

Abstract
UV radiation poses a significant risk to human health. The mechanisms that help repair UV-damaged cells have recently been more clearly defined with the observation that Toll-like receptor 3 can sense self RNA released from necrotic keratinocytes following UV damage. TLR3 activation in the skin induces inflammation and increases expression of genes involved in skin barrier repair. Activation of TLR2 in the skin by commensal microbial products prevents excessive inflammation by blocking downstream TLR3 signaling. This review highlights how UV damage induced inflammation in the skin is propagated by host products and regulated by host inhabitants.
Excessive exposure to ultraviolet (UV) radiation is dangerous and has significant negative effects on human health. In the year 2000, excessive exposure to UV light led to 60,000 deaths worldwide with 1.5 million disability-adjusted life years (DALYs) also lost (Lucas et al. 2006). A majority of the reported morbidity and mortalities were linked to skin cancer; including melanoma, basal cell carcinoma, and squamous cell carcinoma, though sunburn also had a significant contribution to the 1.5 million DALYs lost. Furthermore, the economic and psychological impact of solar aging remains unquantified.
Despite the great impact on human health, relatively little research has been dedicated towards understanding the biological events associated with this common process. Much more work has been applied to understanding the benefits of solar exposure. UV light is needed to synthesize Vitamin D, which is necessary for human health (Institute of Medicine 2011; Ross et al. 2011). Complete sun avoidance would result in diseases related to Vitamin D deficiency and it is predicted that these diseases would lead to 3,304 million DALYs to be lost (Lucas et al. 2006). In addition to generation of Vitamin D, low levels of UV radiation also enhance permeability barrier function of the skin. Skin exposed to either UVA or UVB radiation prior to chemical irritant application is more resistant to damage as measured by transepidermal water loss (alkali resistance test, dimethylsulfoxide test and sodium lauryl sulfate test) (Lehmann et al. 1991). Barrier repair is also accelerated in skin that has been previously exposed to sub-erythemal broadband UVB radiation prior to tape-stripping barrier disruption. In these studies, it was also observed that antimicrobial peptide production is increased following low level broadband UVB exposure (Hong et al. 2008). Furthermore, UVB radiation can provide therapeutic benefit to patients with certain dermatological conditions. Patients with Psoriasis are commonly treated with narrow band UVB (Picot et al. 1992) and atopic dermatitis patients with either narrow, broad, or combination UVA/UVB (Jekler and Larkö 1988; Grundmann and Beissert 2012) exposure as the beneficial effects typically outweigh the apparent long term negative effects. UVB damage to the skin results in inflammation characterized by increased NF-κB activation, increased inflammatory cytokines including TNF and IL-6, increases in cis-urocanic acid, prostaglandins, ROS, and DNA damage (Ichihashi et al. 2003; Black et al. 2008). As is the case for the negative effects of UV on the skin, the mechanisms behind the beneficial effects of UVB are still unclear. However, new insights into the functions of the innate immune system have opened a window of opportunity to better understand these important interactions.
Innate immune receptors recognize products of cell death
The primary function of the innate immune system has historically been described as a system to detect pathogens and to rid them from the body. These microbes have unique physical characteristics termed microbe associated molecular patterns (MAMPs) that allow them to be recognized as foreign by the host’s array of pattern recognition receptors (PRRs) that are present on various immune cells as well as epithelial cells including keratinocytes (Lebre et al. 2007). At the most fundamental level, it is appropriate to recognize that activation of these PRRs causes an immune response that induces inflammation and eliminates the microbe from the host. However, these same innate immune receptors have the capability to recognize many different chemical structures in addition to those found on microbes. An important class of such non-microbial compounds is made up of certain endogenous molecules made by the host but normally separated from PRR by nature of their compartmentalization. During times of cell death, when compartmentalization of intracellular environments is disturbed, many of these host components are released into an extracellular environment, and this can illicit an immune response. These endogenous molecules that can activate an immune response in a sterile environment, absent of infection, are termed damage associated molecular patterns (DAMPs) and similarly to MAMPs, they activate PRRs resulting in inflammation and recruitment of leukocytes (Kono and Rock 2008).
Regulated cell death in the skin is ongoing as keratinocytes terminally differentiate and undergo the process of cornification in order to produce the stratum corneum (Candi et al. 2005). UV damage however, can cause unintended damage to keratinocytes and subsequent cell death in the skin, often observable in histological sections as “sunburn cells”, which are accepted to be keratinocytes undergoing apoptosis (Young 1987). High doses of UV radiation cause formation of cyclobutane pyrimidine dimers and (6–4) photoproducts in DNA which leads to mutations that can eventually result in skin cancer (Ichihashi et al. 2003) when nucleotide excision repair fails (Rass and Reichrath 2008). For this reason, it is essential for the health of an individual to dispose of these mutated cells by way of apoptosis. It has been shown that mice lacking p53, an important factor for apoptotic signaling, accumulate significantly more skin tumors than WT mice after chronic exposure to broadband UVB (Li et al. 1998). Apoptosis serves an essential role in response to sunburn, though is generally shown to be an anti-inflammatory or immunosuppressive event (Voll et al. 1997; Freire-de-Lima et al. 2006).
While UVB-induced apoptosis has been extensively described as occurring in the skin after UVB damage, much less is known about the extent of necrosis that occurs following UVB damage to keratinocytes in vivo. In vitro studies have shown that exposing keratinocytes to narrowband UVB radiation produces fractions of immunostimulatory, necrotic cells (Annexin V−, PI+) in addition to non-immunostimulatory apoptotic cells (Annexin V+, PI−)(Lai et al. 2009). While in vivo studies have not explicitly shown necrosis occurring in the epidermis, it has also been described that apoptotic cells can progress to secondary necrosis if they are not properly cleared by phagocytic cells (Kono and Rock 2008; Berghe et al. 2010; Tabas 2010). Many mediators of both apoptosis and necrosis, including TNF, are known to be stimulated following UVB exposure (Lai et al. 2009; Bernard et al. 2012; Galluzzi et al. 2012). In this context, it could be speculated that sunburn leads to necrosis in the skin. Interestingly it has recently been shown that HMGB1, a danger signal released from necrotic cells (Scaffidi et al. 2002), is released from both cultured keratinocytes as well as keratinocytes present in murine skin following UVB exposure (Johnson et al. 2013). While this DAMP has also been shown to be actively secreted from monocytes and macrophages, (Wang et al. 1999; Semino et al. 2005), its immunostimulatory potential has been well described following necrosis (Scaffidi et al. 2002; Kono and Rock 2008; Krysko et al. 2008). HMGB1 has also been shown to be released during pyroptosis, a specialized form of proinflammatory cell death (Lu et al. 2012). Whether HMGB1 release from keratinocytes exposed to UVB is an active or passive process remains to be determined.
As necrosis has classically been described as an unregulated form of cell death in which membrane integrity is lost, it has only been observed in vitro in keratinocytes in which membrane permeability can be measured or where morphology of a rupturing cell membrane could be observed. This has made observing necrosis in vivo difficult. In more recent years however, necrotic cell death has been shown to be dependent on the activation of RIPK1 and/or RIPK3 and has earned the name necroptosis when dependence on these kinases is demonstrated (Cho et al. 2009; Kaczmarek et al. 2013). Additionally, another specialized type of proinflammatory cell death known as pyroptosis, which is dependent on caspase-1 activation, may also be occurring after UVB damage to keratinocytes. It has been demonstrated that UVB radiation can stimulate inflammasome dependent IL-1β activation and secretion in keratinocytes (Feldmeyer et al. 2007). This process is dependent on caspase-1 and can be induced by either inflammasome or pyroptosome formation (Fernandes-Alnemri et al. 2007), which can lead to pyroptosis (Galluzzi et al. 2012; Lian et al. 2012). To what extent levels of apoptosis and necrosis or other forms of cell death are mediated after UVB exposure or whether one pathway is more prevalent at different UV doses is yet to be determined. It has been demonstrated however, that increasing UVC and broadband UVB exposure causes dose-dependent increases in apoptosis (Rehemtulla and Hamilton 1997; Mammone et al. 2000). It has also been shown that higher doses of broadband UVB can induce necrosis in keratinocytes (Mammone et al. 2000; Caricchio et al. 2003). As not all cells in the epidermis visably undergo apoptosis, though theoretically receive the same amount of energy from UV radiation, it is possible that nonapoptotic forms of cell death are occurring in the epidermis after UV damage.
Once membrane integrity is lost during necrosis, cellular components from these damaged cells spill into extracellular spaces (Berghe et al. 2010). These intracellular components are ‘foreign’ to an extracellular environment, and thus are treated so by the immune system. A number of cellular products have been observed to stimulate PRRs, especially during necrosis, including HMGB1 (Scaffidi et al. 2002), S100 proteins (Ryckman et al. 2003), heat shock proteins (Basu et al. 2000), ATP (Bours et al. 2006), uric acid (Shi et al. 2003), hyaluronan (Taylor et al. 2004; Scheibner et al. 2006), chromatin (Ishii et al. 2001) and RNA (Karikó et al. 2004; Cavassani et al. 2008; Lai et al. 2009; Bernard et al. 2012). These molecules, some of which are normally confined to the interior of a cell, gain the ability to activate certain PRRs including Toll-like receptors (TLRs). It has been demonstrated that the PRRs TLR2, TLR3, TLR4, and TLR9 can recognize certain DAMPs and induce an immune response.
TLR3 senses cellular damage following sunburn
TLR3 is a PRR that binds double-stranded RNA (dsRNA) (Liu et al. 2008). Though it has traditionally been accepted that dsRNA is a marker of viral infection or replication (Weber et al. 2006), more recent evidence demonstrates that endogenous sources of RNA can also activate TLR3. In 2004 Kariko et al., showed that TLR3 on dendritic cells could be activated by RNA associated with necrotic cells and also by in vitro transcribed mRNA (Karikó et al. 2004). In this study, when necrotic cells or mRNA was treated with benzonase, a nuclease that degrades all DNA and RNA, the necrotic cells no longer showed the ability to activate inflammatory pathways. In 2008, Cavassani et al., confirmed these findings in vivo demonstrating that less inflammation was present in sterile gut injury models in Tlr3−/− mice. They also showed that macrophages treated with necrotic cells needed functional Tlr3 to produce chemokines. Additionally, they demonstrated that macrophages treated with apoptotic cells produced significantly less cytokines than when treated with necrotic cells (Cavassani et al. 2008). This study demonstrates that if RNA is confined to intracellular compartments, as in apoptotic cells, it is unable to activate Tlr3. Only when RNA leaves the confines of the cell membrane, does it become immunostimulatory.
In 2009, Lai et al. demonstrated that in a sterile wound model in the skin, TNF and IL-6 production was diminished in Tlr3−/− mice (Lai et al, 2009). This publication also demonstrated that narrowband UVB-damaged keratinocytes, when added to keratinocyte cultures, could stimulate the inflammatory cytokines TNF and IL-6, through activation of TLR3. When the UVB-damaged NHEK were treated with RNase, they no longer induced inflammatory cytokines (Lai et al. 2009). In 2011, Lin et al. demonstrated that Tlr3 activation was also important for wound healing in the skin. They showed that Tlr3−/− mice displayed a delay in wound healing, showing deficiencies of infiltrating neutrophils and macrophages (Lin et al. 2011). They also were able to demonstrate that by applying the TLR3 ligand Poly (I:C), a dsRNA mimetic, to the wounds of mice as well as humans, wound healing was accelerated (Lin et al. 2012). These studies suggest that RNA released from damaged keratinocytes induces TLR3-dependent inflammation that promotes tissue repair. It has also been observed that activation of TLR3 in keratinocytes leads to increases in epidermal lipid transport and lipid metabolism gene expression, lipid accumulation and increased lamellar bodies (Borkowski et al. 2013). Because epidermal lipids are essential for proper barrier function and must be regenerated following injury to the epidermis, it can be speculated that activation of TLR3 in the skin helps to restore proper barrier function following UV injury.
Although the phenomenon that necrotic cells could stimulate TLR3 had been published multiple times in multiple cell types, it wasn’t until 2012, that an endogenous ligand for TLR3 was discovered. Bernard et al. demonstrated that narrowband UVB damage to keratinocytes released U1 RNA, a noncoding small nuclear RNA (snRNA), and component of the spliceosome, that could act as a DAMP and activate TLR3 to induce the inflammatory cytokines TNF and IL-6. Bernard and colleagues used RNA sequencing to determine that U1 RNA was significantly increased in keratinocytes 24 hours after narrowband UVB exposure. It was also discovered that in addition to U1 RNA, numerous additional noncoding RNAs were increased in keratinocytes after narrowband UVB exposure (Bernard et al. 2012). Although these studies focused on the effects of U1 RNA, it is possible that any of these noncoding RNAs could act as DAMPs.
dsRNA is the required ligand for TLR3 activation (Liu et al. 2008). Therefore, for an endogenous single-stranded RNA to be recognized by TLR3, it must form double-stranded regions. U1 RNA has a secondary structure comprised of four double-stranded stem-loop regions. These double-stranded regions serve to activate TLR3. The addition of U1 RNA or only a single stem-loop region of U1 RNA to keratinocytes was sufficient to induce TNF. Also, U1 RNA injected into ears of mice only induced inflammation when functional Tlr3 was present (Bernard et al. 2012). It is believed that UVB damage to keratinocytes causes necrosis and that RNA released from necrotic keratinocytes can signal in a paracrine manner to induce inflammation in the skin through TLR3 on neighboring cells (Figure 1).
Figure 1. dsRNA from UV-damaged keratinocytes activates TLR3.
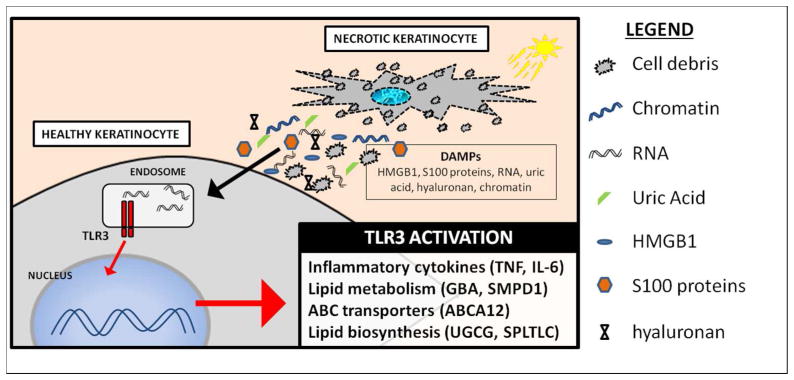
Excessive UV exposure causes necrosis in a population of keratinocytes in the epidermis. Loss of membrane integrity in these necrotic keratinocytes causes cellular contents to be spilled into extracellular space. DAMPs released by necrotic keratinocytes are then taken up by neighboring, healthy keratinocytes. dsRNA from necrotic keratinocytes is trafficked to the endosome where it activates TLR3. Downstream signaling leads to inflammation and increases in lipid biosynthesis, metabolism, and transport.
TLR3 is not the only TLR that has been implicated in the host response to UVB damage. While it is believed that dsRNA released from necrotic keratinocytes plays a role in this response, it is possible that other cells types in the skin can also undergo necrosis and contribute to this phenotype. It has been demonstrated that TLR3 and TLR4 activation can cause necrosis in macrophages that are treated with pan-caspase inhibitors (He et al. 2011) TLR4 which can be activated by numerous DAMPs including hyaluronan (Taylor et al. 2004; Scheibner et al. 2006; Muto et al. 2014), HMGB1, uric acid, heat shock proteins, defensins as well as the bacterial ligand LPS (Kono and Rock 2008), has also been shown to play a role in UVB induced immune suppression, as Tlr4−/− mice fail to exhibit UVB-induced immunosuppression in a contact hypersensitivity model (Lewis et al. 2011). The same phenotype is seen in Tlr3−/− mice, which also fail to show immunosuppression to DNFB contact hypersensitivity when first exposed to narrowband UVB radiation (Bernard et al. 2012). There is also evidence that TLR4 may play a detrimental role in the response to UVB as Tlr4−/− mice show increased nucleotide excision repair in skin (Ahmad et al. 2013) after UVB exposure and higher rates of survival in Tlr4−/− macrophages exposed to UVB (Harberts et al. 2013). The role of TLR4 in UVB-induced cutaneous carcinogenesis remains to be tested and it would be interesting to see whether Tlr4−/− mice had a different incidence of tumor formation. It has been shown however, that chemically induced cutaneous carcinogenesis is in part dependent on Tlr4- (Mittal et al. 2010) and downstream MyD88-signaling (Cataisson et al. 2012). No studies to date have investigated the role of TLR3 on UVB-induced skin cancer.
Regulation of inflammation in the skin after UVB injury
If damage to the epidermis is constantly occurring either by UVB-exposure or mechanical injury, one might pose the question as to why the epidermis is not in a constant inflammatory state. It has been shown that TLR2 activation by commensal microbial ligands can dampen TLR3-induced inflammation (Lai et al. 2009). In these studies, Lai et al. demonstrated that lipoteichoic acid (LTA) from Staphylococcus epidermidis, a major component of the human skin microbiome, can dampen TLR3 signaling in keratinocytes and mouse skin. In these studies, TLR2 activation with LTA increased levels of the signaling molecule TRAF1, which is proteolytically processed by caspase-8 to its active form (N-TRAF1). N-TRAF1 can then bind TRIF to negatively regulate TLR3-TRIF- signaling pathways. The suppressive effects of LTA on TLR3-dependent inflammation were not seen in Tlr2−/− and Traf1−/− mice, showing that TLR2 is essential for mediating TLR3-induced inflammation in the skin. Interestingly, Traf1 levels in skin were significantly lower in germ-free mice than conventionally raised mice (Lai et al. 2009). In this regard, commensal microbes such as S. epidermidis have the potential to suppress excessive inflammation in the skin following UVB damage or mechanical injury and the presence of specific microbes may keep excessive inflammation in check.
Inflammation in the skin serves an important purpose. Inflammation following infection or injury helps to sterilize the wound and promotes wound healing by attracting leukocytes that attack microbes or clear apoptotic and necrotic debris (Martin and Leibovich 2005). On the other hand it has been demonstrated that certain innate immune pathways, those dependent on MyD88 signaling can promote chemically induced skin carcinogenesis (Cataisson et al. 2012). It has also been demonstrated that Il-10−/− mice are resistant to broadband UVB-induced tumorigenesis (Loser et al. 2007). IL-10, which has been shown to be an important immune suppressor, induced following UVB irradiation, has been shown to contribute to UVB-induced skin cancer. While inflammation may help clear infections and resolve wounds in the short term, its role in skin carcinogenesis must be further elucidated as mice deficient in both pro- and anti-inflammatory machinery develop fewer tumors in different models of carcinogenesis. Study of inflammatory pathways should continue to be an important field of research as a balance of short term and long term health effects of UVB are further elucidated.
Conclusions
Because skin serves as the interface of our bodies to the outside world, it constantly comes in contact with microbes and is at highest risk for injury. It is not unfathomable that common receptors exist to deal with distinctly different threats to our well being. As TLR3 has been demonstrated to be important for inflammation in the skin following injury, the downstream pathways of TLR3 signaling must be further elucidated in order to develop therapeutics that take advantage of the beneficial effects without the adverse effects of excessive inflammation. As TLRs continue to be investigated, they are found to be implicated in a growing number of cellular processes. Recently, TLR3 has been linked to itch (Gruber and Holtz 2013) as well as cardiac dysfunction following polymicrobial sepsis (Gao et al. 2012) and will likely continue to be implicated in more disorders in the future. Identifying additional mechanisms of TLR3 activation and further elucidating downstream signaling will give us a better understanding of this pathway and lead to better therapeutics. TLRs and elucidating their expanding roles in various tissues should remain a focal point of future research.
Abbreviations used
- UV
ultraviolet
- DALYs
disability-adjusted life years
- MAMPs
microbe associated molecular patterns
- PRRs
pattern recognition receptorsp
- DAMPs
damage associated molecular patterns
- dsRNA
double-stranded RNA
- TLRs
toll-like receptors
Footnotes
Conflict of Interest
The authors state no conflict of interest.
References
- Ahmad I, Simanyi E, Guroji P, et al. Toll-Like Receptor-4 Deficiency Enhances Repair of UVR-Induced Cutaneous DNA Damage by Nucleotide Excision Repair Mechanism. J Invest Dermatol. 2013:1–8. doi: 10.1038/jid.2013.530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu S, Binder RJ, Suto R, et al. Necrotic but not apoptotic cell death releases heat shock proteins, which deliver a partial maturation signal to dendritic cells and activate the NF-kappa B pathway. Int Immunol. 2000;12:1539–46. doi: 10.1093/intimm/12.11.1539. [DOI] [PubMed] [Google Scholar]
- Berghe TV, Vanlangenakker N, Parthoens E, et al. Necroptosis, necrosis and secondary necrosis converge on similar cellular disintegration features. Cell Death Differ. 2010;17:922–30. doi: 10.1038/cdd.2009.184. [DOI] [PubMed] [Google Scholar]
- Bernard JJ, Cowing-Zitron C, Nakatsuji T, et al. Ultraviolet radiation damages self noncoding RNA and is detected by TLR3. Nat Med. 2012;18:1286–90. doi: 10.1038/nm.2861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black AT, Gray JP, Shakarjian MP, et al. UVB light upregulates prostaglandin synthases and prostaglandin receptors in mouse keratinocytes. Toxicol Appl Pharmacol. 2008;232:14–24. doi: 10.1016/j.taap.2008.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borkowski AW, Park K, Uchida Y, et al. Activation of TLR3 in keratinocytes increases expression of genes involved in formation of the epidermis, lipid accumulation, and epidermal organelles. J Invest Dermatol. 2013;133:2031–40. doi: 10.1038/jid.2013.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bours MJL, Swennen ELR, Di Virgilio F, et al. Adenosine 5′-triphosphate and adenosine as endogenous signaling molecules in immunity and inflammation. Pharmacol Ther. 2006;112:358–404. doi: 10.1016/j.pharmthera.2005.04.013. [DOI] [PubMed] [Google Scholar]
- Candi E, Schmidt R, Melino G. The cornified envelope: a model of cell death in the skin. Nat Rev Mol Cell Biol. 2005;6:328–40. doi: 10.1038/nrm1619. [DOI] [PubMed] [Google Scholar]
- Caricchio R, McPhie L, Cohen PL. Ultraviolet B radiation-induced cell death: critical role of ultraviolet dose in inflammation and lupus autoantigen redistribution. J Immunol. 2003;171:5778–86. doi: 10.4049/jimmunol.171.11.5778. [DOI] [PubMed] [Google Scholar]
- Cataisson C, Salcedo R, Hakim S, et al. IL-1R-MyD88 signaling in keratinocyte transformation and carcinogenesis. J Exp Med. 2012;209:1689–702. doi: 10.1084/jem.20101355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavassani KA, Ishii M, Wen H, et al. TLR3 is an endogenous sensor of tissue necrosis during acute inflammatory events. J Exp Med. 2008;205:2609–21. doi: 10.1084/jem.20081370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho YS, Challa S, Moquin D, et al. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell. 2009;137:1112–23. doi: 10.1016/j.cell.2009.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldmeyer L, Keller M, Niklaus G, et al. The inflammasome mediates UVB-induced activation and secretion of interleukin-1beta by keratinocytes. Curr Biol. 2007;17:1140–5. doi: 10.1016/j.cub.2007.05.074. [DOI] [PubMed] [Google Scholar]
- Fernandes-Alnemri T, Wu J, Yu J-W, et al. The pyroptosome: a supramolecular assembly of ASC dimers mediating inflammatory cell death via caspase-1 activation. Cell Death Differ. 2007;14:1590–604. doi: 10.1038/sj.cdd.4402194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freire-de-Lima CG, Xiao YQ, Gardai SJ, et al. Apoptotic cells, through transforming growth factor-beta, coordinately induce anti-inflammatory and suppress pro-inflammatory eicosanoid and NO synthesis in murine macrophages. J Biol Chem. 2006;281:38376–84. doi: 10.1074/jbc.M605146200. [DOI] [PubMed] [Google Scholar]
- Galluzzi L, Vitale I, Abrams JM, et al. Molecular definitions of cell death subroutines: recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ. 2012;19:107–20. doi: 10.1038/cdd.2011.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao M, Ha T, Zhang X, et al. Toll-like receptor 3 plays a central role in cardiac dysfunction during polymicrobial sepsis. Crit Care Med. 2012;40:2390–9. doi: 10.1097/CCM.0b013e3182535aeb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruber JV, Holtz R. Examining communication between ultraviolet (UV)-damaged cutaneous nerve cells and epidermal keratinocytes in vitro. Toxicol Ind Health. 2013;25:225–30. doi: 10.1177/0748233709103412. [DOI] [PubMed] [Google Scholar]
- Grundmann SA, Beissert S. Modern aspects of phototherapy for atopic dermatitis. J Allergy. 2012;2012:1–8. doi: 10.1155/2012/121797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harberts E, Fishelevich R, Liu J, et al. MyD88 mediates the decision to die by apoptosis or necroptosis after UV irradiation. Innate Immun. 2013:1–11. doi: 10.1177/1753425913501706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He S, Liang Y, Shao F, et al. Toll-like receptors activate programmed necrosis in macrophages through a receptor-interacting kinase-3–mediated pathway. Proc Natl Acadamy Sci. 2011;108:20054–9. doi: 10.1073/pnas.1116302108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong SP, Kim MJ, Jung M-Y, et al. Biopositive effects of low-dose UVB on epidermis: coordinate upregulation of antimicrobial peptides and permeability barrier reinforcement. J Invest Dermatol. 2008;128:2880–7. doi: 10.1038/jid.2008.169. [DOI] [PubMed] [Google Scholar]
- Ichihashi M, Ueda M, Budiyanto a, et al. UV-induced skin damage. Toxicology. 2003;189:21–39. doi: 10.1016/s0300-483x(03)00150-1. [DOI] [PubMed] [Google Scholar]
- Institute of Medicine. Dietary reference intakes for calcium and vitamin D. Natl Acadamies Press; Washingt DC: 2011. p. 1116. [Google Scholar]
- Ishii KJ, Suzuki K, Coban C, et al. Genomic DNA released by dying cells induces the maturation of APCs. J Immunol. 2001;167:2602–7. doi: 10.4049/jimmunol.167.5.2602. [DOI] [PubMed] [Google Scholar]
- Jekler J, Larkö O. UVB phototherapy of atopic dermatitis. Br J Dermatol. 1988:697–705. doi: 10.1111/j.1365-2133.1988.tb03490.x. [DOI] [PubMed] [Google Scholar]
- Johnson KE, Wulff BC, Oberyszyn TM, et al. Ultraviolet light exposure stimulates HMGB1 release by keratinocytes. Arch Dermatol Res. 2013;305:805–15. doi: 10.1007/s00403-013-1401-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaczmarek A, Vandenabeele P, Krysko DV. Necroptosis: the release of damage-associated molecular patterns and its physiological relevance. Immunity. 2013;38:209–23. doi: 10.1016/j.immuni.2013.02.003. [DOI] [PubMed] [Google Scholar]
- Karikó K, Ni H, Capodici J, et al. mRNA is an endogenous ligand for Toll-like receptor 3. J Biol Chem. 2004;279:12542–50. doi: 10.1074/jbc.M310175200. [DOI] [PubMed] [Google Scholar]
- Kono H, Rock KL. How dying cells alert the immune system to danger. Nat Rev Immunol. 2008;8:279–89. doi: 10.1038/nri2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krysko DV, Vanden Berghe T, D’Herde K, et al. Apoptosis and necrosis: detection, discrimination and phagocytosis. Methods. 2008;44:205–21. doi: 10.1016/j.ymeth.2007.12.001. [DOI] [PubMed] [Google Scholar]
- Lai Y, Di Nardo A, Nakatsuji T, et al. Commensal bacteria regulate Toll-like receptor 3-dependent inflammation after skin injury. Nat Med. 2009;15:1377–82. doi: 10.1038/nm.2062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebre MC, van der Aar AMG, van Baarsen L, et al. Human keratinocytes express functional Toll-like receptor 3, 4, 5, and 9. J Invest Dermatol. 2007;127:331–41. doi: 10.1038/sj.jid.5700530. [DOI] [PubMed] [Google Scholar]
- Lehmann P, Hölzle E, Melnik B, et al. Effects of ultraviolet A and B on the skin barrier: a functional, electron microscopic and lipid biochemical study. Photodermatol Photoimmunol Photomed. 1991;8:129–34. [PubMed] [Google Scholar]
- Lewis W, Simanyi E, Li H, et al. Regulation of ultraviolet radiation induced cutaneous photoimmunosuppression by toll-like receptor-4. Arch Biochem Biophys. 2011;508:171–7. doi: 10.1016/j.abb.2011.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G, Tron V, Ho V. Induction of squamous cell carcinoma in p53-deficient mice after ultraviolet irradiation. J Invest Dermatol. 1998:72–5. doi: 10.1046/j.1523-1747.1998.00090.x. [DOI] [PubMed] [Google Scholar]
- Lian L-H, Milora Ka, Manupipatpong KK, et al. The double-stranded RNA analogue polyinosinic-polycytidylic acid induces keratinocyte pyroptosis and release of IL-36γ. J Invest Dermatol. 2012;132:1346–53. doi: 10.1038/jid.2011.482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Q, Fang D, Fang J, et al. Impaired wound healing with defective expression of chemokines and recruitment of myeloid cells in TLR3-deficient mice. J Immunol. 2011;186:3710–7. doi: 10.4049/jimmunol.1003007. [DOI] [PubMed] [Google Scholar]
- Lin Q, Wang L, Lin Y, et al. Toll-like receptor 3 ligand polyinosinic:polycytidylic acid promotes wound healing in human and murine skin. J Invest Dermatol. 2012;132:2085–92. doi: 10.1038/jid.2012.120. [DOI] [PubMed] [Google Scholar]
- Liu L, Botos I, Wang Y, et al. Structural basis of toll-like receptor 3 signaling with double-stranded RNA. Science. 2008;320:379–81. doi: 10.1126/science.1155406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loser K, Apelt J, Voskort M, et al. IL-10 controls ultraviolet-induced carcinogenesis in mice. J Immunol. 2007;179:365–71. doi: 10.4049/jimmunol.179.1.365. [DOI] [PubMed] [Google Scholar]
- Lu B, Nakamura T, Inouye K, et al. Novel role of PKR in inflammasome activation and HMGB1 release. Nature. 2012;488:670–4. doi: 10.1038/nature11290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucas R, McMichael T, Smith W, et al. Solar ultraviolet radiation: global burden of disease from solar ultraviolet radiation. 2006. [Google Scholar]
- Mammone T, Gan D, Collins D, et al. Successful separation of apoptosis and necrosis pathways in HaCaT keratinocyte cells induced by UVB irradiation. Cell Biol Toxicol. 2000;16:293–302. doi: 10.1023/a:1026746330146. [DOI] [PubMed] [Google Scholar]
- Martin P, Leibovich SJ. Inflammatory cells during wound repair: the good, the bad and the ugly. Trends Cell Biol. 2005;15:599–607. doi: 10.1016/j.tcb.2005.09.002. [DOI] [PubMed] [Google Scholar]
- Mittal D, Saccheri F, Vénéreau E, et al. TLR4-mediated skin carcinogenesis is dependent on immune and radioresistant cells. EMBO J. 2010;29:2242–52. doi: 10.1038/emboj.2010.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muto J, Morioka Y, Yamasaki K. Hyaluronan digestion controls DC migration from the skin. J Clin Invest. 2014;1:1–11. doi: 10.1172/JCI67947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picot E, Meunier L, Picot-Debeze MC, et al. Treatment of psoriasis with a 311-nm UVB lamp. Br J Dermatol. 1992;127:509–12. doi: 10.1111/j.1365-2133.1992.tb14850.x. [DOI] [PubMed] [Google Scholar]
- Rass K, Reichrath J. UV damage and DNA repair in malignant melanoma and nonmelanoma skin cancer. Adv Exp Med Biol. 2008;624:162–78. doi: 10.1007/978-0-387-77574-6_13. [DOI] [PubMed] [Google Scholar]
- Rehemtulla A, Hamilton C. Ultraviolet radiation-induced apoptosis is mediated by activation of CD-95 (Fas/APO-1) J Biol Chem. 1997;272:25783–6. doi: 10.1074/jbc.272.41.25783. [DOI] [PubMed] [Google Scholar]
- Ross AC, Manson JE, Abrams SA, et al. The 2011 report on dietary reference intakes for calcium and vitamin D from the Institute of Medicine: what clinicians need to know. J Clin Endocrinol Metab. 2011;96:53–8. doi: 10.1210/jc.2010-2704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryckman C, Vandal K, Rouleau P, et al. Proinflammatory activities of S100: proteins S100A8, S100A9, and S100A8/A9 induce neutrophil chemotaxis and adhesion. J Immunol. 2003;170:3233–42. doi: 10.4049/jimmunol.170.6.3233. [DOI] [PubMed] [Google Scholar]
- Scaffidi P, Misteli T, Bianchi ME. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature. 2002;418:191–5. doi: 10.1038/nature00858. [DOI] [PubMed] [Google Scholar]
- Scheibner Ka, Lutz Ma, Boodoo S, et al. Hyaluronan fragments act as an endogenous danger signal by engaging TLR2. J Immunol. 2006;177:1272–81. doi: 10.4049/jimmunol.177.2.1272. [DOI] [PubMed] [Google Scholar]
- Semino C, Angelini G, Poggi A, et al. NK/iDC interaction results in IL-18 secretion by DCs at the synaptic cleft followed by NK cell activation and release of the DC maturation factor HMGB1. Blood. 2005;106:609–16. doi: 10.1182/blood-2004-10-3906. [DOI] [PubMed] [Google Scholar]
- Shi Y, Evans JE, Rock KL. Molecular identification of a danger signal that alerts the immune system to dying cells. Nature. 2003;425:516–21. doi: 10.1038/nature01991. [DOI] [PubMed] [Google Scholar]
- Tabas I. Macrophage death and defective inflammation resolution in atherosclerosis. Nat Rev Immunol. 2010;10:36–46. doi: 10.1038/nri2675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor KR, Trowbridge JM, Rudisill Ja, et al. Hyaluronan fragments stimulate endothelial recognition of injury through TLR4. J Biol Chem. 2004;279:17079–84. doi: 10.1074/jbc.M310859200. [DOI] [PubMed] [Google Scholar]
- Voll R, Herrmann M, Roth E, et al. Immunosuppressive effects of apoptotic cells. Nature. 1997;534:350–1. doi: 10.1038/37022. [DOI] [PubMed] [Google Scholar]
- Wang H, Bloom O, Zhang M, et al. HMG-1 as a Late Mediator of Endotoxin Lethality in Mice. Science (80- ) 1999;285:248–51. doi: 10.1126/science.285.5425.248. [DOI] [PubMed] [Google Scholar]
- Weber F, Wagner V, Rasmussen SB, et al. Double-Stranded RNA Is Produced by Positive-Strand RNA Viruses and DNA Viruses but Not in Detectable Amounts by Negative-Strand RNA Viruses. J Virol. 2006;80:5059–64. doi: 10.1128/JVI.80.10.5059-5064.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young AR. The sunburn cell. Photodermatol. 1987;4:127–34. [PubMed] [Google Scholar]