

Abstract
Purpose
The ability to successfully treat advanced forms of cancer remains a challenge due to chemotherapy resistance. Numerous studies indicate that NF-κB, a protein complex that controls the expression of numerous genes, as being a key factor in producing chemo-resistant tumors. In this study, the therapeutic potential of transferrin (TF)-targeted mixed micelles, made of PEG-PE and vitamin E co-loaded with curcumin (CUR), a potent NF-κB inhibitor, and paclitaxel (PCL), was examined.
Methods
The cytotoxicity of non-targeted and TF-targeted CUR and PCL micelles as a single agent or in combination was investigated against SK-OV-3 human ovarian adenocarcinoma along with its multi-drug resistant (MDR) version SK-OV-3-PCL-resistant (SK-OV-3TR) cells in vitro.
Results
Our results indicated that the TF-targeted combination micelles were able to improve the net cytotoxic effect of CUR and PCL to clear synergistic one against the SK-OV-3 cells. In addition, even though the non-targeted combination treatment demonstrated a synergistic effect against the SK-OV-3TR cells, the addition of the TF-targeting moiety significantly increased this cytotoxic effect. While keeping CUR constant at 5 and 10 μM and varying the PCL concentration, the PCL IC50 decreased from ~ 1.78 and 0.68 μM for the non-targeted formulations to ~ 0.74 and 0.1 μM for the TF-targeted ones, respectively.
Conclusion
Our results indicate that such co-loaded targeted mixed micelles could have significant clinical advantages for the treatment of resistant ovarian cancer and provide a clear rational for further in vivo investigation.
Keywords: Transferrin, polymeric micelles, curcumin, combination treatment, multi-drug resistance (MDR)
2. Introduction
The acquisition of drug resistance by tumors has been a subject of extensive study for many years and three main mechanisms have been identified through which tumors develop this resistance. First, the tumor microenvironment, such as the high interstitial fluid pressure (1), makes it unfavorable for the easy cellular internalization of drugs. Second, tumors host a collection of mutations and intracellular changes that affect the activity of cytotoxic agents including changes in cell cycle progression or increased repair of damaged DNA. The third and most commonly reported mechanism is the increased energy-dependent efflux of hydrophobic drugs from the cell by ATP-binding cassette (ABC) transporter trans-membrane proteins such as MDR-1 or P-glycoprotein (P-gp) (2). The overexpression of MDR-1 in cancers has been associated with poor prognosis in various tumor types including myeloid leukemia (3), breast cancer (4, 5), ovarian carcinoma (6, 7), osteosarcoma (8), and lung cancer (9, 10).
The nuclear factor kappa-B (NF-κB) is a signaling pathway that is involved in a variety of cellular processes. It has been reported that more than 500 genes are controlled in some form by NF-κB (11). NF-κB upregulation in cancer has been shown to play an antiapoptotic role and is attributed to the development of drug resistance (12, 13). Bentires-Alj et al. demonstrated that the inhibition of NF-κB activity sensitizes resistant colon cancer cells to daunomycin as a result of decreased MDR-1 expression (12). Many other studies have confirmed that NF-κB does indeed induce the expression of the MDR-1 (13–16). CUR, extracted from the perennial herb Curcuma longa, downregulates the NF-κB and Akt pathways and much evidence validates its use as a strong chemosensitizer to improve the therapeutic potential of various chemotherapeutic agents, such as PCL (17, 18). Despite the usefulness of CUR, its use in the clinic has been limited by its hydrophobicity and instability in serum (17). Many drug delivery systems are being sought to tackle these limitations; please see references (19–24) for examples. In this study, we utilized a lipid-based polymeric micellar system to deliver such poorly soluble drugs because of their ease in preparation and the various advantages attributed to them highlighted in this review (25).
In this work, a holo-transferrin (TF)-targeted mixed micellar formulation, comprised of PEG-PE and vitamin E, co-loaded with CUR and PCL was characterized. Then, the cytotoxicity of these micelles was investigated against SK-OV-3 and SK-OV-3TR human ovarian adenocarcinoma cells in vitro. TF is an 80 kDa glycoprotein ligand for the transferrin receptor (TFR) overexpressed on SK-OV-3 cells (26). Recently, the TF ligand has gained popularity as a top choice targeting moiety since it is overexpressed in a variety of cancer cells and has a great potential for in the targeted delivery to tumor cells (27–30). Since the transport of iron predominantly occurs through the TFR, the greater iron demand mediated by fast growth and division is the key factor attributed to the overexpression of TF on cancer cells (31, 32). A study by Kobayashi et el. demonstrated that by using TF as a targeting moiety on doxorubicin(DOX)-loaded liposomes, the P-gp mediated efflux of DOX was bypassed and a 3.5-fold higher toxicity was observed versus free DOX (33). This effect of TF, combined with CUR, could have significant advantages at reversing MDR. Therefore, co-loading CUR and PCL into a TF-targeted mixed micellar formulation of PEG-PE / vitamin E and their simultaneous delivery in one targeted micellar drug delivery system, was hypothesized to be an effective system for the treatment of resistant cancers.
3. Materials and methods
3.1 Materials
1,2-Distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000] (PEG2000-PE) was purchased from CordenPharma International (Plankstadt, Germany);1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE) and 1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine-N-(lissamine rhodamine B sulfonyl) (Rh–PE) were purchased from Avanti Polar Lipids (Alabaster, AL); pNP-PEG3400-pNP was purchased from Laysan Bio (Arab, AL). Curcumin (CUR) was purchased from Sigma (St. Louis, MO, USA catalog #C7727). Paclitaxel was purchased from LC laboratories (Woburn, MA catalog # P-9600). Vitamin E (>96% purity) and human holo-transferrin were purchased from Sigma (St. Louis, MO, USA).
3.2 Methods
3.2.1 Cell cultures
The SK-OV-3 human ovarian adenocarcinoma cell line was purchased from the American Type Culture Collection ATCC (Manassas, VA, USA). SK-OV-3TR, the PCL resistant variant of SK-OV-3, was a kind gift from Dr. Duan Zhenfeng (MGH, Boston, MA). Cell culture media and supplements were purchased from Cellgro (Herndon, VA, USA). SK-OV-3 cells were cultured in McCoy’s media while SK-OV-3TR cells were cultured in RPMI 1640 media both supplemented with 10% fetal bovine serum, 50 U/mL penicillin, and 50 mg/mL streptomycin. Cells were grown at 37°C in a humidified incubator with 5% CO2, and were passaged according to ATCC protocols.
3.2.2 Preparation of drug-loaded micelles
Mixed micelles incorporating PCL and/or CUR were prepared by the lipid thin film hydration method. Various weight % of PCL (10 mg/ml in 0.1% acetic acid methanol solution) and/or CUR (3 mg/ml in 0.1% acetic acid methanol solution) were added a to PEG2000-PE and vitamin E (89:11 molar ratio) solution in chloroform. A concentration of 5 mM micelle forming material was used in all experiments. The organic solvents were removed by the rotary evaporation to form a thin film of drug/micelle-forming material mixture. This film was further dried under high vacuum overnight to remove any remaining organic solvents (Freezone 4.5 Freeze Dry System Labconco, Kansas City, MO). The film was hydrated with 10 mM phosphate buffered saline (PBS) pH 7.4. The mixture was incubated in water bath at 40°C for 10 min and then vortexed for at least 5 minutes to insure proper resuspension of the film. The non-incorporated drugs were separated by centrifugation (13,500 g) for 5 minutes followed by filtration through a 0.2 μm membrane filters. For cell association studies, 1 mole% of the fluorescent probe, Rh–PE was added to the micelle forming material during preparation.
3.2.3 Synthesis of pNP-PEG3400-PE polymer
pNP-PEG3400-PE was synthesized according to standard procedure (34). Briefly, pNP-PEG3400-pNP and DOPE were dissolved in dry chloroform, co-incubated with TEA and then reacted at RT under Argon with continuous stirring overnight. The products of this reaction were then separated on a sepharose (CL4B) column. Fractions were analyzed by TLC to identify the aliquots that contain the pNP-PEG3400-PE product. The pNP-PEG3400-PE containing fractions were then frozen, lyophilized, weighed, and reconstituted with chloroform to a concentration of 10 mg/ml and stored at −80° C for further use.
3.2.4 Preparation of TF-modified micelles
To attach TF to the micelles, the reactive polymer, pNP-PEG3400-PE in chloroform was added to a round bottom flask. Chloroform was evaporated by rotary evaporation to form a thin film. Films were further dried under vacuum overnight to remove any residual solvents and then rehydrated with stock TF solution in PBS (pH 7.4) at a molar ratio of pNP-PEG3400-PE:TF 2:1. The pH of the solution was adjusted with 1.0 N NaOH to 8.5 as needed. Reaction time was 4 hrs at RT to allow sufficient TF conjugation and the complete hydrolysis of the unreacted pNP groups at the higher pH. TF-micelles were then dialyzed using a 100,000 MWCO cellulose ester membrane against PBS (pH 7.4) for 1 hr followed by another 4 hrs of dialysis to insure the complete removal of the unconjugated TF. Targeted mixed micelles were prepared by co-incubating drug-loaded micelles with TF-micelles at various mole ratios of pNP-PEG3400-PE (0.5–5 mol %) to PEG2000-PE. Samples were vortexed and allowed to mix overnight at room temperature before use to form the TF-targeted micelles.
Conjugation efficiency of TF was measured using a micro binichoninic acid (BCA) kit (Pierce, Rockford, IL) according to the manufacturer’s instructions. Protein content was determined by comparing TF-micelles to a known concentration of TF and BCA standards. Signals from TF samples were normalized with empty non-targeted micelle samples at the same lipid concentration.
3.2.5 Characterization of micelles
3.2.5.1 Micelle size
The micelle size (hydrodynamic diameter) was measured by the dynamic light scattering (DLS) using a N4 Plus Submicron Particle System (Beckman Coulter Corporation, Miami, FL, USA). The micelles were diluted with the deionized water until the concentration providing light scattering intensity was between 5×104 and 1×106 counts/second. The particle size distribution of all samples was measured in triplicate.
3.2.5.2 Drug solubilization efficiency
Drug incorporation efficiency was measured by reverse-phase HPLC using an Xbridge C18 (4.6 mm × 250 mm) column (Waters Corporation, Milford, MA) on a Hitachi Elite LaChrome HPLC equipped with an autosampler (Pleasanton, CA) and diode array detector. The column was eluted with 60:40 acetonitrile:water at a flow rate of 1 mL/min. PCL was detected at a wavelength of 227 nm, while CUR was detected at 420 nm. All samples were analyzed in triplicate.
3.2.5.3 Micelle stability
The micelles were stored at 4°C for up to two months. The samples were monitored periodically (once a week) for any changes in appearance, particle size, and drug content. When the hydrophobic drugs are completely solubilized in the core of the micelle, the micellar solution is clear. However, when the drug is present outside of the micelle core, turbidity is observed and precipitation occurs. Therefore, any change in the appearance of the micellar solution indicates instability. In addition, the presence of free drug in the micellar solution alters the size measurements due to the presence of drug crystals so any change in the size distribution also indicates instability. Prior to the weekly determination of the drug content, the micellar solution were centrifugated (13,500 g) for 5 minutes followed by filtration through a 0.2 μm syringe filter before characterization (Nalgene, Rochester, NY) to remove any non-incorporated drug that present in the solution followed by HPLC analysis as previously described.
3.2.6 Targeting potential of the TF-modified micelles by FACS analysis
Cell association of TF-modified micelles was compared to non-modified micelles using fluorescence associated cell sorting (FACS) Becton Dickinson FACScan (Becton, Dickinson and Company, NJ) and data analysis was performed using CellQuest software (Becton, Dickinson and Company, NJ). Briefly, empty non-targeted and TF-targeted micelles were prepared under sterile conditions containing 1 mol% Rh-PE. SK-OV-3 and SK-OV-3TR cells were seeded in 6-well plates at a density of 200,000 cells/well and grown for 24 hrs. Micelles were added to serum complete media at a lipid concentration of 0.3 mg/mL. After incubation with the micellar formulations, cells were washed three times, detached using Accutase (Innovative Cell Technologies, Inc San Diego, CA) and prepared for analysis following 1 hour incubation. The red fluorescence of Rh-PE was recorded at the emission wavelength of 580 nm (channel FL-2). A total of 20,000 gated events were acquired for each sample. To confirm the involvement of the TFR in the increased association of the TF-targeted micelles, free TF was added to the medium at 2 mg/mL before cells were treated with the various micellar formulations. Data analysis was performed using CellQuest software.
3.2.7 Cell viability assays
Viability of cells was measured using the CellTiter Blue® (Promega, Madison, WI) viability assay according to the manufacture’s protocol. Briefly, cells were seeded in 96-well plates at a density of 3,000 cells/well and grown for 24 hrs. Then cells were continuously incubated with the various formulations for 48 hrs in serum complete media. After 48 hrs of treatment, media was removed and the cells were washed with 200 μL serum complete media and then incubated with 100 μL of the media containing 20 μL of the CellTiter Blue® reagent. Cell viability was evaluated after 2 hrs of incubation by measuring the fluorescence (excitation 530 nm, emission 590 nm) using a Synergy HT multi-detection microplate reader (Biotek, Winooski, VT).
3.2.8 Data analysis
Data were generated in multiples of triplicates for proper statistical analysis. The data are expressed as mean ± SD. Student’s t-test was used to determine statistical significance. Two groups were considered statistically significant when P < 0.05.
4. Results and dicussion
4.1 Preparation and characterization of micelles
The solubility of CUR or PCL in water is < 1 μg/mL. PEG2000-PE micelles are able to effectively solubilize PCL and CUR as a single agent without the addition of vitamin E. However, the addition of vitamin E to the formulation increased the solubility of PCL and CUR by a factor of 4 (35). Both poorly soluble compounds were successfully incorporated into the PEG-PE / vitamin E mixed micelles and the solubility was increased by at least 600-fold at a 5 mM lipid concentration. CUR was effectively encapsulated at a concentration of ~1.2 mg/ml (8.7% w/w) and PCL at a concentration of ~600 μg/ml (~4.7% w/w). The same concentrations of both drugs were achieved when co-loaded into the same micellar formulation. The drug loads were retained inside the micelles for at least 2 months at 4°C. This high encapsulation efficiency and stability is attributed to the hydrophobicity of the drugs and the effectiveness of these micelles in solubilizing such lipophilic drugs. Furthermore, size and zeta potential measurements monitored over the same period showed no difference in size or charge (Data not shown). For both the non-targeted and TF-targeted micelles, the hydrodynamic diameter ranged from 15 – 20 nm and therefore the addition of the TF targeting moiety did not significantly alter the micellar size. The zeta potential of the non-targeted and TF-targeted micelles was −27.7 ± 1.7 and −18.9 ± 0.9 mV, respectively. Activated pNP groups were used to attach TF to the distal end of the PEG3400-PE polymer. To remove the unreacted TF from the formulations, an extensive dialysis was performed. The reaction yield after dialysis, as determined by a BCA protein assay, was ~60%. Known amounts of TF-PEG3400-PE were added to the drug-loaded or empty micelles. These mixtures were then incubated overnight to ensure that proper lipid mixing has occurred and to generate the desired concentration of targeted drug-loaded micelles.
4.2 Targeting potential of the TF-modified micelles by FACS analysis
To determine the optimal ligand density on the surface of micelles, the density at which maximum cell association is observed, a series of cell association studies were performed by flow cytometry. Micellar cell association comprises of both the fraction of micelles that were surface attached after the washing step and also the internalized fraction. Figure 1 shows increase in the rhodamine intensity of the various formulations versus non-targeted empty micelles (the control group). The 0% TF-PEG-PE represents the non-targeted Rh-labeled micelles. The non-targeted Rh-labeled micelles have a 20% increase in Rh intensity versus the non-targeted non-Rh containing micelles since non-targeted micelles do associate with the cells without the aid of a targeting moiety. Even though the TF-targeted micelles showed superior association versus non-targeted micelles after one hour incubation at various concentrations, the highest targeting was achieved at 1 mol% pNP-PEG3400-PE concentration against both cell lines (Figure 1). Theoretically, we expected the increase in ligand density would increase cell association but from our experimental data we determined that at concentrations above 1 mol% TF-PEG3400-PE, micelle-cell association significantly decreased probably because the higher density impedes the TF on the micelles from interacting with the TFR. Hence, for the subsequent viability assays, we used 1 mol% fraction of TF-PEG3400-PE which translates to approximately 10 TF molecules per micelle (36). From the competition experiments, treating the cells with free TF, at 2 mg/mL, before performing the cell association studies reversed the targeting effect. The same association was observed for non-targeted and targeted formulations (data not shown). Thus, the association of the TF-targeted micelles was namely dependent of the TFR and not due to non-specific binding.
Figure 1.
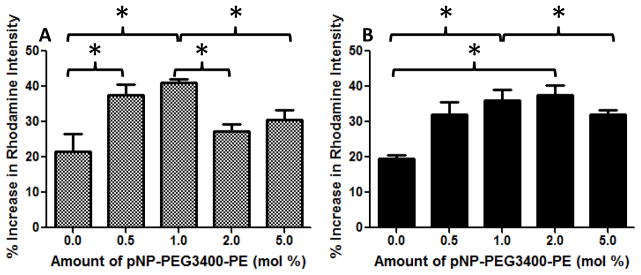
Cell association of the various TF-targeted micelles by flow cytometry to determine optimal ligand density. A- SK-OV-3, B- SK-OV-3TR. Different concentrations of TF-PEG3400-PE (0.5–5 mol %) of TF-targeted Rh-labeled micelles were incubated with the cells for 1 hour at a lipid concentration of 0.3 mg/mL and then analyzed. Values represent % increase in cellular association versus control (empty micelle treated) cells. N=9 + SD, p < 0.05.
4.3 Cell viability assays
The in vitro cytotoxicity of the different micellar formulations was investigated against SK-OV-3 and SK-OV-3TR cell lines. Empty non-targeted and TF-targeted PEG-PE/vitamin E micelles demonstrated very little cytotoxicity against the cells at the concentrations used (data not shown). The results of the dose-response studies with PCL and TF-PCL micelles as a single agent are shown in Figure 2. The PCL-loaded micelle IC50 for SK-OV-3 and SK-OV-3TR cells was determined to be ~10 nM and 2.1 μM, respectively. The resistant cells required >200 fold higher dose of PCL to achieve the same level of cell death as in the sensitive ones. As with other drug delivery systems, the drug delivery potential of the mixed micelles could be improved by attaching a targeting ligand, in this case TF, to the micelle surface. The TFR is overexpressed on proliferating tumor cells because there is a higher need for iron since iron is a cofactor in DNA synthesis. The attachment of TF to the distal tip of the PEG3400-PE polymer was accomplished by using an activated pNP group as previously described in section 3.2.4. The carbamate bond between TF and PEG3400-PE polymer is very stable and ensures the TF presentation on the outer side of the micelle for interaction with target TFR on cells. Using the TF-targeted PCL micelles (TF-PCL), we were able to slightly decrease the IC50 to ~8.5 nM from 10 nM on SK-OV-3 cells and to 1.08 μM from 2.1 μM on the SK-OV-3TR cells. Still, the improvement in toxicity against the resistant cells was almost 2-fold, and this could be attributed to the increase in internalization of the targeted micelles causing a decrease in resistance.
Figure 2.
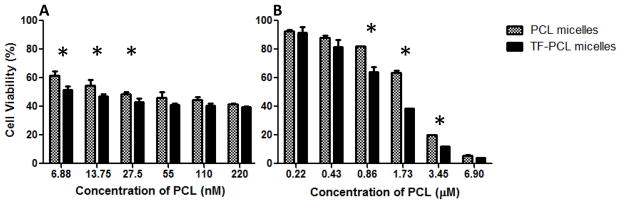
Cell viability of SK-OV-3 (A) and SK-OV-3TR (B) cells after 48hrs of continuous incubation with PCL or TF-PCL micelles at various concentrations. Cell viability was determined using CellTiter Blue cell viability assay. Data shown are representative of 3 independent experiments performed in triplicate, mean + SD * P < 0.05.
Figure 3 shows the in vitro cytotoxicity of the CUR and TF- targeted CUR (TF-CUR) micelles against the SK-OV-3 and SK-OV-3TR cells. The CUR micelles IC50 was determined to be ~21.7 and 23.6 μM, respectively. The TF-CUR micelle IC50 was determined to be ~20.6 and 22.1 μM, respectively. Since the toxicity of CUR is not dependent on cellular resistance to PCL, there was only a very insignificant decrease in the IC50 values. Nonetheless, at the specific 10 μM dose of CUR, we did observe a significant increase in cytotoxicity against both cell lines (Figure 3). No difference in CUR toxicity was observed between SK-OV-3 and SK-OV-3TR cells with concentrations below 10 μM.
Figure 3.
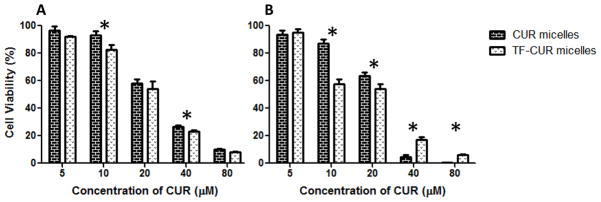
Cell viability of SK-OV-3 (A) and SK-OV-3TR (B) cells after 48hrs of continuous incubation with CUR or TF-CUR micelles at various concentrations. Cell viability was determined using CellTiter Blue cell viability assay. Data shown are representative of 3 independent experiments performed in triplicate, mean + SD * P < 0.05.
To test for synergism between CUR and PCL, we have treated the cells with various concentration of PCL while keeping the CUR concentration constant at 5 and 10 μM. Then the combination index (CI) values were determined using the classic isobologram equation of Chou and Talalay CI = a/A + b/B, where “a” is the PCL IC50 in combination with CUR at concentration “b”; “A” is the PCL IC50 and “B” is the CUR IC50. When the CI is less than 1, a synergistic effect is observed; CI=1 corresponds to an additive effect and when the CI is greater than 1, an antagonistic effect is observed (37).
With respect to the PCL sensitive cells, the non-targeted combination treatment at a CUR concentration of 5 μM yielded a CI of 1.26 at variable concentrations of PCL suggesting that the combination treatment was to some extent antagonistic. At 10 μM concentration of CUR and variable concentrations of PCL, an additive effect was observed in the sensitive cells since the CI was 1. However, the CI values for the TF-targeted combination treatments were 0.70 and 0.51 at 5 and 10 μM CUR, respectively. Numerous studies, highlighted in a review by Dicko et al. (38), have shown that a combination treatment could yield antagonistic, additive, or synergistic effect depending on the drug ratios tested either in vitro or in vivo. The addition of the TF targeting moiety to the mixed micelles dramatically increased the net cytotoxic effect of the combination treatment, and a synergistic effect was observed. Remarkably, at the 5 μM concentration of CUR and variable concentrations of PCL, the effect of the non-targeted formulation transformed from being antagonistic to synergistic with the TF-targeted one with the CI decreasing from 1.26 to 0.70 (Figure 4).
Figure 4.
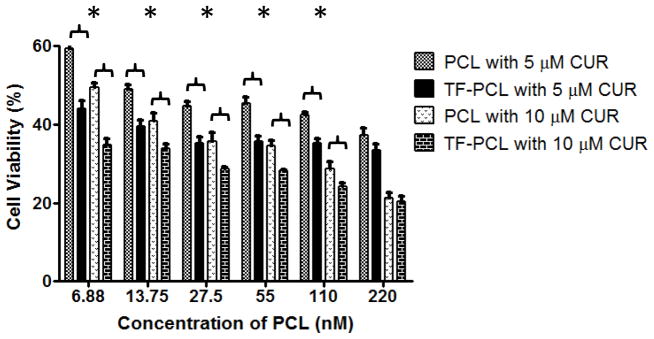
Cell viability of SK-OV-3 cells after 48hrs of continuous incubation with non-targeted and TF-targeted combination treatment of 5 or 10 μM of CUR and various concentrations of PCL. Cell viability was determined using CellTiter Blue cell viability assay. Data shown are representative of 3 independent experiments performed in triplicate, mean + SD * P < 0.05.
On the other hand, the non-targeted micellar co-delivery of CUR and PCL to the resistant cell line resulted in a relatively antagonistic effect at a 5 μM CUR concentration with CI = 1.1. However, at 10 μM of CUR and variable concentrations of PCL, a synergistic effect was observed with a CI = 0.78. Accordingly, a dramatic improvement in the efficacy of PCL was observed at the 10 μM concentration. The IC50 significantly decreased to 0.68 μM from 2.1 μM, a 3-fold decrease in the amount of the drug required to achieve a similar cytotoxic outcome. Therefore reversal of PCL resistance was achieved with the non-targeted CUR and PCL combination treatment in the SK-OV-3TR cells. However, the purpose of this study was to enhance this effect further by utilizing TF-targeting. As expected, the TF-targeted micelles significantly increased the cytotoxic effects of the combination treatment. The IC50 values of the combination treatment when keeping CUR constant at 5 and 10 μM and varying the PCL concentration were ~ 1.78 and 0.68 μM for the non-targeted formulations and ~ 0.74 and 0.1 μM for the targeted ones, respectively (Figure 5). This decrease in the IC50 values corresponded to a change from an additive to a synergistic effect at the 5 μM concentration of CUR (CI decreased from 1 to 0.93) and drastic increase in the synergistic effect at the 10 μM concentration of CUR (CI decreased from 0.78 to 0.49).
Figure 5.
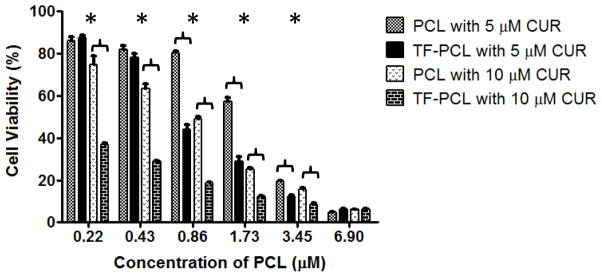
Cell viability of SK-OV-3TR cells after 48hrs of continuous incubation with the non-targeted and TF-targeted combination treatment of 5 or 10 μM of CUR and various concentrations of PCL. Cell viability was determined using CellTiter Blue cell viability assay. Data shown are representative of 3 independent experiments performed in triplicate, mean + SD * P < 0.05.
5. Conclusion
In this study, we designed a stable nano-sized TF-targeted micellar formulation co-loaded with CUR and PCL. This multi-functional mixed-micelle had significantly improved cytotoxic effects in vitro on PCL-sensitive cells and was able to dramatically reverse MDR in the PCL-resistant cells as well at various molar ratios of both drugs. Owing to their ease of preparation and the capability to tailor the drug-load ratio inside the micelles, obtaining synergism in vivo is a realistic possibility and a significant enhancement in therapeutic efficacy could be achieved. Therefore, this formulation could be considered a promising cancer-targeting drug delivery system for the treatment of normal and resistant tumors and deserves preclinical in vivo investigation.
Acknowledgments
This work was supported by the NIH/NCI CCNE grant 5U54CA151881 to Vladimir P. Torchilin.
Footnotes
No conflict of interest is declared.
Contributor Information
Abraham H. Abouzeid, Northeastern University, Department of Pharmaceutical Sciences, Center for Pharmaceutical Biotechnology and Nanomedicine, 140 The Fenway, Room 236, 360 Huntington Ave, Boston, MA 02115
Niravkumar R. Patel, Northeastern University, Department of Pharmaceutical Sciences, Center for Pharmaceutical Biotechnology and Nanomedicine, 140 The Fenway, Room 236, 360 Huntington Ave, Boston, MA 02115
Can Sarisozen, Northeastern University, Department of Pharmaceutical Sciences, Center for Pharmaceutical Biotechnology and Nanomedicine, 140 The Fenway, Room 225, 360 Huntington Ave, Boston, MA 02115.
Vladimir P. Torchilin, Email: V.Torchilin@neu.edu, Distinguished Professor, Northeastern University, Department of Pharmaceutical Sciences, Center for Pharmaceutical Biotechnology and Nanomedicine, 140 The Fenway, Room 211/214, 360 Huntington Ave, Boston, MA 02115
References
- 1.Hofmann M, Guschel M, Bernd A, Bereiter-Hahn J, Kaufmann R, Tandi C, et al. Lowering of tumor interstitial fluid pressure reduces tumor cell proliferation in a xenograft tumor model. Neoplasia. 2006;8(2):89–95. doi: 10.1593/neo.05469. Epub 2006/04/14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Szakacs G, Paterson JK, Ludwig JA, Booth-Genthe C, Gottesman MM. Targeting multidrug resistance in cancer. Nature reviews Drug discovery. 2006;5(3):219–34. doi: 10.1038/nrd1984. Epub 2006/03/07. [DOI] [PubMed] [Google Scholar]
- 3.Borg AG, Burgess R, Green LM, Scheper RJ, Liu Yin JA. Overexpression of lung-resistance protein and increased P-glycoprotein function in acute myeloid leukaemia cells predict a poor response to chemotherapy and reduced patient survival. British Journal of Haematology. 1998;103(4):1083–91. doi: 10.1046/j.1365-2141.1998.01111.x. [DOI] [PubMed] [Google Scholar]
- 4.Gregorcyk S, Kang Y, Brandt D, Kolm P, Singer G, Perry RR. Best clinical research paper p-glycoprotein expression as a predictor of breast cancer recurrence. European journal of surgical oncology : the journal of the European Society of Surgical Oncology and the British Association of Surgical Oncology. 1996;22(1):121. doi: 10.1007/BF02409045. [DOI] [PubMed] [Google Scholar]
- 5.Burger H, Foekens JA, Look MP, Meijer-van Gelder ME, Klijn JGM, Wiemer EAC, et al. RNA Expression of Breast Cancer Resistance Protein, Lung Resistance-related Protein, Multidrug Resistance-associated Proteins 1 and 2, and Multidrug Resistance Gene 1 in Breast Cancer: Correlation with Chemotherapeutic Response. Clinical Cancer Research. 2003;9(2):827–36. [PubMed] [Google Scholar]
- 6.Materna V, Pleger J, Hoffmann U, Lage H. RNA expression of MDR1/P-glycoprotein, DNA-topoisomerase I, and MRP2 in ovarian carcinoma patients: correlation with chemotherapeutic response. Gynecologic oncology. 2004;94(1):152–60. doi: 10.1016/j.ygyno.2004.03.035. [DOI] [PubMed] [Google Scholar]
- 7.Arts HJG, Katsaros D, de Vries EGE, Massobrio M, Genta F, Danese S, et al. Drug Resistance-associated Markers P-Glycoprotein, Multidrug Resistance-associated Protein 1, Multidrug Resistance-associated Protein 2, and Lung Resistance Protein as Prognostic Factors in Ovarian Carcinoma. Clinical Cancer Research. 1999;5(10):2798–805. [PubMed] [Google Scholar]
- 8.Chan HSL, Haddad G, DeBoer G, Ling V, Grogan TM. P-glycoprotein Expression: Critical Determinant in the Response to Osteosarcoma Chemotherapy. Journal of the National Cancer Institute. 1997;89(22):1706–15. doi: 10.1093/jnci/89.22.1706. [DOI] [PubMed] [Google Scholar]
- 9.Kawasaki M, Nakanishi Y, Kuwano K, Takayama K, Kiyohara C, Hara N. Immunohistochemically detected p53 and P-glycoprotein predict the response to chemotherapy in lung cancer. European Journal of Cancer. 1998;34(9):1352–7. doi: 10.1016/s0959-8049(98)00067-7. [DOI] [PubMed] [Google Scholar]
- 10.Yeh JJ, Hsu NY, Hsu WH, Tsai CH, Lin CC, Liang JA. Comparison of Chemotherapy Response with P-Glycoprotein, Multidrug Resistance-Related Protein-1, and Lung Resistance-Related Protein Expression in Untreated Small Cell Lung Cancer. Lung. 2005;183(3):177–83. doi: 10.1007/s00408-004-2532-1. [DOI] [PubMed] [Google Scholar]
- 11.Gupta SC, Sundaram C, Reuter S, Aggarwal BB. Inhibiting NF-κB activation by small molecules as a therapeutic strategy. Biochimica et Biophysica Acta (BBA) - Gene Regulatory Mechanisms. 2010;1799(10–12):775–87. doi: 10.1016/j.bbagrm.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bentires-Alj M, Barbu V, Fillet M, Chariot A, Relic B, Jacobs N, et al. NF-kappaB transcription factor induces drug resistance through MDR1 expression in cancer cells. Oncogene. 2003;22(1):90–7. doi: 10.1038/sj.onc.1206056. Epub 2003/01/16. [DOI] [PubMed] [Google Scholar]
- 13.Thévenod F, Friedmann JM, Katsen AD, Hauser IA. Up-regulation of Multidrug Resistance P-glycoprotein via Nuclear Factor-κB Activation Protects Kidney Proximal Tubule Cells from Cadmium-and Reactive Oxygen Species-induced Apoptosis. Journal of Biological Chemistry. 2000;275(3):1887–96. doi: 10.1074/jbc.275.3.1887. [DOI] [PubMed] [Google Scholar]
- 14.Zhou G, Kuo MT. NF-κB-mediated Induction of mdr1b Expression by Insulin in Rat Hepatoma Cells. Journal of Biological Chemistry. 1997;272(24):15174–83. doi: 10.1074/jbc.272.24.15174. [DOI] [PubMed] [Google Scholar]
- 15.Ogretmen B, Safa AR. Negative Regulation of MDR1 Promoter Activity in MCF-7, but Not in Multidrug Resistant MCF-7/Adr, Cells by Cross-Coupled NF-κB/p65 and c-Fos Transcription Factors and Their Interaction with the CAAT Region†. Biochemistry. 1999;38(7):2189–99. doi: 10.1021/bi982236+. [DOI] [PubMed] [Google Scholar]
- 16.Macus Tien Kuo ZL, Wei Yingjie, Lin-Lee Yen-chiu, Tatebe Shigeru, Mills Gordon B, Unate Hitoshi. Induction of human MDR1 gene expression by 2-acetylaminofluorene is mediated by effectors of the phosphoinositide 3-kinase pathway that activate NF-κB signaling. Oncogene. 2002;21(13):1948–54. doi: 10.1038/sj.onc.1205117. [DOI] [PubMed] [Google Scholar]
- 17.Sreekanth CN, Bava SV, Sreekumar E, Anto RJ. Molecular evidences for the chemosensitizing efficacy of liposomal curcumin in paclitaxel chemotherapy in mouse models of cervical cancer. Oncogene. 2011;30(28):3139–52. doi: 10.1038/onc.2011.23. Epub 2011/02/15. [DOI] [PubMed] [Google Scholar]
- 18.Bava SV, Sreekanth CN, Thulasidasan AK, Anto NP, Cheriyan VT, Puliyappadamba VT, et al. Akt is upstream and MAPKs are downstream of NF-kappaB in paclitaxel-induced survival signaling events, which are down-regulated by curcumin contributing to their synergism. The international journal of biochemistry & cell biology. 2011;43(3):331–41. doi: 10.1016/j.biocel.2010.09.011. Epub 2010/10/05. [DOI] [PubMed] [Google Scholar]
- 19.Shaikh J, Ankola DD, Beniwal V, Singh D, Kumar MNVR. Nanoparticle encapsulation improves oral bioavailability of curcumin by at least 9-fold when compared to curcumin administered with piperine as absorption enhancer. European Journal of Pharmaceutical Sciences. 2009;37(3–4):223–30. doi: 10.1016/j.ejps.2009.02.019. [DOI] [PubMed] [Google Scholar]
- 20.Kakkar V, Muppu SK, Chopra K, Kaur IP. Curcumin loaded solid lipid nanoparticles: An efficient formulation approach for cerebral ischemic reperfusion injury in rats. European Journal of Pharmaceutics and Biopharmaceutics. doi: 10.1016/j.ejpb.2013.02.005. [DOI] [PubMed] [Google Scholar]
- 21.Gangwar RK, Dhumale VA, Kumari D, Nakate UT, Gosavi SW, Sharma RB, et al. Conjugation of curcumin with PVP capped gold nanoparticles for improving bioavailability. Materials Science and Engineering: C. 2012;32(8):2659–63. [Google Scholar]
- 22.Akhtar F, Rizvi MMA, Kar SK. Oral delivery of curcumin bound to chitosan nanoparticles cured Plasmodium yoelii infected mice. Biotechnology advances. 2012;30(1):310–20. doi: 10.1016/j.biotechadv.2011.05.009. [DOI] [PubMed] [Google Scholar]
- 23.Sahu A, Bora U, Kasoju N, Goswami P. Synthesis of novel biodegradable and self-assembling methoxy poly(ethylene glycol)–palmitate nanocarrier for curcumin delivery to cancer cells. Acta biomaterialia. 2008;4(6):1752–61. doi: 10.1016/j.actbio.2008.04.021. [DOI] [PubMed] [Google Scholar]
- 24.Patel NR, Rathi A, Mongayt D, Torchilin VP. Reversal of multidrug resistance by co-delivery of tariquidar (XR9576) and paclitaxel using long-circulating liposomes. International journal of pharmaceutics. 2011;416(1):296–9. doi: 10.1016/j.ijpharm.2011.05.082. Epub 2011/06/28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Torchilin VP. Targeted polymeric micelles for delivery of poorly soluble drugs. CMLS, Cell Mol Life Sci. 2004;61(19–20):2549–59. doi: 10.1007/s00018-004-4153-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang J, Tian S, Petros RA, Napier ME, DeSimone JM. The Complex Role of Multivalency in Nanoparticles Targeting the Transferrin Receptor for Cancer Therapies. Journal of the American Chemical Society. 2010;132(32):11306–13. doi: 10.1021/ja1043177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Indira Chandran V, Matesic L, Locke JM, Skropeta D, Ranson M, Vine KL. Anti-cancer activity of an acid-labile N-alkylisatin conjugate targeting the transferrin receptor. Cancer letters. 2012;316(2):151–6. doi: 10.1016/j.canlet.2011.10.021. [DOI] [PubMed] [Google Scholar]
- 28.Xu S, Liu Y, Tai H-C, Zhu J, Ding H, Lee RJ. Synthesis of transferrin (Tf) conjugated liposomes via Staudinger ligation. International journal of pharmaceutics. 2011;404(1–2):205–10. doi: 10.1016/j.ijpharm.2010.10.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pang Z, Gao H, Yu Y, Chen J, Guo L, Ren J, et al. Brain delivery and cellular internalization mechanisms for transferrin conjugated biodegradable polymersomes. International journal of pharmaceutics. 2011;415(1–2):284–92. doi: 10.1016/j.ijpharm.2011.05.063. [DOI] [PubMed] [Google Scholar]
- 30.Hong M, Zhu S, Jiang Y, Tang G, Pei Y. Efficient tumor targeting of hydroxycamptothecin loaded PEGylated niosomes modified with transferrin. Journal of Controlled Release. 2009;133(2):96–102. doi: 10.1016/j.jconrel.2008.09.005. [DOI] [PubMed] [Google Scholar]
- 31.Daniels TR, Bernabeu E, Rodriguez JA, Patel S, Kozman M, Chiappetta DA, et al. The transferrin receptor and the targeted delivery of therapeutic agents against cancer. Biochimica et biophysica acta. 2012;1820(3):291–317. doi: 10.1016/j.bbagen.2011.07.016. Epub 2011/08/20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gomme PT, McCann KB, Bertolini J. Transferrin: structure, function and potential therapeutic actions. Drug discovery today. 2005;10(4):267–73. doi: 10.1016/S1359-6446(04)03333-1. [DOI] [PubMed] [Google Scholar]
- 33.Kobayashi T, Ishida T, Okada Y, Ise S, Harashima H, Kiwada H. Effect of transferrin receptor-targeted liposomal doxorubicin in P-glycoprotein-mediated drug resistant tumor cells. International journal of pharmaceutics. 2007;329(1–2):94–102. doi: 10.1016/j.ijpharm.2006.08.039. [DOI] [PubMed] [Google Scholar]
- 34.Torchilin VP, Levchenko TS, Lukyanov AN, Khaw BA, Klibanov AL, Rammohan R, et al. p-Nitrophenylcarbonyl-PEG-PE-liposomes: fast and simple attachment of specific ligands, including monoclonal antibodies, to distal ends of PEG chains via p-nitrophenylcarbonyl groups. Biochimica et Biophysica Acta (BBA) - Biomembranes. 2001;1511(2):397–411. doi: 10.1016/s0005-2728(01)00165-7. [DOI] [PubMed] [Google Scholar]
- 35.Sawant RR, Sawant RM, Torchilin VP. Mixed PEG-PE/vitamin E tumor-targeted immunomicelles as carriers for poorly soluble anti-cancer drugs: improved drug solubilization and enhanced in vitro cytotoxicity. European journal of pharmaceutics and biopharmaceutics : official journal of Arbeitsgemeinschaft fur Pharmazeutische Verfahrenstechnik eV. 2008;70(1):51–7. doi: 10.1016/j.ejpb.2008.04.016. Epub 2008/06/28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Torchilin VP, Lukyanov AN, Gao Z, Papahadjopoulos-Sternberg B. Immunomicelles: targeted pharmaceutical carriers for poorly soluble drugs. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(10):6039–44. doi: 10.1073/pnas.0931428100. Epub 2003/04/30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chou TCTP. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Advances in enzyme regulation. 1984;22(0):27–55. doi: 10.1016/0065-2571(84)90007-4. [DOI] [PubMed] [Google Scholar]
- 38.Dicko A, Mayer LD, Tardi PG. Use of nanoscale delivery systems to maintain synergistic drug ratios in vivo. Expert Opinion on Drug Delivery. 2010;7(12):1329–41. doi: 10.1517/17425247.2010.538678. [DOI] [PubMed] [Google Scholar]