

Abstract
Immobilizing highly branched polyamidoamine (PAMAM) dendrimers to the cell surface represents an innovative method of enhancing cell surface loading capacity to deliver therapeutic and imaging agents. In this work, we click hybridized immune cells, i.e., macrophage RAW264.7 (RAW), with PAMAM dendrimer G4.0 (DEN) on the basis of bioorthogonal chemistry. Efficient and selective cell surface immobilization of dendrimers was confirmed by confocal microscopy. Viability and motility of RAW-DEN hybrids remained the same as untreated RAW cells according to WST-1 assay and wound closure assay. Furthermore, Western blot analysis revealed that there were no significant alterations in the expression levels of signaling molecules AKT, p38 and NFκB (p65) and their corresponding activated (phosphorylated) forms in RAW-DEN hybrids, indicating that the hybridization process neither induced cell stress response nor altered normal signaling pathways. Taken together, this work shows the feasibility of applying bioorthogonal chemistry to create cell-nanoparticle hybrids and demonstrates the non-invasiveness of this cell surface engineering approach.
Keywords: bioorthogonal chemistry, cell surface engineering, drug delivery, dendrimers, imaging
1. Introduction
Immune cells such as monocytes and macrophages actively infiltrate the tumor mass in response to tumor invasion, recurrence, and metastasis or tumor hypoxia.[1] Such cell types with their endogenous cancer-targeting and -attacking abilities are promising carriers to deliver therapeutics to the inner core of solid tumors, which is often inaccessible to standard modalities. A few early studies have demonstrated the use of macrophages as carriers to deliver anticancer genes and gold particles via phagocytosis.[1b, 2] However, phagocytosis is not applicable to cell-based anticancer drug delivery. Anticancer drugs cannot be directly loaded into the cellular vehicle or loosely encapsulated on the cell surface because of the detrimental impact of their inherent toxicity on cell viability and functions. Resolution of this issue is critical to the realization of the potential of this therapeutic approach.
Recently, we reported an innovative method of using macrophages as carriers by hybridizing them with polyamidoamine (PAMAM) dendrimers through cell surface modifications.[3] Dendrimers are well-fined highly branched macromolecules possessing a high density of surface groups (e.g., 64 primary amine groups for ethylenediamine (EDA) core PAMAM dendrimer G4.0).[4] As illustrated in Scheme 1, anchoring dendrimer macromolecules at the cell surface would considerably expand cell surface loading capacity. This hybrid vector is envisioned to have several appealing characteristics. Because of the possession of a number of terminal groups by dendrimer, a high payload of anticancer drugs can be covalently conjugated to the cell surface without causing toxicity to the carrier cell as a result of avoidance of burst release. Highly adaptable structures of dendrimers are suitable for delivery of imaging reagents and functional moieties that help with cancer treatment, diagnosis, or understanding of cellular behaviors in the context of tumorigenesis.[5] In this way, nanoparticles become truly “stealth”, sneaking past the body’s immune system. Monocytes/macrophages themselves are native to the human body and take surface-anchored dendrimers to the therapeutically relevant tissue sites and this hybrid vector will serve as personalized medicine to treat individual patients which may also result in enhanced therapeutic efficacy by making more anticancer drugs available to cancer cells or tumor mass.
Scheme 1.

Immune cell-nanoparticle hybrid vector represents a novel platform for delivery of therapeutic and imaging reagents through cell surface modification. Immune cells are removed from the animal, and azides are metabolically introduced into cell-surface glycans. Polyamidoamine (PAMAM) dendrimer undergoes a series of surface functionalization chemistries to possess strain-promoted alkyne, drug, imaging agent and other moieties of interest. Azide-expressing cells are then incubated with functionalized clickable dendrimers to form cell-nanoparticle hybrids following a highly efficient selective bioorthogonal reaction, namely strain-promoted azide–alkyne cycloaddition in water under physiological conditions without using any additional reagents. Following ex vivo cell surface engineering, the hybrid vehicles are injected back to the same animal for therapeutic and/or diagnostic treatment.
In our previous approach, we chemically treated macrophages with sodium periodate to generate aldehyde groups on the cell surface and then made them react with amine-terminated PEG-dendrimers to form transient Schiff base linkages.[3] Further treatment with sodium cyanoborohydride converted the Schiff base linkages to stable amide linkages. This multistep chemical approach relies on existing sialic acids on the cell surface, which can be a potential limit for nanoparticle loading. To hybridize the surface of immune cells with dendrimers, it is critical to enhance cell-nanoparticle hybridization reaction efficiency and avoid extensive chemical modifications or damages to the cellular vehicle. Bioorthogonal chemistry developed by Bertozzi has emerged as a powerful tool for live cell surface labeling because of high efficiency and high selectivity.[6] Strain-promoted azide-alkyne cycloaddition (SPAAC), commonly referred to as copper-free click chemistry, is the best known example. It has been shown that the copper-free click reaction proceeds within minutes on live cells with no apparent toxicity comparing to the copper-catalyzed reaction.[7] Furthermore, it minimally interferes with a biological system.[6b]
For the first time, we have applied this novel chemistry to develop an advanced nano-surface engineering method of creating cell-dendrimer hybrids. In this work, clickable dendrimers were synthesized by coupling dibenzocyclooctyne (DIBO) alkyne to PAMAM dendrimer G4.0 followed by FITC labeling. RAW264.7 macrophages (RAW) were cultured in the presence of azido sugar, in order to express azides on the cell surface. Azide-expressing macrophages were then hybridized with clickable dendrimers following SPAAC reaction. Localization of fluorescently-labeled dendrimers following hybridization was determined using confocal microscopy. To ensure the preservation of cell viability and functions throughout the cell-nanoparticle hybridization process, the effects of the bioorthogonal chemistry-based hybridization process on biological functions of RAW cells were investigated. In particular, cell viability, motility, and key intracellular signaling pathways were examined.
2. Experimental Section
2.1. Materials
EDA core PAMAM dendrimer generation 4.0 (technical grade) was purchased from Dendritech (Midland, MI). Dimethyl sulfoxide (DMSO), peracetylated N-azidoacetyl-mannosamine (Ac4ManNAz), N-hydroxysuccinimide (NHS), 1-ethyl-3-[3-dimethylaminopropyl]carbodiimide hydrochloride (EDC), N,N-diisopropylethylamine (DIPEA) and fluorescein isothiocyanate (FITC) were purchased from Sigma-Aldrich (St. Louis, MO). Click-IT® succinimidyl ester DIBO alkyne (simply referred to as DIBO) was purchased from Life Technologies (Grand Island, NY). 4′,6-diamidino-2-phenylindole (DAPI), sodium hydroxide, paraformaldehyde and phosphate-buffered saline (PBS) were purchased from Fisher Scientific (Pittsburgh, PA). Cell proliferation reagent WST-1 was purchased from Roche Applied Science (Indianapolis, IN). NFκB p65, phospho-NFκB p65 (Ser536), p38 MAPK, phospho-p38 MAPK (Thr180/Tyr182) and phospho-Akt (Ser473) antibodies were purchased from Cell Signaling Technology (Danvers, MA). AKT1 (559028) antibody was purchased from BD Biosciences Pharmingen (Mississauga, ON, Canada). β-actin (ACTBD11B7) antibody was purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Goat anti-rabbit antibody conjugated to horseradish peroxidase and goat anti-mouse antibody conjugated to horseradish peroxidase were purchased from Bio-Rad (Hercules, CA).
2.2. Cell Culture
RAW264.7 (RAW), a mouse monocyte/macrophage cell line, was used in this work. RAW cells were cultured in Dulbecco’s modification of Eagle’s medium (DMEM) (Life Technologies, Grand Island, NY) containing high glucose and supplemented with L-glutamine, 10% (v/v) fetal bovine serum (FBS) (Fisher Scientific, Pittsburgh, PA), 100 units/ml of penicillin and 100 μg/ml of streptomycin (both from Thermo Fisher Scientific, Ashville, NC) at 37 °C in a humid environment with 5% CO2.[8]
2.3. Synthesis of Clickable PAMAM Dendrimers
As illustrated in Scheme 2, the synthesis is a one-step reaction. Briefly, following removal of methanol from the stock solution, PAMAM dendrimer G4.0 (0.135 μmol, 1 eq.) was dissolved in DMSO (300 μl) and then mixed with DIPEA (30 eq.). DIBO (5 eq.) was dissolved in DMSO (100 μl) and then added, dropwise, into the dendrimer solution while stirring. The reaction proceeded at room temperature overnight followed by removal of DMSO under vacuum. The resultant G4.0-DIBO conjugates were purified by dialysis against deionized water and then freeze dried.
Scheme 2.
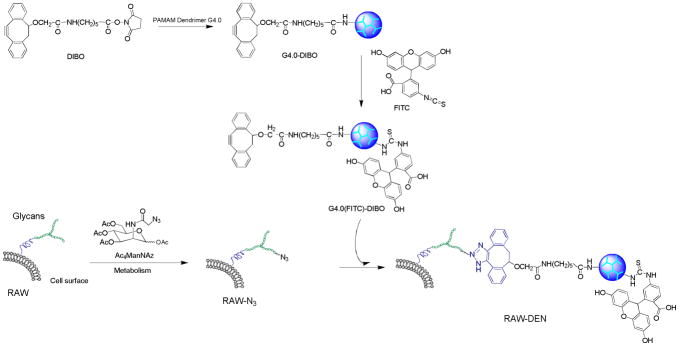
Schematic for hybridization of PAMAM dendrimer and macrophage through bioorthogonal chemistry.
2.4. FITC Labeling
FITC as a fluorescent probe (green) was used for dendrimer labeling. FITC was dissolved in DMSO and then added, dropwise, to DMSO solution of dendrimer (G4.0-DIBO or G4.0) in the presence of DIPEA, in which the molar ratio of DIPEA: FITC: dendrimer was 30:5:1. The reaction mixture was stirred overnight in the dark. Following removal of DMSO under vacuum, the resultant G4.0(FITC) and G4.0(FITC)-DIBO conjugates were dialyzed against deionized water and then freeze-dried.
2.5. General Click Cell-Dendrimer Hybridization Procedures
Prior to surface treatment, azides were metabolically introduced to the surface of RAW cells by incubating RAW cells in culture medium containing azido sugar Ac4ManNAz (50 μM) for 2 days. The resulting RAW cells expressing azides on the surface (i.e., RAW-N3 cells) were washed three times with pH 7.4 PBS. RAW-N3 cells were then treated with G4.0(FITC)-DIBO (25 μM) (1:100 dilution in PBS from a 2.5 mM dendrimer DMSO solution) for 10 min at room temperature to hybridize RAW-N3 cells with clickable dendrimer following copper-free click reaction on the cell surface. The resultant RAW-DEN hybrids were washed with PBS three times and subjected to the various assays described below. For comparison, hybridization between RAW cell and dendrimer devoid of complementarily reactive azide, DIBO, or both), was conducted under the same conditions as for RAW-DEN hybridization (i.e., RAW-N3/G4.0(FITC), RAW/G4.0(FITC), and RAW/G4.0(FITC)-DIBO), and the resulting treatment groups were investigated along with RAW-DEN hybrids.
2.6. Confocal Microscopy
To study uptake and localization of dendrimer nanoparticles, following the treatments described above, RAW cells were fixed with 4% paraformaldehyde immediately and nuclei were counterstained with DAPI (blue). The fixed cells were imaged under a Zeiss LSM 700 confocal laser scanning microscope using a magnification of 630×. Images were analyzed using ImageJ.[9]
2.7. Cell Viability Assessment
Immediately following treatment, cell viability of five groups—RAW(control), RAW-DEN, RAW-N3, RAW/G4.0(FITC)-DIBO, RAW-N3/G4.0(FITC)— was determined by WST-1 cell proliferation assay following the manufacturer’s protocol. The relative cell viability was normalized with respect to the viability of the control group. WST-1 cell proliferation reagent was purchased from Roche Applied Science (Indianapolis, IN). Briefly, the cells were incubated with WST-1 reagent (10 μl) in the cell culture media (100 μl) for 30 min. The absorbance of each sample solution was then measured at 450 nm against a background control as blank. The wavelength of 650 nm was used as the reference wavelength.
2.8. Western Blot Analysis
To examine whether or not key cellular signaling pathways in RAW cells might be affected by azido sugar culture and G4.0(FITC)-DIBO treatment, Western blot analysis of total cellular protein in RAW-N3 and RAW-DEN cellular vehicles was carried out following procedures described previously.[10] Briefly, total cell lysates (30 μg of protein) were separated on a 10% SDS-PAGE gel and transferred onto a polyvinylidene difluoride (PVDF) membrane. The membrane was blocked for 2 h in Tris-buffered saline (TBS) containing 5% non-fat dry milk. The specific proteins on the membrane were determined by incubation with primary antibodies overnight at 4 ºC with shaking. After washing in TBS containing 0.5% Tween 20, the membrane was incubated in a 1:3000 dilution of appropriate secondary antibody at room temperature for 1 h in wash buffer. The specific antigen-antibody interactions were detected using enhanced chemiluminescence (Pierce ECL Western Blotting Substrate) (Thermo Scientific, Rockford, IL). β-actin was used as a loading control.
2.9. Wound Closure Assay
Motility of cellular vehicles (i.e., RAW, RAW-N3, and RAW-DEN, 3 wells for each type of cellular vehicle) was assessed using wound-closure (scratch) assays. Briefly, RAW cells were plated in 12-well cell culture plates. When the cells reached 80% confluence, the cell culture medium was replaced with fresh culture medium either containing 50 μM Ac4ManNAz or free of azido sugar. When the cells reached 100% confluence, one group of RAW cells (3 wells) fed with azido sugar was further treated with G4.0-DIBO (25 μM) in PBS for 10 min to generate RAW-DEN hybrids. The fully confluent cells were washed three times with PBS. A sterile pipette tip was then used to denude the surface of each well. Following complete removal of the cells within the scratch area, each well was washed three times with PBS and then replaced with fresh cell culture medium. The width of the scratch was measured at three different points using a light microscope and AxioVision software (Carl Zeiss Microimaging, Thornwood, NY). Following 20-h culture at 37 °C, the scratch width was measured again at the same positions. Cell migration rate was calculated as follows:
[11]
2.10. Statistical Analysis
All the data were expressed as means ± standard errors and subjected to one way analysis of variance (ANOVA) followed by Student t-test for unpaired samples. A value of p < 0.05 was considered as statistically significant.
3. Results
3.1. Bioorthogonal Chemistry-Based Cell-Nanoparticle Hybridization
Our new bioorthogonal chemistry-based cell-nanoparticle hybridization approach involves two steps: 1) metabolic incorporation of azido sugars into the cell surface with peracetylated N-azidoacetylmannosamine (Ac4ManNAz, 50 μM) for 48 h, and 2) copper-free click reaction on the cell surface in the presence of clickable dendrimers (Scheme 2). Different from our previous approach using anionic carboxylate-terminated PAMAM dendrimer,[3] we used cationic amine-terminated PAMAM dendrimer G4.0 as a model to investigate the hybridization efficiency and to explore the adaptability of dendrimers used for cell-dendrimer hybridization because both types of PAMAM dendrimers have been commonly utilized in drug and gene delivery applications.[5e, 12] To this end, a different synthetic route has been developed to apply bioorthogonal chemistry for cell-nanoparticle hybridization. In particular, succinimidyl ester DIBO alkyne (DIBO) was coupled to amine terminated PAMAM dendrimer G4.0 to make clickable dendrimers. The resultant G4.0-DIBO conjugates were further labeled with FITC to allow fluorescent imaging for localization of nanoparticles. Except for the untreated cells (control), the other three groups were incubated with equimolar amounts of FITC.
As expected, untreated RAW cells lack the fluorescent label, whereas RAW cells treated with FITC show uniform distribution of fluorescence inside the cell due to non-specific uptake (Figure 1a). FITC-labeled PAMAM dendrimer G4.0 was found to be taken up more prominently by RAW-N3 cells and accumulated primarily in the cytoplasm. This observation was attributed to a high density of cationic charges on the dendrimer surface responsible for promoting nonspecific cellular uptake of FITC-labeled dendrimer. There was no obvious accumulation of dendrimers on the cell surface. Although RAW-N3 cells present azide groups on the surface, the click reaction did not take place because the dendrimer did not possess complementarily reactive alkynes on the surface. In contrast, a significant increase in fluorescence intensity was observed on the surface of the RAW-N3 cells following incubation with G4.0(FITC)-DIBO, indicating successful hybridization of dendrimer nanoparticles with the cell surface (Figure 1b). Significant reduction in uptake of the nanoparticles into the cell reaffirms the efficiency of bioorthogonal chemistry.
Figure 1.



Hybridization of PAMAM dendrimer and macrophage through bioorthogonal chemistry and confirmation by confocal microscopy. (a) Colocalization assay of FITC-labeled G4.0 (green) with nuclei (blue) by confocal microscopy following different surface treatments. (b) Quantitative analysis of cell fluorescence intensities using ImageJ. (c) Representative confocal images of a single RAW-DEN cell. (original magnification, 630×).
It is critical to ensure that RAW cells are minimally affected by the hybridization process. Therefore, cell morphology was monitored throughout the process. No abnormal morphological cell changes were observed. Some RAW-DEN cells undergoing mitosis were observed, suggesting a normal cell cycle. Detailed examination of single hybrid cells (Figure 1c) clearly illustrates that the fluorescence is mainly localized on the cell surface as opposed to in the cytoplasm for those dendrimer particles uptaken by cells via endocytosis. [13] The heterogeneity of a dendrimer-immobilized cell surface reflects the distribution of azide groups that were metabolically integrated into the cell surface. RAW cells maintained good viability throughout the process, as shown in Figure 2. The doses of azido sugar and dendrimers used in the hybridization process had negligible toxicity effects on the cells.
Figure 2.

Cell viability of RAW cells following various treatments as determined by WST-1 cell proliferation assay following the manufacturer’s protocol. The relative cell viability was normalized with respect to the viability of the control group. The data are expressed as mean ± SEM. No statistically significant difference was noted for comparisons between subgroups.
3.2. Intracellular Signaling Pathways in the Hybrid Cell Vehicles
Cells utilize signaling pathways to regulate their biological functions as well as their interactions with the microenvironment. Common stress-activated signaling pathways include nuclear factor (NF) κB (p65), p38 mitogen-activated protein kinases (MAPKs) and AKT. In this work, we primarily examined whether or not these signaling pathways had been altered by the hybridization process. As judged by Western blot analysis, expression levels of AKT, p65 and p38 in RAW cells were similar before and after 48 h culture in the presence of Ac4ManNAz at various concentrations up to 50 μM (Figures 3a and b). The levels of the phosphorylated forms of these key signaling molecules were also unchanged, indicating that azido sugar likely had not altered the signaling pathways involving AKT, p65, and p38. Consistent with the cytotoxicity assay, azido sugar is biocompatible with RAW cells.
Figure 3.




Biological effects of azido sugar and PAMAM dendrimers on intracellular signaling pathways in RAW cells during the hybridization process. Western blot analysis and quantitative densitometry of signaling molecules AKT, p65 (NFκB), p38 MAPK and their corresponding phosphorylated forms in RAW cells treated with Ac4ManNAz for 48 h at the indicated concentrations (a, b) and in RAW cells treated with PAMAM dendrimer G4.0 for 10 min at the indicated concentrations (c, d).
The second step in the cell-nanoparticle hybridization process is to anchor polycationic PAMAM dendrimers at the cell surface. Inherent phagocytosis of macrophages and the cationic surface of dendrimers would be expected to enable entry of dendrimers into the cell, although bioorthogonal chemistry employed in this work has greatly reduced inadvertent dendrimer uptake by RAW cells. PAMAM dendrimers show dose- and generation-dependent toxicity.[14] The dendrimer concentrations used for cell-nanoparticle hybridization did not cause a decrease in cell viability. However, the effects of amine-terminated PAMAM dendrimers on intracellular signaling pathways are unknown. Therefore, we evaluated intracellular AKT, p65 and p38 signaling pathways in RAW cells following 10-min treatment of PAMAM dendrimer G4.0 at various concentrations up to 100 μM.
The Western blot results showed that the acute exposure of PAMAM dendrimer G4.0 to RAW cells did not alter intracellular p65 and p38 signaling pathways (Figures 3C and 3D), indicating that no acute stress in RAW cells was induced by PAMAM dendrimer G4.0. Furthermore, normal activation of intracellular AKT was preserved although there was a minimal decrease in AKT and p-AKT levels, suggesting that the macrophages were able to survive and retain proliferative capacity, consistent with the results of viability assays. These observations are also consistent with the previous reports that dendrimers are biocompatible at relatively low concentrations and over a short exposure time.[15]
3.3. Assessment of Cell Motility
RAW cells are adherent cells. Assessment of their migration speed allows one to evaluate whether reaction at the cell surface causes loss of cell motility. To this end, we performed a 2D wound closure assay. We first introduced a “wound” by denuding confluent monolayers of RAW cells with a pipette tip, and determined their motility over 20 h (Figure 4a). As summarized in Figure 4b, untreated RAW cells migrated at a mean rate of 7.2 μm/h. The mean migration rates were 6.3 μm/h and 6.9 μm/h for RAW-N3 and RAW-DEN hybrids, respectively. No significant difference was observed between the control group and the surface modified RAW cells. These results confirm that surface modification of macrophages via copper-free click chemistry does not reduce cell motility.
Figure 4.


Wound closure assay to assess the motility of RAW cells following different treatments. (a) After RAW cells 100% confluence, the cell monolayer was denuded. The distance across the denuded area was measured at 0 h and at 20 h (Original magnification, 50×). (b) Motility is presented as mean ± SEM (n=18). Data are representative of experiments conducted on three independent occasions.
4. Discussion
There are many advantages about nanoparticle-based anticancer drug delivery. The composition, shape, size, charge, and morphology of nanoparticles can be finely tuned to achieve some desirable pharmacokinetic and pharmacodynamic profiles for a drug such as prolonged release, reduced systemic toxicity, tumor-specific efficacy.[16] However, this approach has two major limitations. First, because of heterogeneities of vascular permeability and the complex microenvironment of cancer biology, delivery of drug-carrying nanoparticles including those decorated with tumor-specific ligand relies heavily on passive mechanisms such as the enhanced permeability and retention (EPR) effect. Second, only a small percentage of the injected drug can finally accumulate in the tumor with the aid of nanoparticles, in large part, due to rapid clearance by immune cells in the liver and spleen, which is commonly known as reticuloendothelial system (RES). Immune cells including T cells and macrophages have been explored as drug carriers for cancer therapy because of their endogenous cancer-targeting or-attacking abilities. Unfortunately, loading therapeutic components into the cell via phagocytosis is the primary means for drug delivery.[1b, 2] Covalently conjugating drug-loaded particles to the surface of immune cell such as T cell has emerged as an appealing method for cancer immunotherapy.[17] The highly localized drug on the cell surface can continuously and directly exert pseudoautocrine stimulation of transferred cells in vivo and avoid its systemic toxicity.[17] Nonetheless, none of the approaches mentioned above is applicable to cell-based anticancer drug delivery.
We proposed a new concept to takes advantage of the best aspects of both types for anticancer drug delivery. Using the macrophage surface to covalently deliver anticancer drugs would prevent phagocytosis of those inherently toxic therapeutic agents, thus avoiding significant loss of cell viability and functions such as motility. High payloads of functional moieties such as drugs and fluorescent probes at cell surfaces can be achieved by virtue of the high loading capacity of dendrimers.[18] Although various techniques have been developed to engineer cell surfaces,[19] coupling nanoparticles such as dendrimers[3] and lipid-coated PLGA nanoparticles [17] to the cell surface represents a new way of utilizing cell surfaces, thus generating a great need to develop efficient chemistries for cell surface modifications to avoid inadvertent particle uptake by the cell and minimize cell function loss. Recently, a series of bioorthogonal reactions has been developed based on the Staudinger ligation and SPAAC.[6b] SPAAC reactions employ cyclooctynes to boost click reactions in the absence of copper catalyst,[20] a source of toxicity in copper-catalyzed alkyne-azide cycloaddition (CuAAC) due to production of copper-induced reactive oxygen species (ROS).[6b, 21] Therefore, bioorthogonal chemistry, which has been acclaimed as a safe and noninvasive method to probe molecules in cells and live organisms,[7, 22] has great potential for live cell surface modification with dendrimers.
An essential criterion for developing a clinically acceptable cell surface engineering protocol is to maintain cell viability and functions.[23] We examined the biological functions of the cellular vehicle via analysis of several key signaling molecules based on the following justification. p65 is a key mediator of inducible transcription in the innate immune system. It plays a central role in regulating cellular responses to a variety of stimuli, such as stress, cytokines, free radicals, ultraviolet irradiation, inflammation and infection.[24] p38 MAPKs are a class of mitogen-activated protein kinases responding to stress stimuli, such as cytokines, ultraviolet irradiation, heat shock, and osmotic shock, but also play important roles in cell differentiation, apoptosis and immune response.[25] As reported, p38 can be rapidly phosphorylated in response to lipopolysaccharide (LPS) stimulation, resulting in the production of pro-inflammatory cytokines such as interleukin-1 (IL-1) and tumor necrosis factor α (TNFα).[26] AKT, also known as protein kinase B, is a serine/threonine-specific protein kinase that is involved in metastatic pathways and survival signaling pathways. Activation of AKT kinase may cause cell detachment, proliferation, invasion, angiogenesis and protection against apoptosis.[27]
We provided evidence to prove the proof-of-concept of click hybridizing macrophages with dendrimers and its non-invasiveness to the engineered cells.[23] It is also important to evaluate cell surface stability of dendrimers following copper-free click chemistry and study potential effects of the factors including cell type, immobilized molecules, and immobilization technique on the fate of cell-surface immobilized moieties (i.e., dendrimers). In addition to these factors, dynamic intracellular dendrimer distribution and additional parameters including particle size, surface charge, particle geometry, cell type, cell membrane turnover, hybridization conditions (e.g., surface densities of azide and dendrimer concentrations) may affect dendrimer stability. Those factors will be investigated systematically in future work to gain insight into the cell surface engineering optimization for achieving targeted durability for cell-surface immobilized dendrimers. Future work also includes in vivo validation of macrophage homing following hybridization and assessment of therapeutic and diagnostic functions of the cell-dendrimer hybrids. Drugs or imaging probes can be covalently conjugated to or complexed with the dendrimer prior to cell-dendrimer hybridization. The linkages between dendrimer and cell and the linkages between drug/imaging probe and dendrimer can be engineered to be cleavable in response to external stimuli such as pH or enzymes or be stable. We envision that the whole hybrid system is modular and capable of controlled drug release using molecular engineering approaches.
5. Conclusion
PAMAM dendrimer G4.0 was successfully immobilized to the RAW cell surface via bioorthogonal chemistry and confirmed by confocal microscopy. Both azido sugar and PAMAM dendrimer G4.0 are cytocompatible under the conditions used for cell-nanoparticle hybridization. The viability, intracellular signaling pathways, and motility of RAW cells remained unaltered. Although internalization of nanoparticles by macrophages seems to be an inevitable process because of their innate phagocytic capability, the application of bioorthogonal chemistry provides an efficient non-invasive method for cell surface modification and hybridization with nanoparticles, and it has greatly reduced the intracellular uptake of nanoparticles during the hybridization process.
Acknowledgments
This work was supported by the National Science Foundation (CAREER Award CBET0954957). Confocal microscopy was performed in the Department of Neurobiology and Anatomy Microscopy Facility at VCU, supported, in part, with funding from NIH-NINDS Center core grant (5P30NS047463).
Contributor Information
Leyuan Xu, Department of Biomedical Engineering, Virginia Commonwealth University, Richmond, VA 23284, USA.
Dr. Olga Yu. Zolotarskaya, Department of Biomedical Engineering, Virginia Commonwealth University, Richmond, VA 23284, USA
Prof. W. Andrew Yeudall, Department of Biomedical Engineering, Virginia Commonwealth University, Richmond, VA 23284, USA, Philips Institute of Oral and Craniofacial Molecular Biology, Virginia Commonwealth University, Richmond, VA 23298, USA, Massey Cancer Center, Virginia Commonwealth University, Richmond, VA 23298, USA
Prof. Hu Yang, Email: hyang2@vcu.edu, Department of Biomedical Engineering, Virginia Commonwealth University, Richmond, VA 23284, USA, Massey Cancer Center, Virginia Commonwealth University, Richmond, VA 23298, USA
References
- 1.a) Fridman WH, Pages F, Sautes-Fridman C, Galon J. Nat Rev Cancer. 2012;12:298. doi: 10.1038/nrc3245. [DOI] [PubMed] [Google Scholar]; b) Brown JM, Wilson WR. Nat Rev Cancer. 2004;4:437. doi: 10.1038/nrc1367. [DOI] [PubMed] [Google Scholar]
- 2.a) Griffiths L, Binley K, Iqball S, Kan O, Maxwell P, Ratcliffe P, Lewis C, Harris A, Kingsman S, Naylor S. Gene Ther. 2000;7:255. doi: 10.1038/sj.gt.3301058. [DOI] [PubMed] [Google Scholar]; b) Paul S, Snary D, Hoebeke J, Allen D, Balloul JM, Bizouarne N, Dott K, Geist M, Hilgers J, Kieny MP, Burchell J, Taylor-Papadimitriou J, Acres RB. Hum Gene Ther. 2000;11:1417. doi: 10.1089/10430340050057495. [DOI] [PubMed] [Google Scholar]; c) Choi MR, Stanton-Maxey KJ, Stanley JK, Levin CS, Bardhan R, Akin D, Badve S, Sturgis J, Robinson JP, Bashir R, Halas NJ, Clare SE. Nano Lett. 2007;7:3759. doi: 10.1021/nl072209h. [DOI] [PubMed] [Google Scholar]
- 3.Holden CA, Yuan Q, Yeudall WA, Lebman DA, Yang H. Int J Nanomedicine. 2010;5:25. [PMC free article] [PubMed] [Google Scholar]
- 4.a) Tomalia DA, Baker H, Dewald J, Hall M, Kallos G, Martin S, Roeck J, Ryder J, Smith P. Macromolecules. 1986;19:2466. [Google Scholar]; b) Bosman AW, Janssen HM, Meijer EW. Chem Rev (Washington, DC US) 1999;99:1665. doi: 10.1021/cr970069y. [DOI] [PubMed] [Google Scholar]
- 5.a) Arima H, Yoshimatsu A, Ikeda H, Ohyama A, Motoyama K, Higashi T, Tsuchiya A, Niidome T, Katayama Y, Hattori K, Takeuchi T. Mol Pharmaceutics. 2012;9:2591. doi: 10.1021/mp300188f. [DOI] [PubMed] [Google Scholar]; b) Majoros IJ, Myc A, Thomas T, Mehta CB, Baker JR., Jr Biomacromolecules. 2006;7:572. doi: 10.1021/bm0506142. [DOI] [PubMed] [Google Scholar]; c) Maruyama-Tabata H, Harada Y, Matsumura T, Satoh E, Cui F, Iwai M, Kita M, Hibi S, Imanishi J, Sawada T, Mazda O. Gene Ther. 2000;7:53. doi: 10.1038/sj.gt.3301044. [DOI] [PubMed] [Google Scholar]; d) Papagiannaros A, Dimas K, Papaioannou GT, Demetzos C. Int J Pharm. 2005;302:29. doi: 10.1016/j.ijpharm.2005.05.039. [DOI] [PubMed] [Google Scholar]; e) Zhu S, Hong M, Zhang L, Tang G, Jiang Y, Pei Y. Pharm Res. 2010;27:161. doi: 10.1007/s11095-009-9992-1. [DOI] [PubMed] [Google Scholar]; f) Zong H, Thomas TP, Lee KH, Desai AM, Li MH, Kotlyar A, Zhang Y, Leroueil PR, Gam JJ, Banaszak Holl MM, Baker JR., Jr Biomacromolecules. 2012;13:982. doi: 10.1021/bm201639c. [DOI] [PubMed] [Google Scholar]
- 6.a) Saxon E, Bertozzi CR. Science. 2000;287:2007. doi: 10.1126/science.287.5460.2007. [DOI] [PubMed] [Google Scholar]; b) Sletten EM, Bertozzi CR. Acc Chem Res. 2011;44:666. doi: 10.1021/ar200148z. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Sletten EM, Bertozzi CR. Angew Chem Int Ed Engl. 2009;48:6974. doi: 10.1002/anie.200900942. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Chang PV, Prescher JA, Hangauer MJ, Bertozzi CR. J Am Chem Soc. 2007;129:8400. doi: 10.1021/ja070238o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Baskin JM, Prescher JA, Laughlin ST, Agard NJ, Chang PV, Miller IA, Lo A, Codelli JA, Bertozzi CR. Proc Natl Acad Sci U S A. 2007;104:16793. doi: 10.1073/pnas.0707090104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lei B, Zha W, Wang Y, Wen C, Studer EJ, Wang X, Jin F, Wang G, Zhang L, Zhou H. Mol Pharmaceutics. 2010;7:844. doi: 10.1021/mp100003r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.W. S. Rasband. ImageJ. U. S. National Institutes of Health; Bethesda, Maryland, USA: 1997–2012. http://imagej.nih.gov/ij/ [Google Scholar]
- 10.Xu L, Shen S, Ma Y, Kim JK, Rodriguez-Agudo D, Heuman DM, Hylemon PB, Pandak WM, Ren S. Am J Physiol: Endocrinol Metab. 2012;302:E788. doi: 10.1152/ajpendo.00337.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yeudall WA, Wrighton K, Deb S. In: p53 Protocols. Deb S, Deb SP, editors. Vol. 962. Humana Press; 2013. p. 135. [Google Scholar]
- 12.a) Perumal OP, Inapagolla R, Kannan S, Kannan RM. Biomaterials. 2008;29:3469. doi: 10.1016/j.biomaterials.2008.04.038. [DOI] [PubMed] [Google Scholar]; b) Sk UH, Kambhampati SP, Mishra MK, Lesniak WG, Zhang F, Kannan RM. Biomacromolecules. 2013;14:801. doi: 10.1021/bm3018615. [DOI] [PubMed] [Google Scholar]; c) Lesniak WG, Mishra MK, Jyoti A, Balakrishnan B, Zhang F, Nance E, Romero R, Kannan S, Kannan RM. Mol Pharmaceutics. 2013;10:4560. doi: 10.1021/mp400371r. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Kailasan A, Yuan Q, Yang H. Acta Biomater. 2010;6:1131. doi: 10.1016/j.actbio.2009.08.036. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Yuan Q, Lee E, Yeudall WA, Yang H. Oral Oncol. 2010;46:698. doi: 10.1016/j.oraloncology.2010.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Yuan Q, Fu Y, Kao WJ, Janigro D, Yang H. ACS Chem Neurosci. 2011;2:676. doi: 10.1021/cn200078m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Navath RS, Kurtoglu YE, Wang B, Kannan S, Romero R, Kannan RM. Bioconjug Chem. 2008;19:2446. doi: 10.1021/bc800342d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.a) Yang H, Kao JW. Int J Nanomedicine. 2007;2:89. doi: 10.2147/nano.2007.2.1.89. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Yang H, Lopina ST, DiPersio LP, Schmidt SP. J Mater Sci: Mater Med. 2008;19:1991. doi: 10.1007/s10856-007-3278-0. [DOI] [PubMed] [Google Scholar]
- 15.a) Duncan R, Izzo L. Adv Drug Deliv Rev. 2005;57:2215. doi: 10.1016/j.addr.2005.09.019. [DOI] [PubMed] [Google Scholar]; b) Jevprasesphant R, Penny J, Jalal R, Attwood D, McKeown NB, D’Emanuele A. Int J Pharm. 2003;252:263. doi: 10.1016/s0378-5173(02)00623-3. [DOI] [PubMed] [Google Scholar]
- 16.a) Goldberg M, Langer R, Jia X. J Biomater Sci, Polym Ed. 2007;18:241. doi: 10.1163/156856207779996931. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Freitas RA., Jr Nanomedicine. 2005;1:2. doi: 10.1016/j.nano.2004.11.003. [DOI] [PubMed] [Google Scholar]; c) LaVan DA, McGuire T, Langer R. Nat Biotechnol. 2003;21:1184. doi: 10.1038/nbt876. [DOI] [PubMed] [Google Scholar]
- 17.Stephan MT, Moon JJ, Um SH, Bershteyn A, Irvine DJ. Nat Med. 2010;16:1035. doi: 10.1038/nm.2198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.a) Liu M, Frechet JMJ. Pharm Sci Technol Today. 1999;2:393. doi: 10.1016/s1461-5347(99)00203-5. [DOI] [PubMed] [Google Scholar]; b) Esfand R, Tomalia DA. Drug Discovery Today. 2001;6:427. doi: 10.1016/s1359-6446(01)01757-3. [DOI] [PubMed] [Google Scholar]
- 19.a) Gahmberg CG, Tolvanen M. Methods Enzymol. 1994;230:32. doi: 10.1016/0076-6879(94)30005-4. [DOI] [PubMed] [Google Scholar]; b) Srivastava G, Kaur KJ, Hindsgaul O, Palcic MM. J Biol Chem. 1992;267:22356. [PubMed] [Google Scholar]; c) Keppler OT, Stehling P, Herrmann M, Kayser H, Grunow D, Reutter W, Pawlita M. J Biol Chem. 1995;270:1308. doi: 10.1074/jbc.270.3.1308. [DOI] [PubMed] [Google Scholar]; d) Burchenal JE, Deible CR, Deglau TE, Russell AJ, Beckman EJ, Wagner WR. J Thromb Thrombolysis. 2002;13:27. doi: 10.1023/a:1015364024487. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Panza JL, Wagner WR, Rilo HL, Rao RH, Beckman EJ, Russell AJ. Biomaterials. 2000;21:1155. doi: 10.1016/s0142-9612(99)00283-5. [DOI] [PubMed] [Google Scholar]; f) De Bank PA, Kellam B, Kendall DA, Shakesheff KM. Biotechnol Bioeng. 2003;81:800. doi: 10.1002/bit.10525. [DOI] [PubMed] [Google Scholar]
- 20.a) Codelli JA, Baskin JM, Agard NJ, Bertozzi CR. J Am Chem Soc. 2008;130:11486. doi: 10.1021/ja803086r. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Gordon CG, Mackey JL, Jewett JC, Sletten EM, Houk KN, Bertozzi CR. J Am Chem Soc. 2012;134:9199. doi: 10.1021/ja3000936. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Jewett JC, Bertozzi CR. Org Lett. 2011;13:5937. doi: 10.1021/ol2025026. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Sletten EM, Nakamura H, Jewett JC, Bertozzi CR. J Am Chem Soc. 2010;132:11799. doi: 10.1021/ja105005t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hong V, Steinmetz NF, Manchester M, Finn MG. Bioconjugate Chem. 2010;21:1912. doi: 10.1021/bc100272z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.a) Dehnert KW, Beahm BJ, Huynh TT, Baskin JM, Laughlin ST, Wang W, Wu P, Amacher SL, Bertozzi CR. ACS Chem Biol. 2011;6:547. doi: 10.1021/cb100284d. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Laughlin ST, Baskin JM, Amacher SL, Bertozzi CR. Science (New York, NY) 2008;320:664. doi: 10.1126/science.1155106. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Laughlin ST, Bertozzi CR. ACS Chem Biol. 2009;4:1068. doi: 10.1021/cb900254y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stephan MT, Irvine DJ. Nano Today. 2011;6:309. doi: 10.1016/j.nantod.2011.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gupta SC, Sundaram C, Reuter S, Aggarwal BB. Biochim Biophys Acta, Gene Regul Mech. 2010;1799:775. doi: 10.1016/j.bbagrm.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cuadrado A, Nebreda AR. Biochem J. 2010;429:403. doi: 10.1042/BJ20100323. [DOI] [PubMed] [Google Scholar]
- 26.del Barco Barrantes I, Nebreda AR. Biochem Soc Trans. 2012;40:79. doi: 10.1042/BST20110676. [DOI] [PubMed] [Google Scholar]
- 27.Sheng S, Qiao M, Pardee AB. J Cell Physiol. 2009;218:451. doi: 10.1002/jcp.21616. [DOI] [PubMed] [Google Scholar]