

Abstract
A standard conservation strategy for plant genetic resources integrates in situ (on-farm or wild) and ex situ (gene or field bank) approaches. Gene bank managers collect ex situ accessions that represent a comprehensive snap shot of the genetic diversity of in situ populations at a given time and place. Although simple in theory, achieving complementary in situ and ex situ holdings is challenging. Using Trifolium thompsonii as a model insect-pollinated herbaceous perennial species, we used AFLP markers to compare genetic diversity and structure of ex situ accessions collected at two time periods (1995, 2004) from four locations, with their corresponding in situ populations sampled in 2009. Our goal was to assess the complementarity of the two approaches. We examined how gene flow, selection and genetic drift contributed to population change. Across locations, we found no difference in diversity between ex situ and in situ samples. One population showed a decline in genetic diversity over the 15 years studied. Population genetic differentiation among the four locations was significant, but weak. Association tests suggested infrequent, long distance gene flow. Selection and drift occurred, but differences due to spatial effects were three times as strong as differences attributed to temporal effects, and suggested recollection efforts could occur at intervals greater than fifteen years. An effective collecting strategy for insect pollinated herbaceous perennial species was to sample >150 plants, equalize maternal contribution, and sample along random transects with sufficient space between plants to minimize intrafamilial sampling. Quantifying genetic change between ex situ and in situ accessions allows genetic resource managers to validate ex situ collecting and maintenance protocols, develop appropriate recollection intervals, and provide an early detection mechanism for identifying problematic conditions that can be addressed to prevent further decline in vulnerable in situ populations.
Introduction
Since utilization of plant genetic resource (PGR) conservation is an important aim, it’s long been recognized that effective strategies need to integrate in situ (on-farm or in the wild) and ex situ (gene or field bank) approaches. In situ conservation allows the natural trajectory of evolution to continue to mold genetic variation, and ensures sustained access to genetically adapted populations. An important strength of ex situ conservation is that it readily provides the diverse germplasm needed by plant breeders, natural research managers and basic and applied researchers. In contrast to in situ populations, ex situ accessions provide a genetic snapshot, reflecting a wild (or on-farm) population’s adaptation to the biotic and abiotic conditions it was collected in. The static nature of ex situ conservation is frequently cast as a disadvantage; however, there is evidence that these snapshots conserve alleles that can be lost from in situ populations. For example, in a comparison of barley (Hordeum vulgare L.) landraces conserved in situ (on farm) in Morocco, and ex situ (since 1985), for resistance to current strains of powdery mildew (Blumeria graminis f.sp. hordei), ex situ accessions had more qualitative resistance than in situ accessions, for some pathotypes of powdery mildew, due to rare resistance genes being preserved ex situ, but lost in situ [1]. Other studies have reported the conservation of molecular marker bands in ex situ samples that have disappeared from current in situ populations [2], [3]. These studies reaffirm the general consensus that the most effective PGR conservation strategies need to integrate both an in situ and ex situ approach [4]–[6].
Gene bank curators and managers generally aim to have ex situ samples that represent the inherent diversity of in situ populations at the time they are sampled. The literature suggests this might not be a straight forward proposition. In situ and ex situ accessions were compared in maize (Zea mays L.) landraces using morphological [7] and SSR markers [8]. Ex situ accessions of American Indian Hopi landraces differed significantly from their in situ counterparts that had been maintained on farm in Arizona and the differences were attributed to original collecting bottlenecks and seed multiplication in a different environment (Iowa) [7]. In contrast, similar levels of genetic diversity and insignificant differentiation between in situ and ex situ samples of Jala, a Mexican maize race, were attributed to good initial sampling and effective regeneration practices [8]. Bean (Phaseolus vulgare L.) landraces conserved ex situ and on farm were compared using SSR markers [3] and phenotypic and development traits [9]. Both studies found that gene diversity was significantly less in ex situ subpopulations. SSR data indicated significant genetic differentiation in ex situ subpopulations as well as loss of alleles, gain of new alleles, and reduction of rare alleles and increase of common alleles [3]. Significant changes in yield, 100 seed weight, maturity and leaf area were also reported [9]. Both studies concluded that changes in genetic and phenotypic makeup in ex situ subpopulations could be attributed to regeneration practices. Using ISSR markers similar results were found in Oca (Oxalis tuberosa Mol), a tuber crop species grown in the Andes [10].
The challenges of maintaining complementary samples of wild species using ex situ and in situ methods are also evident. RAPDs were used to examine three remaining natural populations and one ex situ (conserved in a botanical garden field bank) subpopulation of Vatica guangxiensis (X. L. Mo), a rare endemic of southwestern China. The ex situ subpopulation contained 88.31% of the total genetic variation and was thought to adequately represent extant natural variation [11]. In contrast, RAPDs were used to examine Parashorea chinensis H. Wang, a rare endemic timber tree of southwest China and adjacent areas of Laos and Vietnam. The ex situ subpopulation (conserved in a field bank) was found to contain only 77.1% of the total variation found in samples taken from seven in situ sites. Additional sampling was recommended [12]. RAPDs were also used to assess ex situ samples (cultivated as ornamentals) of Berberidopsis corallina Hook. f., a threatened vine endemic to southern Chile, and found to only represent the northern part of the natural range of the species [13]. All three studies emphasized the importance of adequate sampling to ensure ex situ accessions represent the genetic diversity of wild species. In contrast, in situ populations of Agropyron cristatum (L.) Gaertn., a widely distributed wind pollinated grass species, were compared with gene bank counterparts collected 28–30 years previously, using SSRs. Although there were individual loci differences for number of alleles and genetic diversity, when averaged across all loci there was no significant difference among recent recollections and ex situ stored accessions [14]. RAPDs were used to examine recollections of Solanum jamesii Torrey (diploid outcrosser) and Solanum fendleri A. Gray (tetraploid inbreeder), crop wild relative (CWR) species of potato (Solanum tuberosum L.). Subpopulations were originally sampled in 1958, and recollected in 1978 and 1992. Significant genetic differences were found between all ex situ and re-collected in situ subpopulations of S. jamesii, and 12 of 16 comparisons of S. fendleri. Relative differences were attributed to mating system, vulnerability of small populations (<100 plants) to genetic change, and difference in original sampling procedures [2].
These studies emphasize two factors that are important to obtain and maintain ex situ samples that reflect an accurate snapshot of in situ populations. One, comprehensive initial sampling is imperative at both population and taxon level; and two, changing selection regimes–from either the ex situ regeneration/field site, or in situ site itself, can contribute to the divergence of in situ and ex situ accessions. Although a large body of literature outlines collecting parameters to ensure adequate sampling of PGR [15]–[17], and regeneration strategies to minimize genetic change [18]–[20], the literature also reflects how challenging it is to obtain ex situ samples that represent in situ populations. Recollection, rather than regeneration, may be a useful approach. This has been proposed for wild species that are difficult to regenerate ex situ, because natural conditions are too costly to replicate, or the material itself is difficult to propagate [20]. An area that has had little investigation is the frequency that recollection should occur. In other words, how frequently should a snapshot be taken? Recollection frequency is usually driven by declining viability of ex situ samples. Should ex situ managers also consider how quickly the genetic structure of in situ PGR might be changing? Seven farmer varieties of rice, conserved ex situ and in situ (on farm) over a period of time when production practices changed, were compared using allozymes, agronomic, stress resistance and morphological traits. Although yield and genetic diversity remained the same, traits associated with adaptation to new production practices significantly changed between ex situ and on farm subpopulations in a period of seventeen years [21]. Considering that climate change is occurring at a faster pace than predicted [22] and that models predict environmental change will not only drive changes in distribution and species extinction [23], but impact genetic diversity [24]. It is timely investigating how quickly in situ populations change over time, and how this may impact ex situ resampling.
Trifolium thompsonii Morton (2n = 2x = 16) is a restricted endemic herbaceous species that grows on the east slopes of the Cascade Mountains, in central Washington State, USA. The species is ranked as globally imperiled [25] and wild clover species, especially native to the US, have been identified as important to conserve in the USDA National Plant Germplasm System collection [26]. The species is a dominant forb in early seral communities that are disturbed by fire or grazing [27]. Fire has been excluded for nearly a century in the Cascade Mountains and species rarity has been attributed to increased shading due to overstory trees and competition with ground-layer species [28]–[30]. In 1988 the Dinkleman Fire swept through much of the range of T. thomsonii, effectively returning the area consumed to an early seral community. We used this species as an insect-pollinated, strongly out-crossing model to study population genetic change, since it is diploid, occurs in diverse environments that are undergoing successional change, occurs in populations greater than 10,000 plants, and is readily accessed and easily sampled. The aims of our study were to examine four distinct populations of T. thompsonii using Amplified Fragment Length Polymorphism (AFLP) markers to i) understand general patterns of population genetic diversity and structure, including the relative influence of gene flow, selection and genetic drift, and ii) examine the extent that diversity and structure changed over time. Specifically, this information would help us understand how adequate our efforts have been to conserve T. thomsonii using in situ and ex situ methods and help us determine if ex situ resampling intervals should be driven by declining seed viability in storage, or the occurrence of significant change in the genetic structure of in situ populations.
Materials and Methods
Ethics Statement
Collecting permits for populations growing on US Forest Service land were obtained from the Entiat District Ranger of the Okanogan-Wenatchee National Forest. Verbal permission to collect on state land was obtained from the Natural Areas Ecologist of the Washington Department of Natural Resources. Verbal permission from the landowner (Weaver family) was obtained to collect from private land.
Species, Study Locations and Sampling
T. thompsonii is reported to occur at 9–14 sites within 187 km2 on the east slopes of the Cascade Mountains, in southeastern Chelan and adjacent Douglas County in central Washington State, USA [25]. In 1977, the USDA Forest Service (USFS) established the 81 ha Thompson Clover Research Natural Area (RNA) and in 1993, the Washington Department of Natural Resources (DNR) established the 777 ha Entiat Slope Natural Area Preserve (KEYSTONE). Four sites, including RNA and KEYSTONE were sampled (Fig. 1). Area of each study location was mapped using a GPS (Global Positioning System) in 1995. Study location latitude and longitude were reported in decimal degrees, and elevation, in meters above sea level (masl). Badger Mountain (BADGER) (47.534, −120.2057, 1068 masl) was the only site that lies east of the Columbia River and occurred on private land. RNA (47.566, −120.324, 687 masl) and TENAS (47.581, −120.299, 982 masl) were located due west and northwest (respectively) of BADGER, on the USFS Okanogan-Wenatchee National Forest. KEYSTONE (47.635, −120.278, 1176 masl) was the northern most site. Size of area, habitat classification, and estimated census size of the four populations can be found in Table 1.
Figure 1. Location of studied populations of Trifolium thompsonii in Washington State, USA.

Inset map shows Douglas and Chelan county, where Thompson clover is endemic. Four populations were studied: KEYSTONE (red), TENAS (green), RNA (blue) and BADGER (yellow, located east of the Columbia river, and south of RNA).
Table 1. Size and habitat of study locations, population census estimate based on mean plant density, and number of plants analyzed using AFLP markers.
| Location | Area (km2) | Habitata | Population census | No. plants analyzed | |||
| <1998 | 2008 | 1995 | 2004 | 2009 | |||
| BADGER | 0.04 | Artemisia tridentata/Agropyron spicatum | 193,368b | 166,666d | 18 | 64 | 60 |
| RNA | 1.01 | Artemisia tridentata-vasayana/A. spicatum | 6,075828c | 1,630,785e | 19 | 63 | 56 |
| TENAS | 0.36 | Pseudotsuga menziesii/Calmagrostis rubescens | 7,174,060c | 5,564,140f | 0 | 64 | 60 |
| KEYSTONE | 0.82 | Artemisia tridentata-vasayana/A. spicatum | 1,481,738b | 1,545,595g | 35 | 64 | 48 |
Habitat description based on classification of vegetation [27].
[27].
[30].
Washington Natural Heritage Program Report Trifolium thompsonii EO 2342-SF 1856.
Washington Natural Heritage Program Report Trifolium thompsonii EO 6503.
Washington Natural Heritage Program Report Trifolium thompsonii EO 3235.
Washington Natural Heritage Program Report Trifolium thompsonii EO 2342.
In 1995 and again in 2004, seed was sampled between July 22 and August 10 from the four sites, and placed in ex situ storage (−18°C). In 2009, leaf tissue was sampled from in situ populations growing at the four sites. For the purposes of this study, each site was designated as a population, and samples collected from a specific site during a specific year were considered subpopulations. Seed and leaf tissue samples were collected by walking multiple haphazard linear transects of 150 m throughout the study site. Two inflorescence or three young and healthy leaves were sampled from a single plant if it occurred at 5 m intervals along thetransect. This distance was considered sufficient to minimize within family sampling based on observations of seed dispersal [28]. A total of 150 plants were sampled per site. Sampling protocols, which also included bulking seed from maternal lines, were in accordance to standard collecting protocols for efficient ex situ conservation of plant genetic resources. Seed was cleaned and stored in a −18°C freezer. For the analysis, seed was scarified and subjected to an 18 hour water rinse that we have found effective in overcoming seed dormancy. Seeds were germinated in petri dishes and fresh and healthy seedlings were transported to the molecular laboratory where seedling tissue was frozen at −80°C and lyophilized in a Vertis Lyo-Centre Freeze Dryer (SP Industries, Gardiner NY). A third of the 1995 seedlings were inadvertently frozen during transit and had to be discarded. The 2009 leaf samples were placed in envelops, kept at 4°C for 24 hours then placed in a −80°C freezer. Leaf tissue was lyophilized using the same procedure as the seedling tissue.
DNA Isolation and AFLP marker generation
Although less informative than codominant markers, AFLP markers have been used widely to investigate population genetic structure in insect-vectored out crossing species (i.e. [31]–[34]). As with any molecular marker system, care needs to be taken to minimize genotyping errors so methods used to isolate DNA and generate AFLPs incorporated rigorous protocols, internal standards and replication to ensure genotypes determined by the marker analysis correspond to the real genotypes being examined [35].
Freeze-dried seedlings or leaf tissues were pulverized in a SPEX SamplePrep 2000 Geno/Grinder, and DNA extraction was automated using the Wizard Magnetic 96 DNA Plant System (Promega). The AFLP analysis was based on individual plants germinated from seed collected at the same locations in 1995 and 2004, and on leaf samples of individual plants collected from the same locations in 2009.
AFLP markers were generated using locally developed procedures based on technology by [36]. Double restriction digestion was done in a 25 ul reaction containing 250 ng of DNA, 1X NE Buffer 4, 1X Purified BSA and 5.0 U each of EcoRI and MseI restriction enzymes (New England BioLabs). Fifteen ul of the restriction digest reaction was run on a 1.5% agarose gel to verify the completion of digestion.
Adapter sequences (EcoRI-Fwd, 5′- ctc gta gac tgc gta cc; EcoRI-Rev, 5′- aat tgg tac gca gtc tac; MseI - Fwd, 5′- gac gat gag tcc tga g, and MseI-Rev, 5′- tac tca gga ctc at) were obtained through Eurofins MWG/Operon (Huntsville, Alabama). Each adapter pair was adjusted to 100 pM/ul (EcoRI) and 200 pM/ul (MseI), mixed in equal amounts, and allowed to anneal for 1 hour at 37°C and cooled to room temperature. The annealed pairs were then diluted to 5 pM/ul (EcoRI) and 50 pM/ul (MseI), aliquoted to 100 ul amounts and frozen for future use.
Ligation was done at 20°C for 2 hours in a 20 ul reaction containing 10 ul of the remaining restriction digest, 1X T4 Ligase Buffer (New England BioLabs), 5 pMoles EcoRI adapter, 50 pMoles MseI adapter, 0.5 mM ATP, and 80 cohesive end Units of T4-ligase (New England Biolabs). The completed ligation reaction was diluted 10∶1 and used for the Pre-amplification. Pre-amplification and selective amplification were done on an ABI 9700 thermocycler using cycling programs prescribed by [36] but in 10 ul reactions. The pre-amplification product was diluted 10∶1 and 2 ul used for selective amplification. Four separate primer pairs were used for selective amplification (Eacg/Mctg, Eaca/Mctc, EacaMcag, and EaggMctg, where the last 3 letters indicate the selective nucleotides following the EcoRI and MseI primer sequences) since they provided clear reproducible bands and were sufficiently polymorphic to show variation within and between populations. Marker fragments were separated on a LI-LOR 4300 DNA Analyzer (LI-COR Biosciences). Unambiguous bands were identified and scored as either present or absent. To estimate genotyping error, four replicates were run on eight individual plants each, from two contrasting sites. Genotyping error was estimated at less than 1%.
Data Analysis
Since we had unequal sample sizes (Table 1.), a subsampling approach was used to determine if uneven sample sizes would bias our analysis [37]–[38]. Sub sampling was performed in R (script available upon request) by modifying the Diversity function in AFLPdat [39] to sub sample a random sample of 18 individuals (the minimal sample size), from each subpopulation and calculate Nei’s gene diversity and proportion of polymorphic markers. This was reiterated 100 times and means and standard deviation were calculated. Since observed and sub sampled data were consistently the same (Table S1.), and suggested that bias due to unequal sample size was low, the subsequent analysis was based on the observed data.
For each year, band patterns were calculated for overall population and subpopulations for the following: total number of bands (NB), number of common bands (CB) (frequency >0.05), number of rare bands (RB), (frequency <0.05), number of not widely shared bands (NWS), (common bands found in 25% or fewer populations) and number of private bands (PB). Allele frequencies were estimated using AFLPsurv 1.0 [40] based on a Bayesian method with non-uniform prior distribution [41]. Hardy-Weinberg equilibrium was assumed since Thompson clover is a highly out crossing wild species. Allele frequencies were used to estimate percent polymorphic loci at the 5% level and Nei’s gene diversity and its standard error was estimated for each subpopulation. T tests were carried out to determine if genetic diversity was significantly different among the four populations for each sampled year (i.e BADGER04, RNA04, TENAS04, KEYSTONE04), and across the three sampled years for each population (i.e. BADGER95, BADGER04, BADGER09). A Bonferroni correction was made to ensure an overall critical p-value of 5%. To examine genetic differentiation, AFLPsurv 1.0 was used to estimate overall population Fst at the four locations. It was also used to test the null hypothesis that there was no genetic differentiation among the four populations for each sampled year, and across the three sampled years for each population. Five thousand permutations were used.
To further examine population differentiation and partitioning of genetic variance, a euclidean distance metric was estimated and used for an analysis of molecular variance (AMOVA) to calculate Phi statistics (which is analogous to Fst), using GENALEX 6.5 ([42]–[43]. Pairwise PhiPT was estimated among the four populations by combining the 2004 and 2009 data. Probability values were based on 999 permutations. The AMOVA was also used to examine the distribution of variation among location (PhiRT) (i.e. BADGER, RNA, KEYSTONE), and among subpopulations (i.e. 1995, 2004, 2009) within location (PhiPR). Since TENAS lacked data for 1995, it was not included in this analysis. Significance of the AMOVA was tested using 999 permutations.
To examine genetic distance among the spatially and temporally sampled populations, Nei’s DAin [44] which does not involve an evolutionary model, was estimated using PowerMarker 3.25 [45]. A cluster analysis was conducted on all subpopulations using the neighbor joining algorithm to construct a tree from the distance matrix. The program was also used to calculate 1000 bootstrap distance matrices which were imported into the CONSENSE program of PHYLIP [46] to build a majority rule consensus tree.
Population structure was also examined using the software STRUCTURE v2.3.3 [47]–[50]. Ten replications with a burn-in of 20,000 iterations followed by 20,000 additional iterations were used at each K level until results indicated lowered and erratic values for P(X|K). The parameter set included the ADMIXTURE model with allele frequencies correlated, RECESSIVE ALLELES model, USEPOPINFO and STARTATPOPINFO turned on with 11 subpopulations labeled (each location/year), and the LOCPRIOR model. Average Q-plots over the ten replications were calculated using the ancillary software CLUMPP [51], and graphic displays of population structure were developed from the q-frequencies of the mean of ten runs using DISTRUCT software [51].
To examine gene flow on a current timescale we carried out two different assignment tests. The first used AFLPOP 2.0 [52] which uses a modified allocation method [53] to compute the log likelihood of an unknown individual’s allelic phenotype in each subpopulation based upon the frequency of the dominant band (presence) at each locus. Each unknown individual is then allocated into a source subpopulation with the greatest log likelihood. The minimal log-likelihood difference for the allocation of an individual to a population was set at 0.5. This is a conservative threshold in that assignment of an individual to a subpopulation was not made unless the probability of the given assignment was 5 times more likely than the next most probable assignment. If this threshold was not met, the individual was not assigned to any subpopulation and were designated as ‘criteria not met’ (CNM). The CNM category does not imply that an individual does not belong to a subpopulation, but that there are two or more subpopulations with similar probabilities of assignment, and hence minimal log-likelihood difference is less than the designated threshold. We also used STRUCTURE v2.3.3 to identify potential migrants between populations taking into account the source population of the sample. We set GENSBACK = 2, which tests for evidence of ancestry for two generations.
To examine the presence of historic gene flow, we tested for isolation by distance among the 4 populations for each year separately using GENALEX 6.5 [43]. A geographic distance matrix was computed based on latitude and longitude coordinates and correlations with the genetic distance matrix were tested using a Mantel test with 9999 permutations [54].
Although genetic differentiation can result from limited gene flow, forces of selection can play an important role in determining the rate of population differentiation. Selection can be detected by identifying loci that have Fst values that fall outside the Fst range for neutral loci. We used the outlier method of BayeScan 2.1 [55], that uses a Bayesian method to infer the posterior probability of each locus being under selection by defining and comparing two alternative models; including and excluding the effects of selection [56]–[57]. We used 20 pilot runs of 2000 iterations followed by a burn-in of 50 000 iterations and 100,000 iterations, and used a thinning interval of 10. Loci were considered under selection if the posterior odds (PO) was greater than 10 [56]. Subpopulations were examined since the main assumption of the underlying model is that populations exchanging migrants contribute to a common pool of migrants [56]. This assumption would have been violated if we combined data across years. We also dropped out loci whose overall frequency was <0.05, as recommended by the manual. We also used DFDIST as a second outlier detection method, implemented in MCHEZA [58]. MCHEZA allows for the use of dominant markers and models neutral distribution through coalescent simulations under a symmetric island model parameterized by the observed data of neutral loci (i.e. non-neutral loci are dropped out). The simulated neutral distribution is then used to identify individual loci that fall outside the neutral range. For each program we conducted three independent runs and corrected for multiple testing by setting a false discovery rate (FDR) <0.1.
The occurrence of genetic drift can also influence genetic diversity, the rate unfavorable alleles are fixed, and the efficiency of selection [3]. Variance effective population size (Ne
V) is an important parameter used to assess genetic drift [59]. Since we had temporally spaced samples, we estimated contemporary Ne
V using a method developed for dominant markers by [60]. Fixed alleles, alleles identified as under selection, and those with a frequency ≤0.2 were eliminated prior to estimation of Ne
V
[60]. Because Thompson clover requires several years of establishment prior to seed set, the samples were estimated to be 3 (1995–2004) and 2 (2004–2009) breeding generations apart. Since sample sizes were small and Ne reasonably large, 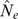 was estimated using
was estimated using 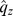 in all cases.
in all cases.
Results
Patterns of genetic diversity and differentiation
The four primer pairs revealed a total of a 129 readily discernible polymorphic bands. Table 2 summarizes the AFLP alleles from the ex situ samples collected in 1995 and 2004 and the in situ sample from the same populations in 2009. Across all populations, the number of common alleles was the same in all three sampling periods; however, more rare alleles were sampled in 2004 and 2009, compared to 1995. In the individual populations, the proportion of rare alleles was greater in TENAS and KEYSTONE, compared to BADGER and RNA, and this pattern was observed in all three sample years. The occurrence of private alleles was very low; however, there were the occurrence of common bands that were not widely shared among populations. KEYSTONE had the lowest proportion of polymorphic loci, followed by TENAS. BADGER and RNA had the highest percentage of polymorphic loci and were comparable. For all populations, percent polymorphic loci did not change over the fifteen years, with the exception of BADGER, where 2004 (79.8) was greater than 1995 (75.2) and 2009 (76.0).
Table 2. AFLP band patterns and % polymorphic loci for four populations and for the combined populations (POP), sampled for three time periods.
| BAND | BADGER | RNA | TENAS | KEYSTONE | POP |
| 1995 | |||||
| NB | 110 | 109 | – | 109 | 121 |
| CB | 110 | 109 | – | 102 | 109 |
| RB | 0 | 0 | 7 | 12 | |
| NWS | 2 | 0 | – | 0 | 0 |
| PB | 1 | 0 | – | 0 | 1 |
| %P | 75.2 | 76.7 | – | 69.0 | 73.6 |
| 2004 | |||||
| NB | 111 | 118 | 112 | 114 | 126 |
| CB | 105 | 109 | 99 | 100 | 109 |
| RB | 6 | 9 | 13 | 14 | 17 |
| NWS | 2 | 0 | 3 | 1 | 0 |
| PB | 0 | 0 | 0 | 0 | 0 |
| %P | 79.8 | 77.5 | 72.9 | 69.0 | 74.8 |
| 2009 | |||||
| NB | 109 | 118 | 113 | 109 | 127 |
| CB | 107 | 112 | 102 | 97 | 109 |
| RB | 2 | 6 | 11 | 12 | 18 |
| NWS | 3 | 3 | 2 | 1 | 0 |
| PB | 0 | 0 | 0 | 0 | 1 |
| %P | 76.0 | 77.5 | 72.1 | 67.4 | 73.2 |
NB = total number of bands; CB = Common bands (number of different bands with a frequency >0.05); RB = Rare bands (number of different bands with a frequency <0.05); NWS = Not widely shared bands, (common bands found in 25% or fewer populations); PB = number of private bands; %P = percent polymorphic loci at the 5% level, expressed as a percentage.
Across all populations there was no significant change in gene diversity (P<0.05) from 1995 to 2009. Table 3 summarizes the gene diversity statistics for each subpopulation. In 1995, KEYSTONE had significantly less diversity than BADGER and RNA. TENAS and KEYSTONE also had significantly less diversity in 2004 and 2009 compared to BADGER and RNA. Interestingly, the BADGER 1995 subpopulation, based on eighteen individual plants, had significantly greater gene diversity than 2004 and 2009 subpopulations based on >50 individual plants. Diversity measurements were the same for RNA, TENAS and KEYSTONE regardless of the year sampled. The four populations showed significant but low genetic differentiation (P<0.001). Populations located close together had less differentiation than populations located further apart. The overall estimate for Fst was 0.05. Pairwise Fst between geographically close sites ranged from.03 in 1995 and 2004 to.04 in 2009. In contrast, Fst between BADGER and KEYSTONE, which were the furthest away, ranged from.07 in 1995, .05 in 2004 and.06 in 2009. Population pairwise PhiPT values showed a similar result (Table 4).
Table 3. Expected heterozygosity (He) under Hardy-Weinberg genotypic proportions for ex situ samples collected in 1995, recollected in 2004, and in situ populations sampled in 2009.
| Location | 1995 | 2004 | 2009 |
| BADGER | 0.292 aa | 0.276 ab | 0.275 ab |
| RNA | 0.278 aa | 0.271 aa | 0.274 aa |
| TENAS | – | 0.247 ba | 0.250 ba |
| KEYSTONE | 0.250 ba | 0.250 ba | 0.242 ba |
Differences in first letter correspond to significant differences in He among the four locations within the same year. Differences in second letter correspond to significant differences in He within location across the three years sampled (Bonferroni correction for testing seven hypotheses based on overall critical p-value of 5% is ≤0.007).
Table 4. Geographic distance (km) between each location (above diagonal) and population pairwise PhiPT values (below diagonal) based on samples collected in 2004 and 2009 (Values significant at P<0.001).
| Population | BADGER | RNA | TENAS | KEYSTONE |
| BADGER | – | 10.07 | 9.09 | 13.21 |
| RNA | 0.068 | – | 2.47 | 8.21 |
| TENAS | 0.081 | 0.046 | – | 6.31 |
| KEYSTONE | 0.099 | 0.087 | 0.085 | – |
Spatial and temporal structure
Based on the AMOVA analysis, 90% of the variation occurred within populations. Seven percent of the variation occurred among populations and 3% occurred among subpopulations collected within each location (P-value <0.01 with 999 permutations) (Table 5). The neighbor-joining tree based on Nei’s genetic distance is shown in Fig. 2. Similar to the AMOVA, the tree suggested that spatial structure was more significant than temporal structure. Subpopulations collected from the same location clustered together; however, populations sampled in 1995 were less similar to populations sampled in 2004 and 2009, which were more closely related. Genetic structure associated with geographic distance was also evident. Subpopulations collected at KEYSTONE, the northern-most site, were genetically more distant compared to the subpopulations collected at TENAS, RNA, and BADGER. However, KEYSTONE subpopulations were more closely related to TENAS, which is closer, geographically, and least related to BADGER, which is the site furthest away.
Table 5. Nested analysis of molecular variance (AMOVA) based on 129 polymorphic loci based on populations sampled at three locations (BADGER, RNA, and KEYSTONE), for three different years (1995, 2004, 2009).
| AMOVA | df | MS | Variation | % variation | Phi | P-value |
| Location | 2 | 225.538 | 1.229 | 7 | 0.069 | 0.01 |
| Year/location | 6 | 40.166 | 0.529 | 3 | 0.032 | 0.01 |
| Subpopulation | 431 | 16.085 | 16.085 | 90 | 0.099 | 0.01 |
| Total | 439 | 17.844 | 100 |
Figure 2. Neighbor-joining tree showing genetic relationship among Thompson clover.

Relatedness of four populations of Thompson clover sampled in 1995, 2004, and 2009, based on Nei’s genetic distance estimated using AFLPs.
The STRUCTURE analysis also supported strong spatial structure. The plot of P(X|K) indicated that K = 2 and K = 4 were the most likely groupings (Figure S1). Fig. 3 shows Q-plots for each K group which are useful for visualizing population genetic structure and the presence of admixture. At K = 2, the KEYSTONE population stood out as distinct from BADGER, RNA and TENAS, in all three years. As with the cluster analysis, geographic structure was apparent. Looking at the level of admixture, the proportion of alleles belonging to the KEYSTONE group decreased from TENAS to BADGER, reflecting increasing geographic distance. At K = 4, geographic differentiation between the four populations was apparent. Although not as distinct, variation across sampling year was apparent. For example in BADGER 1995, admixture with RNA and TENAS was apparent, but the proportion of alleles belonging to RNA was much less in BADGER 2004 and BADGER 2009. The proportion of alleles belonging to TENAS was somewhat less in BADGER 2004, and much less in BADGER 2009.
Figure 3. Q-plots based on AFLP analyses.
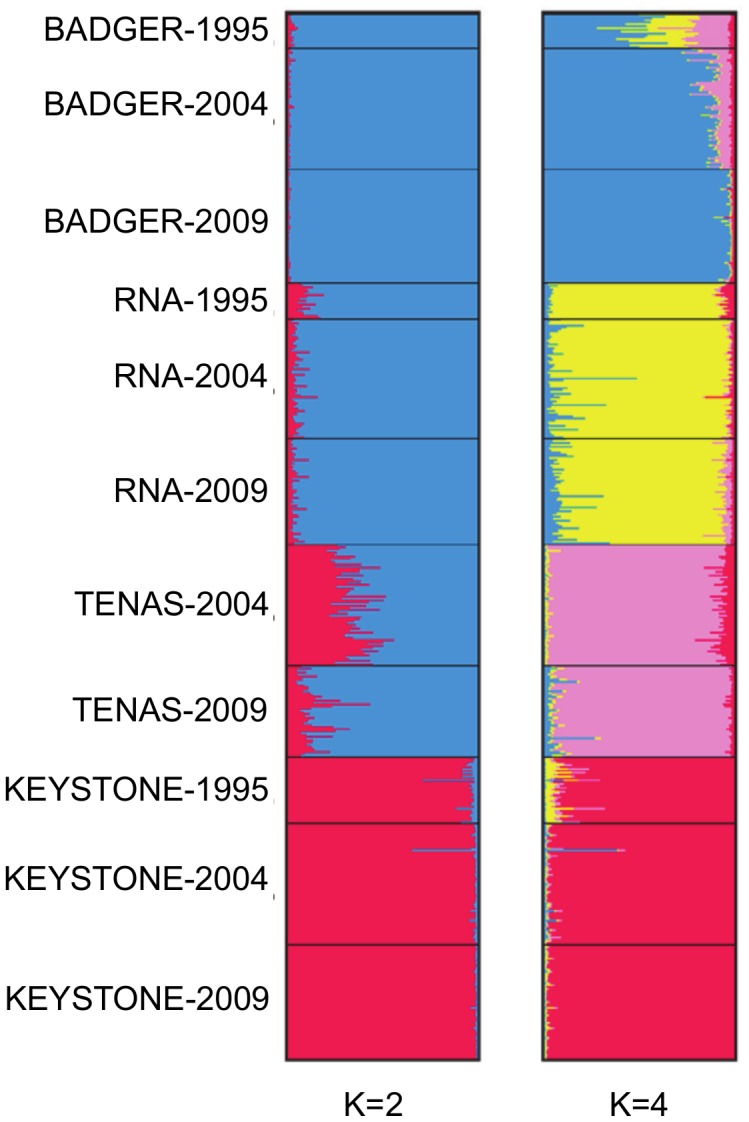
Bayesian analysis using the software STRUCTURE suggested most likely groups of K = 2 and K = 4.
Evidence for gene flow
The assignment tests suggested that limited gene flow did occur between the four populations. With AFLPOP, (Table 6), individuals were assigned to the correct source population at a rate that ranged from 80 to 32%. Generally individuals from subpopulations collected at the same location were assigned with a similar level of success to the correct population. TENAS had the highest success, averaging 71% across 2004 and 2009. RNA had the lowest success rate, averaging 43% across the three sampling years. Seventy six percent of assignment events between locations were between locations that were adjacent. However, there was evidence for long distance gene flow from BADGER to RNA, RNA to KEYSTONE, BADGER to TENAS and TENAS to BADGER. Migration between populations did not appear symmetrical. There were twice as many genotypes from RNA assigned to BADGER, then from BADGER to RNA. There were twice as many genotypes from TENAS assigned to RNA, as there were from RNA to TENAS. Only one genotype from KEYSTONE was assigned to a location other than KEYSTONE. Mantel tests for isolation by distance found a significant and positive relationship between genetic distance and geographic distance (P<0.001, 9999 permutations) which suggested that gene flow between the four populations was limited, but did occur based on the evidence provided by the assignment tests.
Table 6. Assignment of 551 individuals from seven subpopulations (columns) to source populations (rows) to detect gene flow between four locations over 15 years.
| Allocated to | BAD95 | BAD04 | BAD09 | RNA95 | RNA04 | RNA09 | TENAS04 | TENAS09 | KEY95 | KEY04 | KEY09 | Total |
| BAD 1995 | 11 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 11 |
| BAD 2004 | 0 | 40 | 7 | 0 | 1 | 2 | 0 | 0 | 0 | 0 | 0 | 50 |
| BAD 2009 | 0 | 8 | 33 | 0 | 2 | 1 | 0 | 1 | 0 | 0 | 0 | 45 |
| RNA 1995 | 1 | 0 | 0 | 8 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 10 |
| RNA 2004 | 0 | 1 | 0 | 3 | 31 | 9 | 2 | 0 | 0 | 0 | 0 | 46 |
| RNA 2009 | 0 | 0 | 1 | 0 | 8 | 22 | 0 | 4 | 0 | 0 | 0 | 35 |
| TEN 2004 | 1 | 1 | 0 | 0 | 0 | 0 | 51 | 1 | 1 | 0 | 0 | 55 |
| TEN 2009 | 0 | 0 | 0 | 0 | 1 | 2 | 6 | 30 | 0 | 0 | 0 | 39 |
| KEY 1995 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 19 | 4 | 0 | 25 |
| KEY 2004 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 5 | 21 | 3 | 29 |
| KEY 2009 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 14 | 35 | 50 |
| CNMa | 5 | 14 | 19 | 8 | 18 | 20 | 3 | 11 | 10 | 25 | 22 | 155 |
| # IND | 18 | 64 | 60 | 19 | 63 | 56 | 63 | 48 | 35 | 64 | 60 | 551 |
Criteria not met (CNM) to meet minimal log-likelihood difference of 0.5 for the allocation of an individual to a population.
Extent of selection and genetic drift
Outlier loci detection suggested that most of the markers we examined were neutral but there was some signature of selection. BayeScan 2.1 identified no significant outliers among the 4 K groups at either prior odds of 10∶1 or 1∶1 for all three years sampled. However, DFDIST showed a high probability (>95%) for divergent selection for 9, 7 and 9 loci in 1995, 2004 and 2009, respectively. In one instance, the same locus experienced selection in all three years, while a second locus experienced selection in 1995 and 2004, and a third, experienced selection in 2004 and 2009.
When we estimated effective population size, we found that NeV declined in RNA from 163 to 113. However, it increased in BADGER and KEYSTONE (Table 7). This coincided with the census population numbers (Table 1), that suggested a drop from 6 million plants in 1998 to slightly more than 1.5 million plants in 2008 at RNA while BADGER and KEYSTONE remained relatively stable.
Table 7. Estimated population sizes (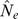 ) of the four locations over the three sampling years.
) of the four locations over the three sampling years.
| Years | BADGER | RNA | TENAS | KEYSTONE |
| Loci | 66 | 77 | 83 | 77 |
| 1995–2004 | 30 | 163 | – | 105 |
| 2004–2009 | 44 | 113 | 57 | 156 |
Discussion
General patterns of genetic diversity and population structure
Although T. thompsonii is a narrowly distributed endemic species, our results showed that it did not have a narrow genetic base. The overall level of genetic diversity (0.26) was similar to comparable AFLP studies on widespread perennial insect-pollinated congeners, T. montanum L., (0.23), [61], and T. alpinum L., (0.24), [62], and higher than T. repens, (0.1) [63]. Average percent polymorphic loci was higher (73%) than reports for T. montanum (58%) [61] and T. repens (31%) [63]. Diversity estimates were also similar to comparable AFLP studies in non related herbaceous perennial insect pollinated species, such as Silene chlorantha (Willd.) (0.20) [34], Abronia alpina Brandegee (.28) [64], and Echinacea laevigata (Boynton and Beadle) Blake (0.26) [65].
Among the four populations, KEYSTONE and TENAS, the northern most populations, had less overall diversity and a lower percent of polymorphic loci compared to BADGER and RNA. They also had about twice as many rare bands. An explanation may be that these populations are relicts of the leading north edge of the species during the Pleistocene glaciations. These populations lie due south of the furthest extent of the Okanogan lobe of the Cordillera ice sheet, and although major valley glaciers extended eastward from the Cascade Range, the closest stopped at the upper Entiat river, approximately 10 km west of KEYSTONE [28], [66]. Simulation studies have shown that neutral genetic variation can be lower along the leading edge of a species range due to founder effects and allele surfing [67], [68].
BADGER was the only population that showed a significant decline in genetic diversity from 1995 to 2004. It was also the only population where a private band was detected. Considering that this population is located in open shrub steppe habitat, as opposed to the other three populations that are located in the forest transition zone, it may contain unique alleles that help it persist in a drier, hotter environment. Although resampling suggested that uneven sampling sizes did not bias our genetic estimates, we would expect if bias was present, sampling too few plants would have underestimated diversity. Although we sampled fewer plants in 1995, compared to 2004 and 2009, 20 individuals were determined to be adequate to assess genetic variation in T. repens, also an insect-pollinated outcrossing perennial species [69]. Our results were also consistent with field observations. Although the census population size of BADGER has remained relatively stable, we observed that the population was protected from grazing in 1995, but was not protected when we returned in 2004 and 2009. Because BADGER is in a unique habitat, compared to other populations, and does not have State or Federal protection, close monitoring is warranted to ensure this population does not continue to decline in the future.
Relative influence of gene flow, selection and genetic drift
Although genetic differentiation between the four populations was significant (albeit weak), as was our test of isolation by distance, there was sufficient evidence to suggest that gene flow did occur among the studied populations.
The assignment test suggested infrequent gene flow occurred, despite the distance between populations. The asymmetrical flow of genes was concordant with site specific characteristics, in terms of climate and topography. The occurrence of twice as many migration events from RNA to BADGER, compared to BADGER to RNA supported the hypothesis that prevailing northwesterly winds may disperse fruits of T. thompsonii or contribute to the long distance movement of insect pollinators, since RNA is located northwest of BADGER [28]. Gene flow also occurred more frequently from TENAS to RNA than from RNA to TENAS. Although separated by 2.5 km, TENAS and RNA occur along the same road. However, TENAS is at a higher elevation, so seed and pollen would more likely move downhill, from TENAS to RNA. Results from the assignment tests reflected the influence of distance as well. Gene flow appeared to have occurred between KEYSTONE and TENAS, separated by 6.31 km, but was restricted between KEYSTONE and RNA, (8.21 km), and did not occur between KEYSTONE and BADGER (13.21 km). The usefulness of topographic data to infer gene flow in wild Trifolium populations has been reported and has been concordant with data generated from morphologic [70], isozyme [71], RAPD [70] and AFLP [63] markers. This supports the use of maps and satellite images to infer gene flow due to the occurrence of landscape barriers or corridors that influence the connectivity between populations.
Although there was some evidence that selective forces were responsible for influencing Fst values for some of the markers we examined, it was only for a few. However one marker was identified as being under divergent selection in all three years examined, and may be associated with an adaptive trait. Although succession has been occurring in the area where RNA, TENAS and KEYSTONE are located since the 1988 fire, our marker set did not detect any overall temporal trends in changing selection pressure. Studies similar to [72] would be useful to examine this in further detail since it has direct relevance to recollecting intervals.
There was limited evidence for genetic drift although we did detect a decline in effective population size in RNA, which was concordant with consensus data. However, we did not detect a decrease in genetic diversity. Our study suggested that further monitoring is warranted in RNA to ensure population decline does not have a negative impact since the population is the largest and oldest in situ reserve of Thompson clover.
Altogether, we tracked the genetic structure of 4 different populations of Thompson clover over 15 years. With the exception of BADGER, the populations did not appear to have undergone substantial change during the time period of our study. Changes in genetic structure of the RNA, TENAS and KEYSTONE populations due to succession following the 1988 fire, were not evident. Although gene flow was evident, admixture tended to decline over time, especially in BADGER and KEYSTONE. This suggested that despite the occasional influx of migrants, individual populations tended to return to equilibrium, as unadapted genotypes were selected against. Overall, spatial differences among the 4 study locations accounted for more than twice the variation as temporal differences within study locations. These results suggested that recollecting intervals for ex situ samples of Thompson clover could be greater than 15–20 years.
Conclusion
In this paper, we report the spatial and temporal genetic variation and population substructure revealed by AFLP markers in T. thompsonii populations sampled from four diverse locations over a 15 year time period. Although there was some evidence for gene flow, selection and drift, there was no difference in overall diversity between ex situ samples collected in 1995 and 2004 and in situ samples collected in 2009. Only one population showed a decline in genetic diversity over the 15 years studied. Our results echo other reports that have found that perennial out crossing species generally showed less difference in genetic diversity between in situ and ex situ accessions compared to annual, self-pollinated species [73].
We can use the lessons we learned in our study to formulate some general guidelines to help ensure ex situ samples of insect-pollinated herbaceous perennial species provide clear snap shots of in situ populations. Our study suggested that a successful ex situ collecting strategy is to collect from a large number of plants (>150 plants), equalize maternal contribution by sampling the same number of heads per plant, and sample along random transects with sufficient space between plants to minimize intrafamilial sampling. We also learned that long distance dispersal can occur, and appeared to play an important role in limiting the extent that our four populations diverged from one another. Long distance gene flow by insect pollinators not only maintained genetic connectivity, but can also help in maintaining and increasing effective population size [74], [75]. Only one immigrant every other generation is needed to prevent differentiation due to genetic drift [76], [77]. Long distance dispersal of pollen by insects can be highly variable since it is dependent upon plant species, pollination vector and the environment [74], so the effective conservation of insect pollinated species requires conserving not only the plant species, but pollinator diversity and abundance [78]. Our results suggested that as a general guideline, for a given geographic area, the benefits of long distance gene flow may allow us to conserve the intraspecific diversity of insect pollinated species (especially populations that occur in similar habitats), with fewer populations, since populations are likely to be weakly divergent. In contrast, self pollinated species, especially annuals, may need more populations protected, since we would expect individual populations to be strongly divergent, due to isolation by distance.
The most important lesson we can apply to the effective ex situ and in situ conservation of PGR is the value of comparing the genetic structure of ex situ and in situ samples. Quantifying genetic change over time not only allows genetic resource managers to validate conservation protocols, develop appropriate recollection intervals, but provides them with an early detection mechanism for identifying problematic conditions that can be addressed to prevent the local extinction of vulnerable in situ populations.
Supporting Information
The plot of P(X|K) indicated that K = 2 and K = 4 were the most likely groupings.
(TIF)
Results from resampling to determine unequal sample bias.
(DOCX)
Acknowledgments
We gratefully acknowledge the assistance of Greg Sinton, Adam Smolenski, Andrew Bell, Alexandr Afonin, Francisco González, Jamie Calley, Estela Cervantes and Jesse Prieto in surveying and collecting. Thanks to Lisa Taylor for AFLP lab work and Gwen Pentecost for producing Fig. 1. We wish to thank Jinguo Hu for helpful comments and suggestions.
Funding Statement
Sources of funding included USDA Agricultural Research Service National Program 301 and 215 and the International Treaty on Plant Genetic Resources for Food and Agriculture - FAO. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Jensen HR, Dreiseitl A, Sadiki M, Schoen DJ (2012) The Red Queen and the seed bank: Pathogen resistance of ex situ and in situ conserved barley. Evol Appl 5: 353–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Del Rio AH, Bamberg JB, Huaman Z, Salas A, Vega SE (1997) Assessing changes in the genetic diversity of potato gene banks. 2. In situ vs ex situ . Theor Appl Genet 95: 199–204. [Google Scholar]
- 3. Negri V, Tiranti B (2010) Effectiveness of in situ and ex situ conservation of crop diversity. What a Phaseolus vulgaris L. landrace case study can tell us. Genetica 138: 985–998. [DOI] [PubMed] [Google Scholar]
- 4. Maxted N, Kell S, Ford-Lloyd B, Dulloo E, Toledo Á (2012) Toward the systematic conservation of global crop wild relative diversity. Crop Sci 52: 774–785. [Google Scholar]
- 5.Maxted N, Ford-Lloyd BV, Kell SP (2008). Crop wild relatives: establishing the context. In: Maxted N, Ford-Lloyd BV, Kell, SP, Iriondo J, Dullo E, Turok J, editors. Crop wild relative conservation and use. Wallingford: CAB International. 3–30.
- 6. Bretting PK, Duvick DN (1997) Dynamic Conservation of Plant Genetic Resources. Advances in Agronomy 61: 1–51. [Google Scholar]
- 7. Soleri D, Smith SE (1995) Morphological and phenological comparisons of two hopi maize varieties conserved in situ and ex situ . Econ Bot 49: 56–77. [Google Scholar]
- 8. Rice EB, Smith ME, Mitchell SE, Kresovich S (2006) Conservation and change: A comparison of in situ and ex situ conservation of Jala maize germplasm. Crop Sci 46: 428–436. [Google Scholar]
- 9. Gómez OJ, Blair MW, Frankow-Lindberg BE, Gullberg U (2005) Comparative study of common bean (Phaseolus vulgaris L.) landraces conserved ex situ in genebanks and in situ by farmers. Genet Res Crop Evol 52: 371–380. [Google Scholar]
- 10. Malice M, Martin N, Pissard A, Rojas-Beltran JA, Gandarillas A, et al. (2007) A preliminary study of the genetic diversity of Bolivian oca (Oxalis tuberosa Mol.) varieties maintained in situ and ex situ through the utilization of ISSR molecular markers. Genet Res Crop Evol 54: 685–690. [Google Scholar]
- 11. Li Q, Xu Z, He T (2002) Ex situ genetic conservation of endangered Vatica guangxiensis (Dipterocarpaceae) in China. Biol Conserv 106: 151–156. [Google Scholar]
- 12. Li Q, He T, Xu Z (2005) Genetic evaluation of the efficacy of in situ and ex situ conservation of Parashorea chinensis (Dipterocarpaceae) in Southwestern China. Biochem Genet 43: 387–406. [DOI] [PubMed] [Google Scholar]
- 13. Etisham-Ul-Haq M, Allnutt TR, Smith-Ramírez C, Gardner MF, Armesto JJ, et al. (2001) Patterns of genetic variation in in and ex situ populations of the threatened chilean vine Berberidopsis corallina, detected using RAPD markers. Ann Bot 87: 813–821. [Google Scholar]
- 14. Che YH, Yang YP, Yang XM, Li XQ, Li LH (2011) Genetic diversity between ex situ and in situ samples of Agropyron cristatum (L.) Gaertn. based on simple sequence repeat molecular markers. Crop Pasture Sci 62: 639–644. [Google Scholar]
- 15.Marshall DR, Brown AHD (1975) Optimal sampling strategies in genetic conservation In: Frankel OH, Hawkes JG, editors. Crop genetic resources for today and tomorrow, International Biological Programme 2. Cambridge:Cambridge University Press. 53–80.
- 16.Marshall DR, Brown AHD (1995) A basic sampling strategy: theory and practice. In: Guarino L, Ramanathan Rao V, Reid R editors. Collecting Plant Genetic Diversity: Technical Guidelines UK:Cab International. 75–92.
- 17.Von Bothmer R, Seberg O (1995) Strategies for collecting wild species. In: Guarino L, Ramanathan Rao V, Reid R editors. Collecting Plant Genetic Diversity: Technical Guidelines UK: Cab International. 93–112.
- 18.Breese EL (1989) Regeneration and multiplication of germplasm resources in seed gene banks: the scientific background. Rome: IBPGR.
- 19.Sackville Hamilton NR, Chorlton KH (1997) Regeneration of accessions in seed collections: a decision guide. Handbook for genebanks No. 5. Rome:International Plant Genetic Resources Institute. 75 p. [Google Scholar]
- 20. Brown AHD, Brubaker CL, Grace J P (1997) Regeneration of Germplasm Samples: Wild Versus Cultivated Plant Species. Crop Sci 37: 7–13. [Google Scholar]
- 21. Tin HQ, Berg T, Bjørnstad Å (2001) Diversity and adaptation in rice varieties under static (ex situ) and dynamic (in situ) management. Euphytica 122: 491–502. [Google Scholar]
- 22.Osborn TJ, Briffa KR (2006) The spatial extent of 20th-century warmth in the context of the past 1200 years Science 311, 841–844. [DOI] [PubMed]
- 23. Jarvis A, Lane A, Hijmans RJ (2008) The effect of climate change on crop wild relatives. Agric Ecosyst Environ 126: 12–23. [Google Scholar]
- 24. Pauls SU, Nowak C, Bálint M, Pfenninger M (2013) The impact of global climate change on genetic diversity within populations and species. Mol Ecol 22: 925–946. [DOI] [PubMed] [Google Scholar]
- 25.NatureServe (2011) NatureServe Explorer: An online encyclopedia of life [web application]. Version 7.1. NatureServe, Arlington, Virginia. Available: http://www.natureserve.org/explorer. Accessed: 30 January 2012.
- 26. Morris JB, Greene SL (2001) Defining a multiple-use germplasm collection for the genus Trifolium . Crop Sci 41: 893–901. [Google Scholar]
- 27. Scherer G, Zamora B, Everett R, Roch B Jr (1996) Habitat characteristics and morphological differences of Trifolium thompsonii populations. Northwest Sci 70: 242–251. [Google Scholar]
- 28.Canfield JE (1977) The ecological life history of Trifolium thompsonii Morton, with reference to its restricted distribution. [Masters Thesis]: University of Washington. 145p.
- 29. Harrod RJ, Halpern CB (2005) Effects of experimental shading and removal of competitors on vegetative and reproductive performance of Trifolium thompsonii Morton, a rare endemic herb of eastern Washington. Nat Areas J 25: 156–164. [Google Scholar]
- 30. Harrod RJ, Halpern CB (2009) Effects of experimental burning on individual performance and population structure of two rare plants of north central Washington. Restoration Ecol 17: 215–225. [Google Scholar]
- 31. Rivers MC, Brummitt NA, Lughadha EN, Meagher TR (2011) Genetic variation in Delonix s.l. (Leguminosae) in Madagascar revealed by AFLPs: Fragmentation, conservation status and taxonomy. Conserv Genet 12: 1333–1344. [Google Scholar]
- 32. Baskauf CJ, Burke JM (2009) Population genetics of Astragalus bibullatus (Fabaceae) using AFLPs. J Hered 100: 424–431. [DOI] [PubMed] [Google Scholar]
- 33. Garrido JL, Fenu G, Mattana E, Bacchetta G (2012) Spatial genetic structure of Aquilegia taxa endemic to the island of Sardinia. Ann Bot 109: 953–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lauterbach D, Burkart M, Gemeinholzer B (2012) Rapid genetic differentiation between ex situ and their in situ source populations: An example of the endangered Silene otites (Caryophyllaceae). Bot J Linn Soc 168: 64–75. [Google Scholar]
- 35. Bonin A, Bellemain E, Eidesen PB, Pompanon F, Brochmann C, et al. (2004) How to track and assess genotyping errors in population genetics studies. Mol Ecol 13: 3261–3273. [DOI] [PubMed] [Google Scholar]
- 36. Vos P, Hogers R, Bleeker M, Reijans M, van de Lee T, et al. (1995) AFLP: A new technique for DNA fingerprinting. Nucleic Acids Res 23: 4407–4414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bomblies K, Yan L, Laitinen RA, Kim S-T, Hollister JD, et al. (2010) Local-Scale Patterns of Genetic Variability, Outcrossing, and Spatial Structure in Natural Stands of Arabidopsis thaliana. PLoS Genet 6(3): e1000890 doi:10.1371/journal.pgen.1000890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Gomaa NH, Montesinos-Navarro A, Alonso-Blanco C (2011) Temporal variation in genetic diversity and effective population size of Mediterrean and subalpine Arabidopsis thaliana populations. Mol Ecol 20: 3540–3554. [DOI] [PubMed] [Google Scholar]
- 39. Ehrich D (2006) AFLPdat: a collection of R functions for convenient handling of AFLP data. Mol Ecol Notes 6: 603–604. [Google Scholar]
- 40. Vekemans X, Beauwens T, Lemaire M, Roldan-Ruiz I (2002) Data from amplified fragment length polymorphism (AFLP) markers show indication of size homoplasy and of a relationship between degree of homoplasy and fragment size. Mol Ecol 11: 139–151. [DOI] [PubMed] [Google Scholar]
- 41. Zhivotovsky LA (1999) Estimating population structure in diploids with multilocus dominant DNA markers. Mol Ecol 8: 907–913. [DOI] [PubMed] [Google Scholar]
- 42. Peakall R, Smouse PE (2006) GENALEX 6: genetic analysis in Excel. Population genetic software for teaching and research. Mol Ecol Notes 6: 288–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Peakall R, Smouse PE (2012) GENALEX 6.5: Genetic analysis in Excel. Population genetic software for teaching and research-an update. Bioinformatics 28: 2537–2539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Nei M, Takezaki N (1983) Estimation of genetic distances and phylogenetic trees from DNA analysis. Proc. 5th World Cong. Genet. Appl. Livestock Prod. 21: 405–412. [Google Scholar]
- 45. Liu K, Muse SV (2005) PowerMarker: an integrated analysis environment for genetic marker analysis. Bioinformatics 21: 2128–2129. [DOI] [PubMed] [Google Scholar]
- 46.Felsenstein J (1993) Phylogeny Inference Package (PHYLIP). Version 3.5. Seattle: University of Washington.
- 47. Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155: 945–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Falush D, Stephens M, Pritchard JK (2003) Inference of population structure using multilocus genotype data: Linked loci and correlated allele frequencies. Genetics. 164: 1567–1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Falush D, Stephens M, Pritchard JK (2007) Inference of population structure using multilocus genotype data: dominant markers and null alleles. Mol Ecol Notes 7: 574–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Evanno G, Regnaut R, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Mol Ecol 14: 2611–2620. [DOI] [PubMed] [Google Scholar]
- 51. Rosenberg NA (2004) DISTRUCT: a program for the graphical display of population structure. Mol Ecol Notes 4: 137–138. [Google Scholar]
- 52. Duchesne P, Bernatchez L (2002) AFLPOP: A computer program for simulated and real population allocation based on AFLP data. Mol Eco Notes 3: 380–383. [Google Scholar]
- 53. Paetkau D, Calvert W, Stirling I, Strobeck C (1995) Microsatellite analysis of population structure in Canadian polar bears. Mol Ecol 4: 347–354. [DOI] [PubMed] [Google Scholar]
- 54. Mantel N (1967) The detection of disease clustering and a generalized regression approach. Cancer Res. 27: 209–220. [PubMed] [Google Scholar]
- 55. Foll M, Fischer MC, Heckel G, Excoffier L (2010) Estimating population structure from AFLP amplification intensity. Mol Ecol 19: 4638–4647. [DOI] [PubMed] [Google Scholar]
- 56. Foll M, Gaggiotti O (2008) A Genome-Scan method to identify selected loci appropriate for both dominant and codominant markers: A Bayesian perspective. Genetics 180: 977–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Fischer MC, Foll M, Excoffier L, Heckel G (2011) Enhanced AFLP genome scans detect local adaptation in high-altitude populations of a small rodent (Microtus arvalis). Mol. Ecol. 20: 1450–1462. [DOI] [PubMed] [Google Scholar]
- 58. Antao T, Beaumont MA (2011) Mcheza: A workbench to detect selection using dominant markers. Bioinformatics 27: 1717–1718. [DOI] [PubMed] [Google Scholar]
- 59. Leberg P (2005) Genetic approaches for estimating the effective size of populations. J Wildl Manage 69: 1385–1399. [Google Scholar]
- 60. Jorde PE, Palm S, Ryman N (1999) Estimating genetic drift and effective population size from temporal shifts in dominant gene marker frequencies Mol Ecol. 8: 1171–1178. [DOI] [PubMed] [Google Scholar]
- 61.Hahn T, Kettle CJ, Ghazoul J, Frei ER, Matter P, et al.. (2012) Patterns of genetic variation across altitude in three plant species of semi-dry grasslands PLoS ONE, 7(8) art. no. e41608. [DOI] [PMC free article] [PubMed]
- 62. Meirmans PG, Goudet J, Gaggiotti OE (2011) Ecology and life history affect different aspects of the population structure of 27 high-alpine plants. Mol Ecol 20: 3144–3155. [DOI] [PubMed] [Google Scholar]
- 63.Hargreaves S, Maxted N, Hirano R, Abberton M, Skøt L, et al. (2010) Islands as refugia of Trifolium repens genetic diversity. Conserv Genet: 1317–1326.
- 64. Jabis MD, Ayers TJ, Allan GJ (2011) Pollinator-mediated gene-flow forsters genetic variability in a narrow alpine endemic, Abronia alpine (Nyctaginaceae). Am J Bot 98: 1583–1594. [DOI] [PubMed] [Google Scholar]
- 65. Peters MD, Xiang Q, Thomas DT, Stucky J, Whiteman NK (2009) Genetic analysis of the federally endangered Echinacea laevigata using amplified fragment length polymorphisms (AFLP)- inferences in population genetic structure and mating system. Conserv Genet 10: 1–14. [Google Scholar]
- 66.Waitt J, Thorson (1983) The Cordilleran Ice Sheet in Washington, Idaho, and Montana. In: Wright J. editor. Late-Quaternary Environments of the United States, Volume 1: The Late Pleistocene St Paul:University of Minnesota Press, 407 p. [Google Scholar]
- 67. Cobben MMP, Verboom J, Opdam PFM, Hoekstra RF, Jochem R, et al. (2011) Projected climate change causes loss and redistribution of genetic diversity in a model metapopulation of a medium-good disperser. Ecography 34: 920–932. [Google Scholar]
- 68. McInerny GJ, Turner JRG, Wong HY, Travis JMJ, Benton TG (2009) How range shifts induced by climate change affect neutral evolution. Proc R Soc Edinb Biol Sci 276: 1527–1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Khanlou KM, Vandepitte K, Asl LK, van Bockstaele E (2011) Towards an optimal sampling strategy for assessing genetic variation within and among white clover (Trifolium repens L.) cultivars using AFLP. Int J Genet Mol Biol 34: 252–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Greene SL, Gritsenko M, Vandemark G (2004) Relating morphologic and RAPD marker variation to collection site environment in wild populations of red clover (Trifolium pratense L.) Gen Res Crop Evol. 51: 643–653. [Google Scholar]
- 71. Mosjidis JA, Greene SL, Klingler KA, Afonin A (2004) Isozyme diversity in wild red clover populations from the Caucasus. Crop Sci 44: 665–670. [Google Scholar]
- 72. Bothwell H, Bisbing S, Therkildsen NO, Crawford L, Alvarez N, et al. (2013) Identifying genetic signatures of selection in a non-model species, alpine gentian (Gentiana nivalis L.), using a landscape genetic approach. Conserv Genet 14: 467–481. [Google Scholar]
- 73. Brütting C, Hensen I, Wesche K (2013) Ex situ cultivation affects genetic structure and diversity in arable plants. Plant Biol 15: 505–513. [DOI] [PubMed] [Google Scholar]
- 74. Ellstrand N (1992) Gene flow by pollen: implications for plant conservation genetics. Oikos 63: 77–86. [Google Scholar]
- 75. Trakhtenbrot A, Nathan R, Perry G, Richardson DM (2005) The importance of long-distance dispersal in biodiversity conservation. Diver Distrib 11: 173–181. [Google Scholar]
- 76. Wright S (1931) Evolution in Mendelian populations. Genetics 16: 97–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Slatkin M (1985) Gene flow in natural populations. Annu Rev Ecol Syst 16: 393–430. [Google Scholar]
- 78. Steffan-Dewenter I, Westphal C (2008) The interplay of pollinator diversity, pollination services and landscape change. J Appl Ecol 45: 737–741. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The plot of P(X|K) indicated that K = 2 and K = 4 were the most likely groupings.
(TIF)
Results from resampling to determine unequal sample bias.
(DOCX)