

Abstract
Chronic electrical activation of the carotid baroreflex produces sustained reductions in sympathetic activity and arterial pressure and is currently being evaluated as antihypertensive therapy for patients with resistant hypertension. However, the influence of variations in salt intake on blood pressure lowering during baroreflex activation has not been determined. As sensitivity of arterial pressure to salt intake is linked to the responsiveness of renin secretion, we determined steady-state levels of arterial pressure and neurohormonal responses in 6 dogs on low, normal, and high salt intakes ( 5, 40, 450 mmol/day, respectively) under control conditions and during a 7-day constant level of baroreflex activation. Under control conditions, there was no difference in mean arterial pressure at low (92±1) and normal (92±2 mmHg) sodium intakes, but pressure increased 9 ±2 mmHg during high salt. Plasma renin activity (2.01±0.23, 0.93±0.20, 0.01±0.01 ng ANGI/mL/hr) and plasma aldosterone (10.3±1.9, 3.5±0.5, 1.7±0.1ng/dL) were inversely related to salt intake, whereas there were no changes in plasma norepinephrine. Although mean arterial pressure (19-22 mmHg) and norepinephrine (20-40%) were lower at all salt intakes during baroreflex activation, neither the changes in pressure nor the absolute values for plasma renin activity or aldosterone in response to salt were different from control conditions. These findings demonstrate that suppression of sympathetic activity by baroreflex activation lowers arterial pressure without increasing renin release and indicate that changes in sympathetic activity are not primary mediators of the effect of salt on renin secretion. Consequently, blood pressure lowering during baroreflex activation is independent of salt intake.
Keywords: arterial pressure, baroreflex, salt intake, salt sensitivity, sympathetic nervous system, renin-angiotensin-aldosterone system, renal nerves
Introduction
Recent technology for chronic electrical activation of the carotid baroreflex has provided a non-pharmacological approach for the treatment of resistant hypertension and, at the same time, a unique experimental tool for determining the mechanisms that account for the lowering of arterial pressure during chronic suppression of central sympathetic outflow.1-2 One unexamined aspect of baroreflex activation (BA) therapy is the influence of changes in salt intake on blood pressure lowering during BA. This is a matter of importance to BA therapy because many patients with resistant hypertension have inappropriate volume expansion that has been managed to varying degrees of success using diuretic therapy and judicious consumption of dietary salt.3-6 Moreover, this issue raises an even more fundamental unresolved question, which is the focus of this study: whether neural mechanisms make an important contribution to the normal regulation of arterial pressure during variations in salt intake. Because electrical stimulation of the carotid sinus has sustained effects to suppress sympathetic activity and, therefore, disrupt the normal physiological changes in autonomic activity to the peripheral circulation, BA provides a unique approach to address this basic question.7-8 Elucidation of the importance of neural mechanisms in the normal regulation of arterial pressure during changes in salt intake may have relevance to BA therapy in the more complex disorder of resistant hypertension.
The renin-angiotensin system is a powerful hormonal system for controlling sodium excretion and plays a major role in regulating body fluid volumes and arterial pressure. Normally, chronic changes in salt intake have relatively little influence on arterial pressure because the activity of the renin-angiotensin system changes inversely with the amount of salt ingested.9-10 In contrast, when plasma levels of angiotensin are maintained at inappropriately elevated or suppressed levels, arterial pressure varies directly with the level of salt intake.9-10 That is, arterial pressure becomes salt sensitive.
It is well established that there is an important interaction between the renal nerves and nonneural mechanisms in the control of renin secretion.11-17 The renal nerves have a tonic stimulatory effect on renin secretion and blockade of β-adrenergic receptors decreases basal levels of renin secretion.13-15,18-19 Furthermore, renal denervation attenuates the increase in renin secretion seen in response to both reductions in sodium intake and in response to acute and chronic reductions in renal perfusion pressure.11-13 However, the quantitative importance of the interactions between neural and nonneural mechanisms in the control of renin secretion and arterial pressure during complex physiological disturbances such as changes in salt intake is difficult to discern and is unresolved.
Based on the above, it is plausible that if suppression of sympathetic activity does impair the ability of the renin-angiotensin system to respond normally to changes in salt intake, then blood pressure would be expected to be salt sensitive during BA. On the other hand, if suppression of sympathetic activity does not influence the mechanisms that normally control renin release during variations in sodium intake, then normal salt-induced changes in blood pressure would likely occur with BA. If this is correct, we hypothesize that increased sodium sensitivity is not expected during BA, although arterial pressure would be lower at all levels of sodium intake. This hypothesis was tested by determining steady-state levels of arterial pressure and the activity of the renin-angiotensin-aldosterone system in response to chronic variations in salt intake under control conditions and during a constant degree of electrical activation of the afferent limb of the carotid baroreflex.
Methods
Animal Preparation
All experimental protocols were performed according to the “Guide for the Care and Use of Laboratory Animals” from the National Institutes of Health and approved by the University of Mississippi Medical Center Institutional Animal Care and Use Committee. Surgical procedures were conducted under isoflurane anesthesia (1.5% to 2.0%) after premedication with acepromazine (0.15 mg/kg, SC) and induction with thiopental (10 mg/kg, IV). Carprofen (Ramadyl), 4 mg/kg, was administered for 3 days postoperatively for analgesia.
Experiments were conducted in 6 chronically instrumented mongrel dogs weighing 22 to 25 kg. The procedures for implantation of vascular catheters in the aorta and vena cava, and implantation of stimulating electrodes around each carotid sinus have been described previously.7-8,20-21 The electrodes and the pulse generator for electrical stimulation of the carotid sinus were provided by CVRx, Inc. (Minneapolis, MN).
Experimental Protocol
Following recovery from surgery the dogs were fitted with a specially designed harness and maintained in metabolic cages as previously reported.7-8,20-22 During a 3-4 week postoperative period and throughout the study, the dogs were given free access to water and maintained on a fixed daily diet of two 15.5 oz. cans of prescription heart diet (H/D; Hill's Pet Products) supplemented with 5 ml of vitamin syrup. Two cans of H/D provide ∼5 mmol of sodium and ∼50 mmol of potassium. Additionally, the dogs received a continuous intravenous infusion of isotonic saline at a rate of 300 mL/day, thus providing a total daily sodium intake of ∼45 mmol. During the postoperative period the dogs were trained to lie quietly in their cages each morning to allow blood sampling under resting conditions. Throughout the study, arterial pressure and heart rate were measured continuously.
After the training period, sodium intake was maintained for 3 weeks at each of the following levels: ∼5, 45, and 450 mmol/day. This was achieved by continuous intravenous infusion of 250 mL 5% dextrose, 250 mL isotonic saline, or 3000 mL isotonic saline/day, respectively. The 3 levels of salt intake were randomized and during week 2 at each salt intake, the carotid sinus was continuously stimulated for 7 days before discontinuing baroreflex activation and initiating a 7 day recovery period. The pre-BA, BA, and recovery periods were maintained for 7 days at each salt intake to achieve stable neurohormonal and hemodynamics responses, and balance between sodium intake and urinary sodium excretion.
For the 7 days of BA, the pulse generator was programmed to deliver continuous impulses using the following parameters: 3-7 V, 30 Hz, and 0.5 msec pulse duration. The intensity of activation was selected by adjusting the voltage to achieve a chronic decrease in MAP of ∼20 mmHg. To achieve this goal, small adjustments in voltage were needed during the first 24-48 hours, but no changes in the intensity of activation were made after the first 48 hours of stimulation.
On the last 2 days of the pre-BA, BA, and recovery periods, arterial blood samples (∼10 ml) were taken while the dogs were recumbent and in a resting state. Blood samples were analyzed for hematocrit, plasma renin activity (PRA), and the plasma concentrations of aldosterone, cortisol, sodium, potassium, protein, and norepinephrine (NE). Water consumption was monitored daily and 24-h urine samples were collected between 11 AM and noon each day, at the time of feeding.
Analytical Methods
The daily hemodynamic values for mean arterial pressure (MAP) and heart rate were averaged from the 20-hour period extending from 11:30-7:30 AM. The data excluded from the 24-hour recordings comprised the time required for flushing catheters, calibrating pressure transducers, feeding, and cleaning cages. Steady-state relationships between MAP and urinary sodium excretion were plotted based on the average values determined during the last 48 hours at each level of sodium intake.
Plasma renin activity and the plasma concentrations of aldosterone and cortisol were measured by radioimmunoassay.7-8,20-21 Plasma concentrations of NE were determined by high-performance liquid chromatography (HPLC) with electrochemical detection (Agilent 1100).8,20-21 Hematocrit and the plasma concentrations of sodium, potassium, and protein were measured by standard techniques.
Statistical Analysis
Results are expressed as mean±SE. Two-way repeated measures ANOVA followed by the Bonferroni post hoc t test for multiple comparisons was used to compare LS and HS to NS, and BA to control at each level of sodium intake. Control values were considered as the average of the pre-BA and recovery periods at each sodium intake. Statistical significance was considered to be P<0.05.
Results
Arterial pressure, heart rate, and Urinary Excretory Responses
The steady-state relationships between arterial pressure and sodium excretion are illustrated in Figure 1. As reflected by the urinary excretion of sodium, sodium balance was achieved by days 6-7 at each level of sodium intake (Table). Under control conditions without BA, there was no significant difference in MAP during LS and NS. In contrast, MAP increased 9±2 mm Hg when sodium intake was increased from NS (93±2 mm Hg) to HS (102±2 mm Hg). There were no significant changes in heart rate during alterations in salt intake (Table).
Figure 1.
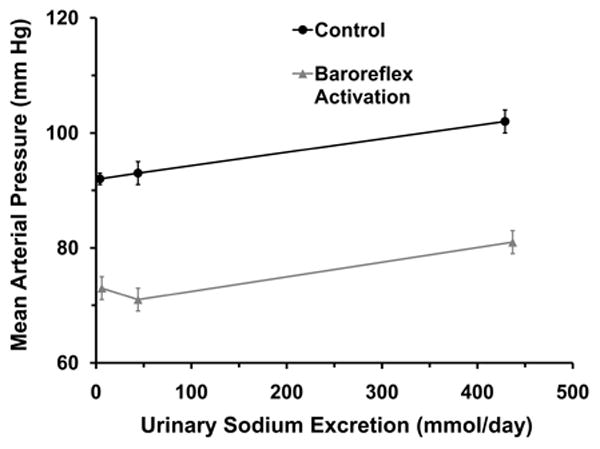
Arterial pressure responses to chronic changes in sodium intake/sodium excretion under control conditions and during chronic baroreflex activation.
Table 1. Responses to Prolonged Baroreflex Activation during Variations in Salt Intake.
| Salt Intake | UNaV (mmol/d) | HR(bpm) | [PNE] (pg/mL) | [PCORT] (μg/dL) | HCT | [PPROT] (g/dL) |
|---|---|---|---|---|---|---|
| LS (5 mmol/d) | 4±1* | 75±4 | 85±8 | 1.2±0.1 | 0.39±0.1 | 6.4±0.1 |
| LS+BA | 6±1* | 61±4† | 52±7† | 1.2±0.1 | 0.35±0.1† | 6.2±0.2 |
| NS (45 mmol/d) | 44±2 | 73±4 | 95±7 | 1.3±0.2 | 0.40±0.1 | 6.4±0.1 |
| NS+BA | 44±3 | 55±4† | 75±7† | 1.3±0.1 | 0.35±0.2† | 6.0±0.1† |
| HS (450 mmol/d) | 429±4* | 72±5 | 93±5 | 1.1±0.1 | 0.38±0.2 | 6.2±0.2 |
| HS+BA | 437±6* | 58±4† | 58±5† | 1.3±0.1 | 0.35±0.2† | 5.8±0.2† |
Values are mean±SEM (n=6). LS indicates low salt intake; NS, normal salt intake; HS, high salt intake; BA, baroreflex activation; UNaV, urinary sodium excretion; HR, heart rate; [PNE], plasma norepinephrine concentration; PCORT, plasma cortisol concentration; HCT, hematocrit, PPROT, plasma protein concentration
P<0.05 vs corresponding NS;
P<0.05 vs before BA at each salt intake
The temporal changes in MAP and sodium excretion associated with BA activation have been illustrated in several of our previous publications.7-8,20-21 As in our previous studies, modest sodium retention occurred on days 1-2 of BA before sodium balance was subsequently achieved. Similarly, significant reductions in MAP and heart rate occurred during the first 48 hours of BA before stabilizing thereafter. There were no further changes in MAP, HR, or sodium balance after day 2 of BA. During BA, steady state reductions in MAP (LS= 19±1; NS= 22±2; HS= 21±3 mm Hg) and HR (LS= 14±1; NS= 18±2; HS= 16±2 bpm) were not significantly different at any level of salt intake. Moreover, salt-dependent changes in MAP were comparable during BA and control conditions. Thus, there was a parallel shift in the relationship between salt intake/excretion and MAP during BA when compared to control conditions (Figure 1). As under control conditions, during BA there were no significant changes in heart rate with variations in salt intake.
Neurohormonal Responses
Neurohormonal responses to changes in salt intake are illustrated in Figure 2 and in the Table. Under control conditions without BA, there were no significant changes in plasma NE concentration during alterations in sodium intake. In contrast, both PRA and plasma aldosterone concentration were inversely related to salt intake, with the most pronounced changes occurring from LS to NS.
Figure 2.
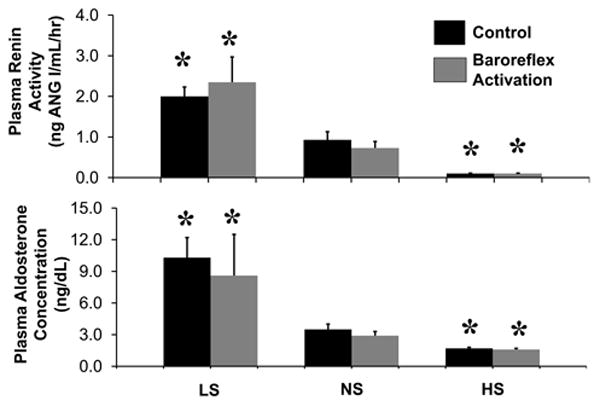
Responses of the renin-angiotensin-aldosterone system to low, normal, and high salt intakes under control conditions and during chronic baroreflex activation. Values are mean ±SEM (n=6). * P<0.05 versus normal salt.
As in our previous studies, there were sustained reductions in plasma NE concentration during BA, reflecting prolonged suppression of central sympathetic outflow.7-8,20-21 In the present study, reductions in plasma NE concentration during BA were not significantly different at any level of salt intake. In addition, as under control conditions, during BA there was an inverse relationship between salt intake and activation of the renin-angiotensin-aldosterone system. Moreover, despite substantial reductions in arterial pressure, values for PRA and plasma aldosterone concentration during BA were not significantly different from those measured under control conditions without BA (Figure 2).
Hematocrit and Plasma Concentrations of Electrolytes and Protein
Under control conditions, there were no significant changes in HCT, or in the plasma concentrations of protein and cortisol during changes in sodium intake (Table). Plasma concentrations of sodium (NS= 148±1 mmol/L) and potassium (NS= 4.2±0.1 mmol/L) were also unchanged with alterations in salt consumption. In contrast, as reported previously, concomitant with the modest fluid retention associated with BA, there were small reductions in HCT and plasma protein concentration (Table) at all salt intakes. In addition, there were no significant changes in plasma sodium, potassium, or cortisol concentration during BA.
Discussion
By maintaining constant suppression of central sympathetic outflow,8 BA provides a novel approach for assessing the role of neural mechanisms in long-term regulation of renin secretion and arterial pressure during variations in salt intake. The two significant findings from this study were the following. First, long-term MAP responses to changes in salt intake were not significantly different during BA when compared to control conditions. Second, despite appreciable steady-state reductions in MAP during BA, absolute values for PRA and plasma aldosterone concentration were similar under the two conditions at all salt levels. Taken together, these findings demonstrate that suppression of central sympathetic outflow by electrical stimulation of baroreceptor afferents lowers arterial pressure without stimulating renin release and does not impair normal salt-intake-dependent renin secretion. They further suggest that physiologic changes in sympathetic activity are not essential for the normal responsiveness of the renin-angiotensin-aldosterone system to variations in salt. Consequently, as reflected by the parallel shift in the relationship between salt intake/excretion and MAP during BA (Figure 1), lowering of blood pressure by BA does not lead to salt sensitivity. Furthermore, concomitant with appropriate suppression of the renin-angiotensin-aldosterone system under both conditions, there was little change in MAP (∼ 10mmHg) during the progressive 90-fold increase in sodium intake (from 5 to 450 mmoL/day).
Changes in activity of the renin-angiotensin system play a critical role in minimizing alterations in arterial pressure during chronic variations in salt intake.9-10 When the renin-angiotensin system is fully functional, chronic changes in salt intake have relatively little influence on arterial pressure. In contrast, when the activity of this system is impaired, arterial pressure is salt sensitive, i.e., there is an exaggerated arterial pressure response to changes in salt intake.9-10 However, the mechanisms regulating renin secretion in response to salt are not clearly defined. Determinants of renin secretion include stretch of renal baroreceptors, sodium chloride transport at the macula densa, and activation of β-adrenergic receptors on juxtaglomerular cells.13,19 Basal levels of RSNA have tonic effects to directly stimulate renin secretion by activation of β-adrenergic receptors on juxtaglomerular cells and to directly stimulate sodium reabsorption by activation of tubular α-adrenergic receptors.13-15,18-19 Because this latter effect includes sodium reabsorption in the proximal tubules and loop of Henle, neurally-mediated changes in sodium chloride delivery to the macula densa may be another potential mechanism whereby changes in RSNA influence renin secretion. Therefore, assuming that physiologically relevant changes in RSNA do occur, neural mechanisms may contribute to the chronic regulation of renin secretion during long-term variations in salt intake.
However, most insight into the importance of neural mechanisms in the control of renin secretion during changes in salt intake has come from studies employing acute intravascular expansion, which activates cardiopulmonary (low pressure) receptors and, at robust salt loads that increase arterial pressure, arterial baroreceptors as well.13,19 These studies indicate that the natriuresis and inhibition of renin secretion following acute volume expansion or activation of cardiopulmonary baroreceptors by atrial distension is mediated, at least in part, by reflex suppression of RSNA. However, because mechanoreceptors undergo adaptation and an appreciable degree of resetting, one view, although not universally accepted, is that the above cardiovascular reflexes are not long-term determinants of volume homeostasis and arterial pressure. Therefore, it is unclear whether cardiopulmonary and arterial baroreceptor-mediated suppression of RSNA plays an important role in the chronic regulation of renin secretion during variations in salt intake.
Experimental limitations have precluded elucidation of the importance of neural mechanisms in the control of renin secretion during chronic alterations in salt intake. One limitation has been the failure to faithfully determine whether changes in RSNA truly occur during variations in salt intake. In conscious animals and in human subjects, determinations of RSNA by direct nerve recordings or by measurement of renal NE spillover during variations in salt intake are inconsistent.13,22-25 These differences likely reflect the duration and magnitude of changes in salt intake and the ability to discern changes in RSNA. Another major limitation has been the inability to achieve reliable and controlled inhibition of prevailing RSNA. An alternate approach taken by Kim et al. was to subject β-adrenergic receptor-deficient mice to low, normal, and high salt intakes.26 Basal levels of plasma renin concentration in β-adrenergic receptor-deficient mice were depressed at all salt levels when compared to values in wild-type mice, consistent with observations indicating that β-adrenergic receptor blockade inhibits renin secretion at spontaneous levels of arterial pressure. Moreover, of greater significance, the normal renin responsiveness to chronic variations in salt intake was maintained, discounting the importance of changes in sympathetic activity through β-adrenergic mechanisms in mediating the effect of salt on renin secretion. Unfortunately, arterial pressure responses to salt were not examined, precluding insight into the importance of β-adrenergic control of renin secretion in the chronic regulation of arterial pressure during variations in salt intake.
In view of the limited and inconclusive observations indicated above, the current findings during BA provide novel insight into the importance of natural changes in central sympathetic outflow in the regulation of renin secretion and arterial pressure during alterations in salt intake. Constant electrical stimulation of the carotid sinus produces an unvarying degree of suppression of central sympathetic outflow, discounting the possibility that unloading of aortic baroreceptors in response to reduced arterial pressure plays a significant role in attenuating the chronic sympathoinhibition produced by BA. The contention that there is stable and sustained suppression of central sympathetic outflow during BA is based on parallel and unvarying reductions in plasma NE concentration and whole body NE spillover (an index of central sympathetic outflow) over a three week period of constant electrical stimulation of the carotid sinus.8 Therefore, because comparable baroreflex-mediated reductions in plasma NE concentration were achieved in the present study irrespective of salt intake, it is likely that suppression of central sympathetic outflow was approximately equivalent at all salt intakes.
A new and important finding in this study was that inhibition of sympathetic activity by BA did not impair the normal responsiveness of renin secretion to variations in salt intake. This finding suggests that salt-induced changes in renin secretion are not primarily dependent on physiological changes in sympathetic activity. Consequently, due to the normal responsiveness of the renin-angiotensin system during BA, the lowering of arterial pressure during BA does not lead to exaggerated changes in arterial pressure during variations in salt intake.9-10 That is, blood pressure lowering during BA is associated with a parallel shift in the relationship between sodium intake and arterial pressure (Figure 1).
This study was not designed to elucidate the nonneural mechanisms that account for changes in renin secretion in response to salt. Further studies are required to clarify these mechanisms. However, a previous study in dogs using a protocol similar to the present one emphasized the importance of progressive increases in GFR in the regulation of sodium excretion during chronic increases in sodium intake.9 Thus, one possible nonneural mechanism for regulation of renin secretion during variations in salt intake is the filtered load of sodium and the attendant sodium chloride delivery to the macula densa, with renin secretion inversely related to GFR. Hormonal mechanisms may also play a role. For example, in keeping with a possible physiological role in the regulation of renin secretion, a study in conscious dogs demonstrated that increments in plasma ANP concentration comparable to those associated with high salt intake inhibit the increase in PRA normally induced by reduced renal perfusion pressure.27-28
An additional significant finding at all levels of salt intake was that PRA did not increase above control levels during BA, despite BA producing reductions in arterial pressure (∼20mmHg) below the threshold pressure (∼15mmHg below control) that normally results in sharp acute increases in renin secretion in conscious dogs.16,29-30 Similarly, in our previous studies in dogs maintained on a normal salt intake, there was no activation of the renin-angiotensin system at even greater baroreflex-mediated reductions in arterial pressure (20-25mmHg).7,21 In conscious dogs, adrenergic receptor blocking studies indicate that activation of β-adrenergic receptors plays the dominant role in neurally-mediated stimulation of renin secretion at renal perfusion pressures above threshold pressure.14-15,18-19 On the other hand, reflex activation of the renal nerves by acute carotid artery occlusion increases the threshold pressure for renin release by stimulation of renal α- but not β-adrenergic receptors.15-16 Accordingly, by suppressing RSNA and attendant tonic stimulation of tubular α-adrenergic receptor-mediated sodium reabsorption, BA may lower the threshold pressure for renin release through a macula densa mechanism. That is, inhibition of sodium reabsorption in the proximal tubules and loop of Henle by BA would be expected to suppress renin secretion by increasing sodium chloride delivery to the macula densa. Although the precise mechanisms that account for the interaction between ambient RSNA and renal perfusion pressure on renin secretion are not clearly defined, reduced activation of both α- and β-adrenergic receptors appears to contribute to inhibition of renin release during the pronounced lowering of arterial pressure by BA. Most significantly, sustained renal sympathoinhibition during chronic BA likely counteracts increases in renin secretion at degrees of pressure reduction that have been shown to lead to striking acute increases in renin secretion. This inhibitory effect of BA on renin secretion plays an important role in permitting substantial and persistent baroreflex-mediated reductions in arterial pressure.21
Perspectives
While the emphasis of this manuscript has been on a mechanistic understanding of the role of the sympathetic nervous system in the regulation of renin secretion and arterial pressure during variations in salt intake, the current findings may also have important clinical implications. Although impressive reductions in arterial pressure have been reported in subjects with resistant hypertension when treated with BA therapy,1,2 the impact of dietary salt intake on baroreflex-mediated blood pressure lowering has not been investigated, and this issue may be particularly relevant to arterial pressure control in this hypertensive population. Subjects with resistant hypertension are exquisitely salt sensitive, in part because drug treatment includes blockade of the renin-angiotensin-aldosterone system.6 They have inappropriate volume expansion that is managed to varying degrees of success by aggressive use of diuretics and judicious dietary salt consumption.3-6 However, poor drug adherence and excess dietary salt ingestion are especially prevalent in resistant hypertension and contribute to erratic volume control and therapy resistance.3-6,31 Despite enhanced salt sensitivity in resistant hypertension, the current findings suggest that the antihypertensive effects of BA in this hypertensive population may be independent of the capricious ingestion of salt. If this notion is true, the antihypertensive effects of BA would be expected to lead to a parallel shift in the relationship between arterial pressure and salt intake and, unlike drug treatment, provide a therapy that is insensitive to salt intake for the resistant hypertensive population.
Novelty and Significance.
What's New?
-
-
A unique medical device for stimulation of the carotid baroreflex was used to determine whether chronic inhibition of central sympathetic outflow alters the normal regulation of renin secretion and blood pressure in response to variations in salt intake.
What is Relevant?
-
-
The renin-angiotensin system is a powerful hormonal system for controlling sodium excretion and body fluid volumes which, when fully functional, prevents changes in salt intake from having much influence on arterial pressure. The sympathetic nervous system is a major controller of renin secretion, but its role in mediating the salt-intake-dependent changes in renin secretion and arterial pressure is unclear.
Conclusions
-
-
Inhibition of sympathetic activity by BA inhibits pressure-dependent renin release but does not impair responsiveness of the renin-angiotensin-aldosterone system to variations in salt intake. Consequently, blood pressure lowering during BA is independent of salt intake.
Acknowledgments
Sources of Funding: National Heart, Lung, and Blood Institute Grant HL-51971
Footnotes
Disclosures: Thomas E. Lohmeier Consultant fees, Scientific Advisory Board, CVRx
Eric D. Irwin Consultant fees, Scientific Advisory Board, CVRx
Adam W Cates Employee, CVRx
References
- 1.Bisognano JD, Bakris G, Nadim MK, Sanchez L, Kroon AA, Schafer J, de Leeuw PW, Sica DA. Baroreflex activation therapy lowers blood pressure in patients with resistant hypertension: results from the double-blind, randomized, placebo-controlled Rheos Pivotal trial. J Am Coll Cardiol. 2011;58:765–773. doi: 10.1016/j.jacc.2011.06.008. [DOI] [PubMed] [Google Scholar]
- 2.Lohmeier TE, Iliescu R. Chronic lowering of blood pressure by carotid baroreflex activation. Mechanisms and potential for hypertension therapy. Hypertension. 2011;57:880–886. doi: 10.1161/HYPERTENSIONAHA.108.119859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kaplan NK. Resistant hypertension. J Hypertens. 2005;23:1441–1444. doi: 10.1097/01.hjh.0000174968.72212.ac. [DOI] [PubMed] [Google Scholar]
- 4.Sarafidis PA, Bakris GL. Resistant hypertension. An overview of evaluation and treatment. J Am Coll Cardiol. 2008;52:1749–1757. doi: 10.1016/j.jacc.2008.08.036. [DOI] [PubMed] [Google Scholar]
- 5.Calhoun DA, Jones D, Textor S, Goff DC, Murphy TP, Toto RD, White A, Cushman WC, White W, Sica D, Ferdinand K, Giles TD, Falkner B, Carey RM. Resistant hypertension: diagnosis, evaluation, and treatment. A scientific statement from the American Heart Association Professional Education Committee of the Council for High Blood Pressure Research. Hypertension. 2008;117:e510–526. doi: 10.1161/CIRCULATIONAHA.108.189141. [DOI] [PubMed] [Google Scholar]
- 6.Pimenta E, Gaddam KK, Oparil S, Aban I, Husain S, Dell'Italia LJ, Calhoun DA. Effects of dietary reduction on blood pressure in subjects with resistant hypertension. Results from a randomized trial. Hypertension. 2009;54:475–481. doi: 10.1161/HYPERTENSIONAHA.109.131235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lohmeier TE, Irwin ED, Rossing MA, Sedar DJ, Kieval RS. Prolonged activation of the baroreflex produces sustained hypotension. Hypertension. 2004;43:306–311. doi: 10.1161/01.HYP.0000111837.73693.9b. [DOI] [PubMed] [Google Scholar]
- 8.Lohmeier TE, Iliescu R, Dwyer TM, Irwin ED, Cates AW, Rossing MA. Sustained suppression of sympathetic activity and arterial pressure during chronic activation of the carotid baroreflex. Am J Physiol Heart Circ Physiol. 2010;299:H402–H409. doi: 10.1152/ajpheart.00372.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hall JE, Guyton AC, Smith MJ, Jr, Coleman TG. Blood pressure and renal function during chronic changes in sodium intake: role of angiotensin. Am J Physiol Renal Electrolyte Physiol. 1980;239:F271–F280. doi: 10.1152/ajprenal.1980.239.3.F271. [DOI] [PubMed] [Google Scholar]
- 10.Guyton AC. Blood pressure control—special role of the kidneys and body fluids. Science. 1991;252:1813–1816. doi: 10.1126/science.2063193. [DOI] [PubMed] [Google Scholar]
- 11.Holmer S, Eckardt KU, Le Hir M, Schricker K, Riegger G, Kurtz A. Influence of dietary NaCl intake on renin gene expression in the kidneys and adrenal glands of rats. Pflügers Arch. 1993;425:62–67. doi: 10.1007/BF00374504. [DOI] [PubMed] [Google Scholar]
- 12.Wagner C, Hinder M, Krämer BK, Kurtz A. Role of renal nerves in the stimulation of the renin system by reduced renal arterial pressure. Hypertension. 1999;34:1101–1105. doi: 10.1161/01.hyp.34.5.1101. [DOI] [PubMed] [Google Scholar]
- 13.DiBona GF, Kopp UC. Neural control of renal function. Physiol Rev. 1997;77:75–197. doi: 10.1152/physrev.1997.77.1.75. [DOI] [PubMed] [Google Scholar]
- 14.Gross R, Hackenberg HM, Hackenthal E, Kirchheim H. Interaction between perfusion pressure and sympathetic nerves in renin release by carotid baroreflex in conscious dogs. J Physiol. 1981;313:237–250. doi: 10.1113/jphysiol.1981.sp013661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kirchheim HR, Hackenthal E, Persson PB. Baroreflex sympathetic activation increases threshold pressure for pressure-dependent renin release in conscious dogs. Pflügers Arch. 1985;405:127–135. doi: 10.1007/BF00584533. [DOI] [PubMed] [Google Scholar]
- 16.Ehmke H, Persson PB, Fischer S, Hackenthal E, Kirchheim H. Resetting of pressure-dependent renin release by intrarenal α1-adrenoceptors in conscious dogs. Pflügers Arch. 1989;413:261–266. doi: 10.1007/BF00583539. [DOI] [PubMed] [Google Scholar]
- 17.Blair ML, Chen YH, Izzo JL., Jr Influence of renal perfusion pressure on α- and β-adrenergic stimulation of renin release. Am J Physiol Endocrinol Metab. 1985;248:E317–E326. doi: 10.1152/ajpendo.1985.248.3.E317. [DOI] [PubMed] [Google Scholar]
- 18.Bie P, Mølstrøm S, Wamberg S. Normotensive sodium loading in conscious dogs: regulation of renin secretion during β-receptor blockade. Am J Physiol Regulatory Integrative Comp Physiol. 2008;296:R428–R435. doi: 10.1152/ajpregu.90753.2008. [DOI] [PubMed] [Google Scholar]
- 19.Damkjaer M, Isaksson GL, Stubbe J, Jensen BL, Assersen K, Bie P. renal renin secretion as regulator of body fluid homeostasis. Pflügers Arch – Eur J Physiol. 2013;465:153–165. doi: 10.1007/s00424-012-1171-2. [DOI] [PubMed] [Google Scholar]
- 20.Lohmeier TE, Iliescu R, Liu B, Henegar JR, Maric-Bilkan C, Irwin ED. Systemic and renal-specific sympathoinhibition in obesity hypertension. Hypertension. 2012;59:331–338. doi: 10.1161/HYPERTENSIONAHA.111.185074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lohmeier TE, Dwyer TM, Hildebrandt DA, Irwin ED, Rossing MA, Sedar DJ, Kieval RS. Influence of prolonged baroreflex activation on arterial pressure in angiotensin hypertension. Hypertension. 2005;46:1194–1200. doi: 10.1161/01.HYP.0000187011.44201.2e. [DOI] [PubMed] [Google Scholar]
- 22.Carroll RG, Lohmeier TE, Brown AJ. Disparity between renal venous norepinephrine and renin responses to sodium depletion. Am J Physiol Fluid Electrolyte Physiol. 1988;254:F754–F761. doi: 10.1152/ajprenal.1988.254.5.F754. [DOI] [PubMed] [Google Scholar]
- 23.DiBona GF, Sawin LL. Renal nerves in conscious rats during volume expansion and depletion. Am J Physiol Fluid Electrolyte Physiol. 1985;248:F15–F23. doi: 10.1152/ajprenal.1985.248.1.F15. [DOI] [PubMed] [Google Scholar]
- 24.Friberg P, Meredith I, Jennings G, Lambert G, Fazio V, Esler M. Evidence for increased renal norepinephrine overflow during sodium restriction in humans. Hypertension. 1990;16:121–130. doi: 10.1161/01.hyp.16.2.121. [DOI] [PubMed] [Google Scholar]
- 25.McBryde FD, Malpas SC, Guild SJ, Barrett CJ. A high-salt diet does not influence renal sympathetic nerve activity: a direct telemetric investigation. Am J Physiol Regulatory Integrative Comp Physiol. 2009;297:R396–R402. doi: 10.1152/ajpregu.90741.2008. [DOI] [PubMed] [Google Scholar]
- 26.Kim SM, Chen C, Faulhaber-Walter R, Oppermann M, Huang Y, Mizel D, Briggs, Schnermann J. Regulation of renin secretion and expression in mice deficient in β1- and β-2 adrenergic receptors. Hypertension. 2007;50:103–109. doi: 10.1161/HYPERTENSIONAHA.107.087577. [DOI] [PubMed] [Google Scholar]
- 27.Scheuer DA, Thrasher TN, Quillen EW, Jr, Metzler CH, Ramsay DJ. Atrial natriuretic peptide blocks renin response to renal hypotension. Am J Physiol Regulatory Integrative Comp Physiol. 1987;251:R423–R427. doi: 10.1152/ajpregu.1987.252.2.R423. [DOI] [PubMed] [Google Scholar]
- 28.Overlack A, Ruppert M, Kolloch R, Göbel B, Kraft K, Diehl J, Schmitt W, Stumpe O. Divergent hemodynamic and hormonal responses to varing salt intake in normotensive subjects. Hypertension. 1993;22:331–338. doi: 10.1161/01.hyp.22.3.331. [DOI] [PubMed] [Google Scholar]
- 29.Farhi ER, Cant JR, Barger AC. Interactions between intrarenal epinephrine receptors and the renal baroreceptor in the control of PRA in conscious dogs. Circ Res. 1982;50:477–485. doi: 10.1161/01.res.50.4.477. [DOI] [PubMed] [Google Scholar]
- 30.Finke R, Gross R, Hackenthal E, Huber J, Kirchheim HR. Threshold pressure for pressure-dependent renin release in the autoregulating kidney of conscious dogs. Pflügers Arch. 1983;399:102–110. doi: 10.1007/BF00663904. [DOI] [PubMed] [Google Scholar]
- 31.Burnier M, Wuerzner G, Struijker-Boudier H, Urquhart J. Measuring, analyzing, and managing drug adherence in resistant hypertension. Hypertension. 2013;62:218–225. doi: 10.1161/HYPERTENSIONAHA.113.00687. [DOI] [PubMed] [Google Scholar]