do not affect the risk for hypospadias in Caucasians. Sex Dev 6: 292–297. [DOI] [PubMed] [Google Scholar]
- 40. Rios M (2013) BDNF and the central control of feeding: accidental bystander or essential player? Trends Neurosci 36: 83–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Biochemical data of patients included in this study. Values outside the age- and sex-specific reference range are given in bold. 1PRA: plasma renin activity; 2AMH: anti-Müllerian hormone; 3Synacthen (250 µg/1.73 m2 BSA); 4hCG: 600 IU/48 h×6; 5hCG: 1000 IU/24 h×3.
(DOC)
Data Availability Statement
The authors confirm that all data underlying the findings are fully available without restriction. All relevant data are within the paper and its Supporting Information files.