

Abstract
Catechol (benzenediol) is present in plant-derived products, such as vegetables, fruits, coffee, tea, wine, areca nut and cigarette smoke. Because platelet dysfunction is a risk factor of cardiovascular diseases, including stroke, atherosclerosis and myocardial infarction, the purpose of this study was to evaluate the anti-platelet and anti-inflammatory effect of catechol and its mechanisms. The effects of catechol on cyclooxygenase (COX) activity, arachidonic acid (AA)-induced aggregation, thromboxane B2 (TXB2) production, lactate dehydrogenase (LDH) release, reactive oxygen species (ROS) production and extracellular signal-regulated kinase (ERK)/p38 phosphorylation were determined in rabbit platelets. In addition, its effect on IL-1β-induced prostaglandin E2 (PGE2) production by fibroblasts was determined. The ex vivo effect of catechol on platelet aggregation was also measured. Catechol (5-25 µM) suppressed AA-induced platelet aggregation and inhibited TXB2 production at concentrations of 0.5–5 µM; however, it showed little cytotoxicity and did not alter U46619-induced platelet aggregation. Catechol (10–50 µM) suppressed COX-1 activity by 29–44% and COX-2 activity by 29–50%. It also inhibited IL-1β-induced PGE2 production, but not COX-2 expression of fibroblasts. Moreover, catechol (1–10 µM) attenuated AA-induced ROS production in platelets and phorbol myristate acetate (PMA)-induced ROS production in human polymorphonuclear leukocytes. Exposure of platelets to catechol decreased AA-induced ERK and p38 phosphorylation. Finally, intravenous administration of catechol (2.5–5 µmole/mouse) attenuated ex vivo AA-induced platelet aggregation. These results suggest that catechol exhibited anti-platelet and anti-inflammatory effects, which were mediated by inhibition of COX, ROS and TXA2 production as well as ERK/p38 phosphorylation. The anti-platelet effect of catechol was confirmed by ex vivo analysis. Exposure to catechol may affect platelet function and thus cardiovascular health.
Introduction
Various benzenediols (e.g., catechol, resorcinol, hydroquinone [HQ]) are present in many plant-derived products, including vegetables, areca nut, fruits, grains, coffee, tea, beer, and wine [1]–[3]. Many catechol (i.e., pyrocatechol) derivatives have been suggested to have therapeutic potential. Phenol and catechol (1,2-benzenediol) are two major metabolites identified in the urine of workers occupationally exposed to benzene. In addition, catechol and HQ (e.g., 1,4-benzenediol) levels in peripheral blood may be a marker of exposure to benzene or cigarette smoke [4]. However, the effect of catechol on human health remains to be investigated.
Catechol scavenges diphenylpicrylhydrazyl radicals and reactive oxygen species (ROS) [5]. The structure of catechin and hydroxychavicol may be responsible for its ROS scavenging and anti-platelet activities [6]. However, Lee and Lin [7] found that catechol, pyrogallol and 1,2,4-benzenetriol may gerenate ROS and exhibit mutagenicity. In addition, catechol and HQ may antagonize transforming growth factor-β (TGF-β)-induced elimination of transformed cells suggesting a co-carcinogenic effect [8].
In many countries, cardiovascular diseases, such as atherosclerosis, stroke and myocardial infarction, are responsible for a great proportion of human morbidity and mortality [9]. Platelets play important roles in thrombosis and hemostasis through platelet adhesion, activation and aggregation; their aggregation may be initiated by various agonists, including adenosine diphosphate (ADP), thrombin, collagen, and prostaglandin endoperoxides. In addition, secretion of the contents of intracellular granules may accompany platelet aggregation, which may further recruit additional platelets to induce full aggregation and subsequent gross thrombus formation. Aberrant platelet activation may directly or indirectly induce blood clot formation, thrombosis and sustained vascular wall inflammation, resulting in cardiovascular diseases (e.g. atherosclerosis and cardiovascular attack) [10], [11]. Although catechol has been shown to inhibit arachidonic acid (AA)-induced platelet aggregation [12], [13], limited mechanistic information is available about its possible anti-platelet and anti-thrombotic effects or toxicity in cardiovascular cells.
Considering the frequency that humans are exposed catechol (pyrocatechol) along with its potential pharmacological or toxicological effects, the anti-platelet and anti-inflammatory effects of catechol and related signaling mechanisms (e.g., cyclooxygenase [COX] inhibition, thromboxane, ROS, ERK/p38 phosphorylation, etc.) were assessed in this study to determine its effects on cardiovascular health.
Materials and Methods
Materials
Catechol (pyrocatechol), arachidonic acid (AA), U46619 (a thromboxane receptor agonist), 2′,7′-diacethyl-chlorofluorescein (DCFH-DA), lactate dehydrogenase (LDH) assay kits, superoxide dismutase (SOD), N-acetyl-L-cysteine (NAC) and sodium citrate were obtained from Sigma (St. Louis, MO, USA). Thromboxane B2 (TXB2) ELISA kits, prostaglandin E2 (PGE2) ELISA kits and a COX inhibitor screening assay were purchased from Cayman Chemical Company (Ann Arbor, MI, USA). Primary antibodies against COX-2, GAPDH (sc-32233) and phospho-extracellular signal-regulated protein kinase (p-ERK) (sc-7383) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). The p-p38 antibody was obtained from Cell Signaling Technology (Beverly, MA, USA).
Platelet aggregation assay
Washed rabbit platelets were isolated from animals in the Animal Center of National Taiwan University Hospital as previously described [14]–[16] and were suspended in Tyrodes solution containing 1 mM calcium and 0.35% bovine serum albumin (BSA). Catechol (final concentration of 0.5–100 µM) was added to the platelets for 3 min after which AA (100 µM) or U46619 (1 µM, a TXA2 receptor agonist) was added, and platelet aggregation was measured using the turbidimetric method described by Born and Cross [17] using an aggregometer (Model 600B, Payton Associates, ON, Canada). The percentage of aggregation inhibition was calculated as described by Teng et al. [18]. All animal studies were approved by the Ethics Committee of the Chang Gung University of Science and Technology and National Taiwan University Hospital.
Lactate dehydrogenase (LDH) activity assay
Following exposure of platelets to catechol (1–200 µM) for 9 min, LDH activity in the supernatant and total cell lysate was determined as an index of platelet damage using LDH activity assay kits [6], [16]. The measurement was dependent on the spectrophotometric method as described by Wroblewski and La Due [19]. LDH generated by injured platelets was compared (in a percentage basis) with the total LDH activity of platelets dissolved in 0.1% Triton X-100 (v/v).
Thromboxane B2 (TXB2) assay
Platelets were preincubated with catechol (0.5–100 µM) for 3 min and then treated with or without AA (100 µM). EDTA (2 mM) and indomethacin (50 µM) were then added to the platelet suspension. After centrifugation at 14,000 rpm for 2 min, TXB2 levels were measured with Cayman ELISA kits according to the manufacturer's instructions [6], [15].
Effects of catechol on COX-1 and COX-2 enzyme activity
The direct effect of catechol on COX-1 and COX-2 enzymatic activities was measured using the COX inhibitor screening assay according to the manufacturer's instructions [6]. In brief, catechol (final 1–50 µM) was incubated with COX-1 or COX-2 in a reaction buffer for 10 min. Thereafter, AA was added as a substrate and incubated for an additional 2 min. This was followed by the addition of 0.1 N HCl and saturated stannous fluoride solution to stop the reaction. COX-1- or COX-2-mediated PGE2 production was then measured using the Cayman ELISA kits.
Fluorimetric assay of ROS production in platelets and PMNs
Platelet ROS production was determined by measuring the platelet 2′,7′-dichlorofluorescein (DCF) fluorescence as described by Iuliano et al. [20] with slight modification. Briefly, washed platelets (5×108/mL) were preincubated with 10 µM 2′,7′-diacethyl- chlorofluorescein (DCFH-DA) at 37°C for 30 min. After centrifugation and washing, 200 µL of platelets (5×108/mL) were added to each well of a 96-well plate containing various concentrations of catechol (final concentrations of 1–50 µM). Then AA (final 100 µM), and the emitted DCF fluorescence was measured immediately by a Gemini XPS microplate spectrofluorometer (Molecular Devices Corporation, Sunnyvale, CA, USA) at an excitation of 485 nm and emission of 535 nm for 15 min. The fluorescence of DCF-labeled platelets without exposure to AA was used as a reference of basal level ROS production.
Human polymorphonuclear leukocytes (PMNs) were isolated as previousy described [16], by the approval of Ethics Committee, National Taiwan University Hospital. Briefly, heparinized blood was mixed with 3% dextran at a ratio of 1∶1 (v/v) in a 50 mL conical tube and placed upright for 20–25 min at room temperature. Then, 10 mL of Ficoll-Hypaque lymphocyte separation medium (LSM1077-PAA Labs, Pasching, Austria) was added, mixed and density-gradient centrifuged at 400×g for 40 min at 4°C. After hypotonic lysis of the erythrocytes, the isolated PMNs (final 4×106 cells/mL) were counted and resuspended in 1× Hanks' balanced salt solution (HBSS) containing 10 µM DCFH-DA for 40 min. Cells were washed with HBSS prior to the addition of phorbol myristate acetate (PMA, final 1 µM) with and without catechol (1–100 µM) pretreatment. The emitted DCF fluorescence was measured using a Gemini XPS microplate spectrofluorometer at an excitation of 485 nm and emission of 535 nm for 20 min. SOD and NAC was used to prevent PMA-induced ROS production by PMNs.
Effect of catechol on AA-induced ERK and p38 phosphorylation in platelets
The prepared platelets (1×109 platelets/mL) were resuspended in Tyrode's solution containing 1 mM CaCl2 and 0.35% BSA, and 0.5 mL of the platelet suspension was placed into an aggregometer with constant stirring. Various amounts of catechol were added and preincubated for 3 min followed by addition of AA (100 µM) for 5 min. After addition of 0.5 mL of Laemmli sample buffer (75 mM Tris-HCl, 2% SDS, 15% glycerol, 3% 2-mercaptoethanol, pH 6.8) to terminate the reaction and for platelet lysis, cell lysates were boiled for 5 min, and 40 µL of cell lysates were subjected to 8% SDS-PAGE. After transfer to a PVDF membrane, blots were probed using anti-GAPDH, anti-p-ERK and anti-p-p38 antibodies as described previously [16]. For some western blot results, Image J software was used for quantitative analysis. The results were expressed as folds of control (as 1).
Ex vivo platelet aggregation of platelet-rich plasma (PRP)
After intravenous injection of catechol (0.5–5 µmole/mouse), blood was drawn from each mouse by heart puncture [21]. Whole blood containing anticoagulant (3.8% sodium citrate in 9∶1 (v/v)) was placed in an Eppendorf tube and centrifuged at 60×g for 4 min to prepare PRP. AA-induced ex vivo platelet aggregability of PRP was measured with an aggregometer as previously described [6], [16], [21].
Effect of catechol on IL-1β-induced PGE2 production and COX-2 expression of dental pulp cells
PGE2 level of dental pulp and its induction by IL-1β is crucial to the pathogenesis of pulpitis and pulpal pain [22], [23]. To know the anti-inflammatory effect, we tested whether catechol may prevent IL-1β-induced COX-2 expression and PGE2 production of pulp cells in this study. Human dental pulp cells were cultured as described previously [14]. For PGE2 analysis, 1×105 pulp cells were inoculated onto 24-well culture plates. After 24 h, medium was changed, and catechol (50 and 100 µM) was added for a 30 min pretreatment followed by addition of IL-1β (5 ng/mL) for an additional 24 h. Culture medium was collected for PGE2 analysis by ELISA. Viability of pulp cells was evaluated by MTT assay as previously [16], because pulp cells are attached to culture wells that is different from platelets floating in the buffer. For analysis of COX-2 expression, 1.5×106 pulp cells were inoculated onto 10-cm culture dishes. After 24 h, medium was changed, and catechol (50 and 100 µM) was added for a 30 min pretreatment, followed by addition of IL-1β (5 ng/mL) for an additional 24 h. Cell lysate of cultured pulp cells were prepared and subjected to western blotting as above (section 2.7.) except that anti-GAPDH and anti-COX-2 antibodies were used.
Statistical analysis
Three or more independent experiments were performed. Results were expressed as percentage of inhibition on AA-induced platelet aggregation and TXB2 synthesis. One-way ANOVA and post-hoc Tukey test were used for statistical analysis.
Results
Effects of catechol on platelet aggregation
The effects of catechol on platelet aggregation were first analyzed. As shown in Figure 1A , catechol suppressed AA-induced platelet aggregation at concentrations ≥10 µM. Quantitative analysis revealed that 5, 10 and 25 µM catechol inhibited AA-induced platelet aggregation by 50%, 71% and 94%, respectively (p <0.05; Figure 1B ), and 100 µM catechol completely inhibited AA-induced platelet aggregation (p <0.05; Figure 1B ). However, it did not significantly induce platelet cytoxicity at concentrations of 1–200 µM as determined by LDH release ( Figure 1C ).
Figure 1. Inhibition of AA-induced platelet aggregation by different concentrations of catechol.
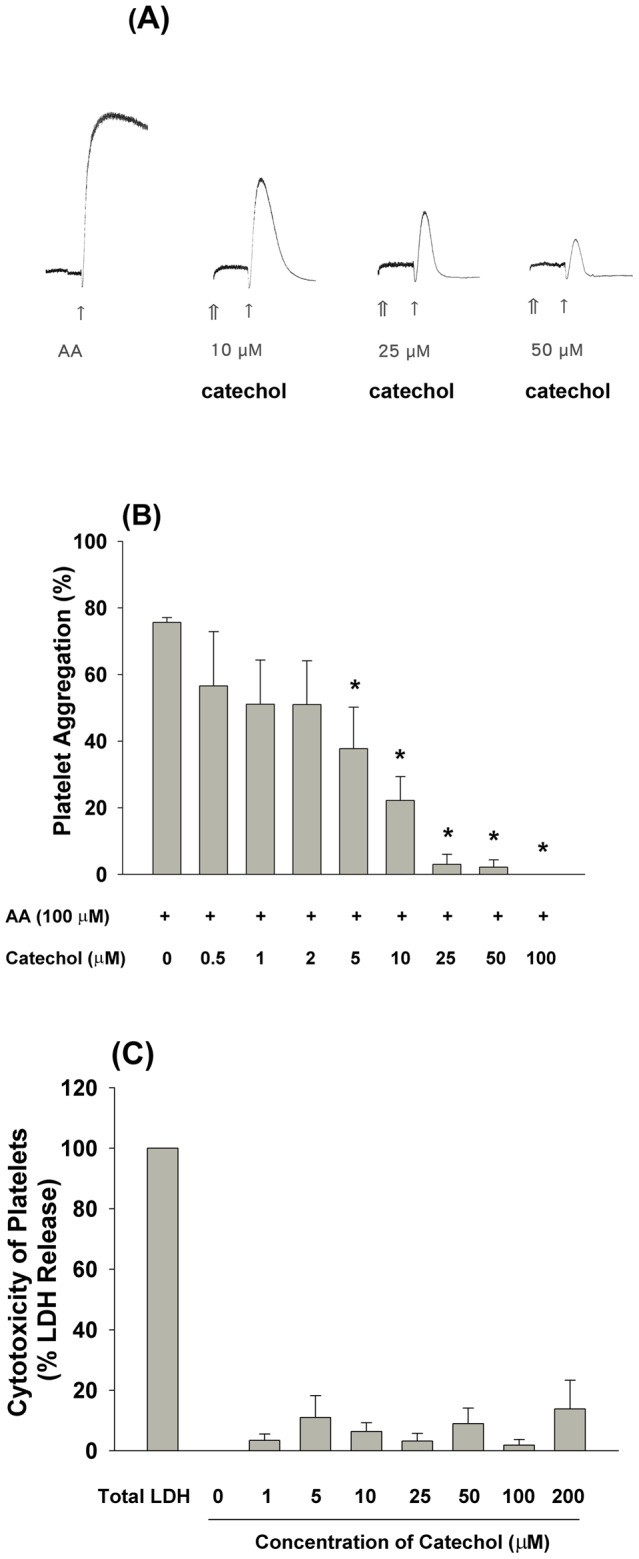
(A) One representative picture of AA-induced platelet aggregation and its inhibition by catechol was shown. (B) Quantitative analysis of AA (100 µM)-induced platelet aggregation and its inhibition by catechol (0.5–100 µM). Results were expressed as % of platelet aggregation (n = 5–8). (C) Cytotoxicity of catechol to platelets. Results were expressed as % of LDH release (n = 3). ↑indicates the addition of AA, and ⇑ indicates the addition of catechol. *indicates significant difference when compared with AA-treated group (P<0.05).
Inhibition of COX-1 and COX-2 activities by catechol
The anti-platelet effect of catechol can be partly explained by its inhibition of COX-1 activity. As shown in Figure 2A , catechol inhibited COX-1 activity by 29%, 38% and 44% at concentrations of 10, 25 and 50 µM, respectively (p <0.05;). Accordingly, catechol also inhibited COX-2 enzymatic activity by 29%, 42% and 50% at concentrations of 10, 25 and 50 µM, respectively (p <0.05; Figure 2B ).
Figure 2. Effect of different concentrations of catechol on the (A) COX-1 enzyme activity (n = 6), and (B) COX-2 enzyme activity (n = 6).
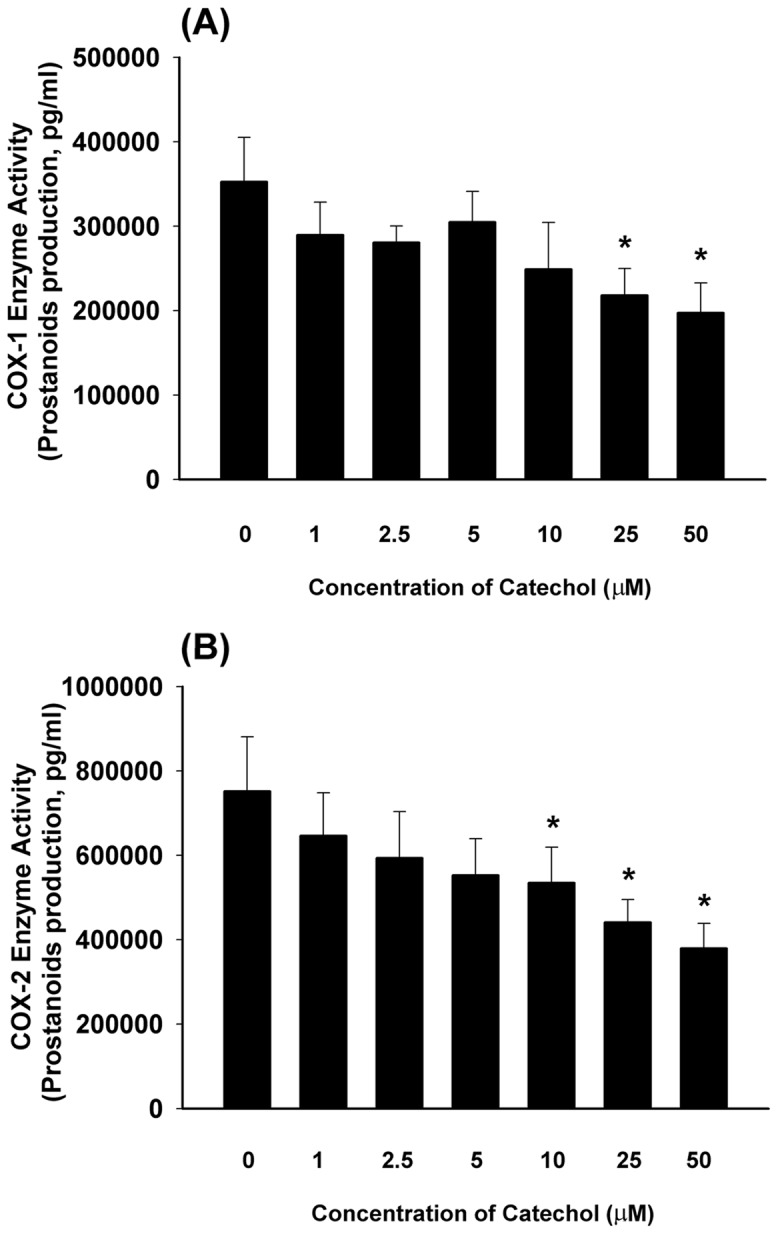
Results were expressed as COX-1 and COX-2 enzyme activity as indicated by PGE2 production (pg/mL). *indicated statistically significant difference when compared with solvent (DMSO) control group (0) (p<0.05).
Effect of catechol on AA-induced platelet TXB2 production and U46619-induced platelet aggregation
We next assessed the effects of catechol on AA-induced TXB2 production by platelets. As shown in Figure 3A , catechol suppressed AA-induced TXB2 production by 64% and 88% at concentrations of 0.5 and 5 µM, respectively (p <0.05).
Figure 3. Effect of catechol on AA-induced TXB2 production in platelets, (A).
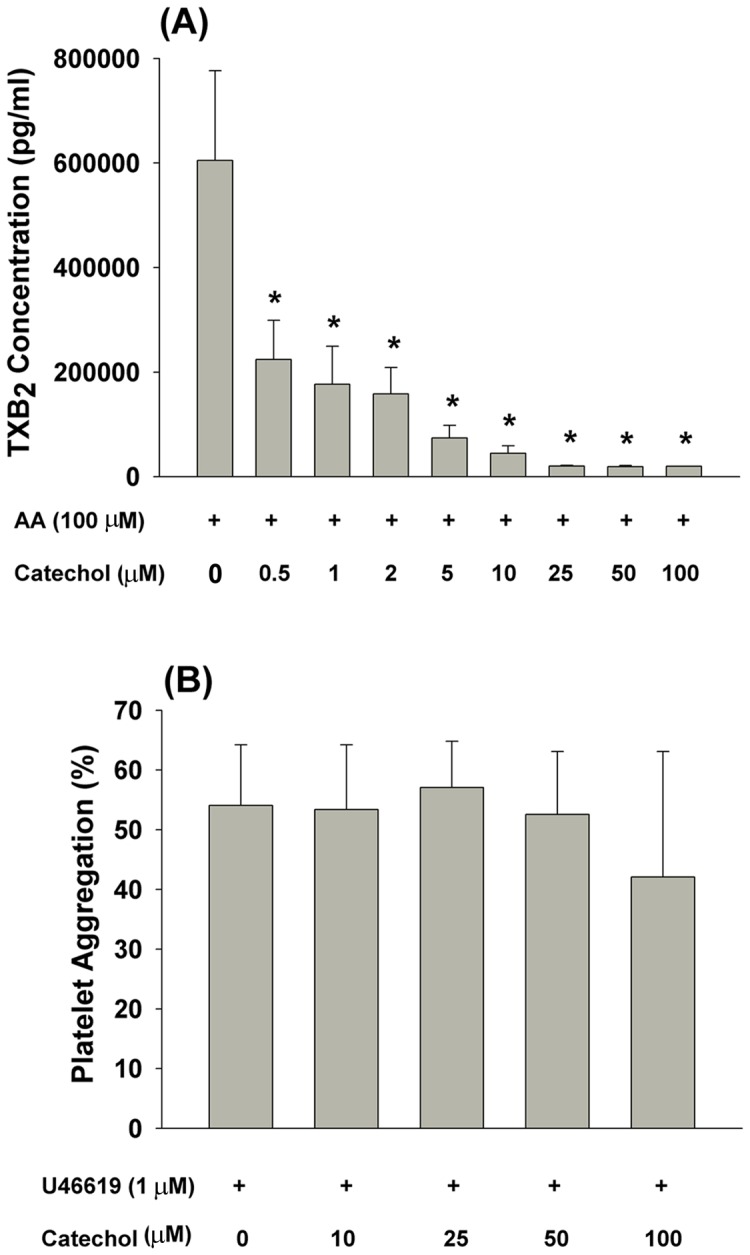
Results were expressed as TXB2 production (n = 5). *denotes statistically significant difference when compared with the group that treated by solvent (DMSO) plus AA, (B) Effect of catechol (10–100 µM) on U46619 (1 µM)-induced platelet aggregation (n = 3).
U46619 (1 µM), a TXA2 receptor agonist, can stimulate platelet aggregation. This effect was not significantly altered by catechol at concentrations ranging from 10–100 µM ( Figure 3B ).
Effects of catechol on AA-induced ROS production by platelets
Exposure of platelets to AA rapidly stimulated cellular ROS production as analyzed by an increase in DCF fluorescence. Pre-incubation with catechol attenuated AA-induced DCF fluorescence in platelets ( Figure 4A ). At concentrations of 1 and 10 µM, catechol effectively suppressed AA-induced ROS production by 35% and 49% (p <0.05; Figure 4B ). Moreover, the PMA-induced ROS production in isolated human PMNs as indicated by an increase in DCF fluorescence. This was inhibited by SOD (500 U/mL), N-acetyl-L-cysteine (NAC, 5 mM) as well as catechol (10–100 µM) (p <0.05; Figure 4C ).
Figure 4. Effect of catechol on AA-induced ROS production in platelets, (A).
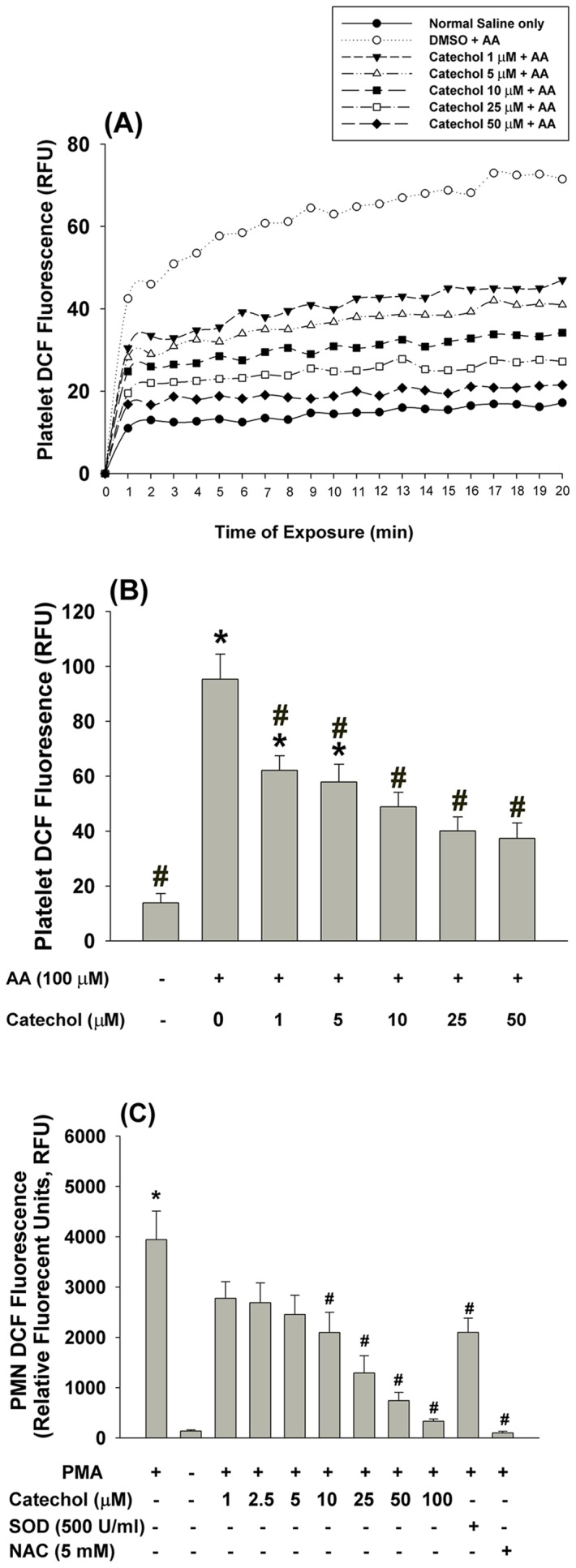
The DCF fluorescence of platelets after exposure to AA with/without catechol was measured. One representative histogram of DCF fluorescence was shown. Results were expressed as relative fluorescent unit (RLU). (B) Quantitative DCF fluorescence of platelets after exposure to AA with/without pretreatment by solvent control (DMSO) or catechol. *denotes significant difference (p<0.05) when compared with normal saline treated group (NC). #denotes statistically significant difference when compared with solvent (DMSO)+AA group (n = 4). (C) Catechol also prevented PMA-induced ROS production in isolated human PMN (n = 6). *denotes significant difference (p<0.05) when compared with control group. #denotes statistically significant difference when compared with vehicle + PMA group
Effects of catechol on AA-induced p38 and ERK phosphorylation in platelets
Mitogen-activated protein kinases (MAPKs), such as ERK and p38, are important for the regulation of platelet aggregation. To determine whether the anti-platelet effect of catechol is due to its attenuation of MAPK signaling, we incubated platelets in AA and found that it rapidly stimulated ERK and p38 phosphorylation ( Figure 5 ). However, pre-incubation of platelets with catechol (5–50 µM) attenuated AA-induced ERK and p38 phosphorylation as analyzed by Western blot analysis. Quantitatively, AA stimulated ERK and p38 phosphorylation to 3.2–fold and 9.4–fold of control, respectively ( Figure 5 ). Catechol (10–50 µM) decreased the AA-induced p-ERK and p-p38 expression to 1.3–1.5 fold and 5.0–6.0 fold of control, respectively ( Figure 5 ).
Figure 5. Effect of catechol on AA-induced p38 and ERK phosphrylation in platelets.
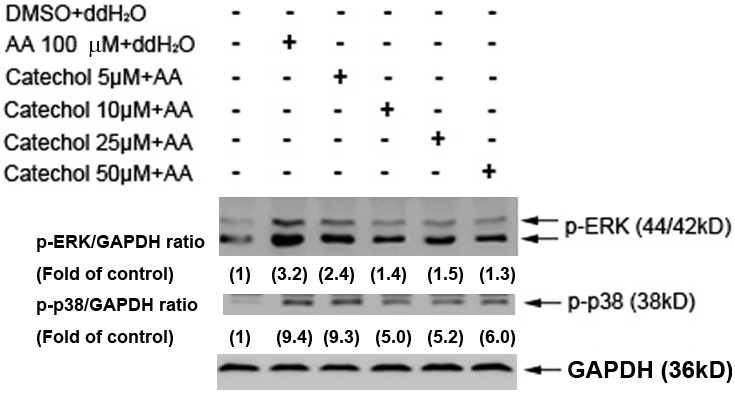
One representative western blotting picture was shown. Expression of GAPDH was used as control. Quantitative results of p-ERK/GAPDH and p-p38/GAPDH ratio were measured by Image J analysis and expressed as fold of control (the value in parenthesis).
Effects of catechol on ex vivo platelet aggregation induced by AA
Intravenous administration of catechol suppressed AA-induced platelet aggregation ex vivo at concentrations higher than 1 µmole/mouse ( Figure 6A ). Specifically, 2.5 and 5 µmole catechol/mouse significantly inhibited ex vivo AA-induced platelet aggregation by 68% and 94%, respectively (p <0.05; Figure 6B ).
Figure 6. Ex vivo platelet aggregation by AA and its inhibition by administration of catechol.
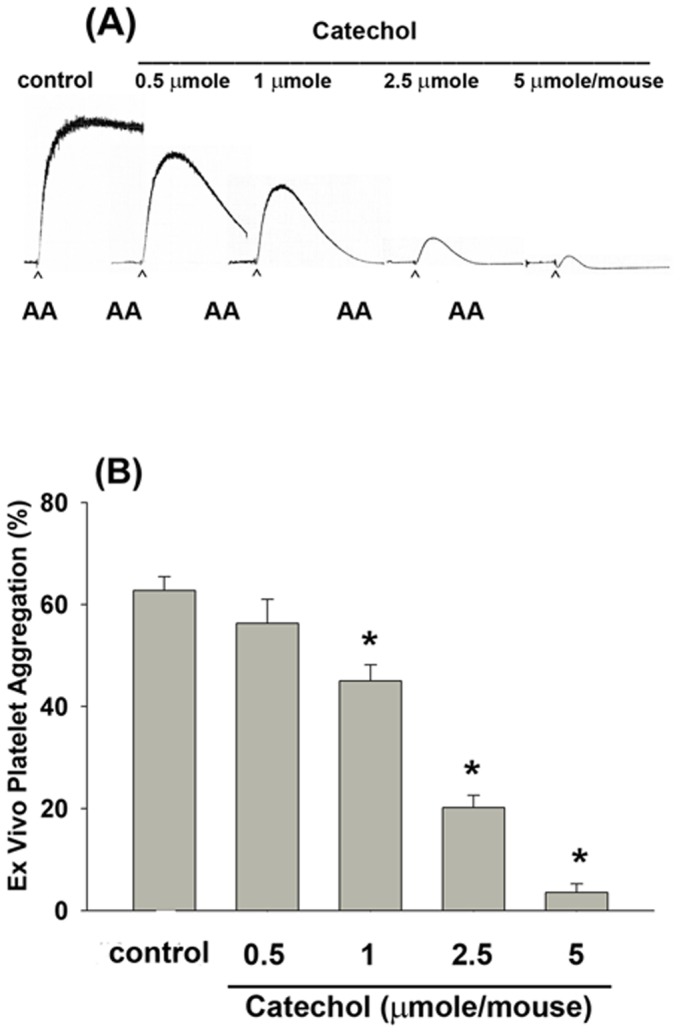
(A) Intravenous administration of catechol (0.5–5 µmole/mouse) inhibited the AA-induced platelet aggregation ex vivo. One representative picture of platelet aggregation was shown with more similar results. (B) Inhibition of AA-induced platelet aggregation ex vivo by intravenous administration of different amounts of catechol (0.5–5 µmole/mouse) into mice. (n = 7). *denotes significant difference when compared with solvent control group (p<0.05).
Effects of catechol on the growth, PGE2 production and COX-2 expression of dental pulp cells
Catechol inhibited COX-1 and COX-2 activities ( Figures 2A and 2B ). Because we have found that IL-1β stimulates COX-2 expression and PGE2 production by pulp cells [23], we further analyzed whether catechol may have anti-inflammatory effect and could inhibit IL-1β-induced PGE2 production of pulp cells. Exposure to 50 and 100 µM catechol with/without IL-1β showed little effect on pulp cell morphology as compared with control pulp cells ( Figure 7A ), and minimal cytotoxicity with 50 and 100 µM catechol was observed ( Figure 7B ). However, catechol (50 and 100 µM) almost completely inhibited the IL-1β-induced PGE2 production by pulp cells ( Figure 7C ). Western blotting analysis further found that catechol (50 and 100 µM) by itself slightly stimulated COX-2 protein expression of pulp cells. In addition, pretreatment by catechol and co-incubation with IL-1β showed no marked stimulation and even enhanced the IL-1β-induced COX-2 expression ( Figure 7D ), suggesting a possible compensatory response.
Figure 7. Catechol inhibited the IL-1β-induced PGE2 production of pulp cells.

Pulp cells were pretreated with catechol with/without further co-incubation by IL-1β for 24-hours. (A) morphology of control fibroblasts, pulp fibroblasts exposed to IL-1β for 24-hours, Pulp fibroblasts incubated with 0.1 mM catechol for 24-hours, and Pulp fibroblasts exposure to 0.1 mM catechol with IL-1β for 24-hours, (B) Viable cells were estimated by MTT assay. MTT reduction was shown as % of control (Mean ± SE) (n = 8). *denotes significant difference (p<0.05) when compared with solvent control group. (C) PGE2 level in the cultured medium was measured and the PGE2 concentration was shown as pg/ml (Mean ± SE) (n = 7). *denotes significant difference when compared with solvent control group; #denotes significant difference (p<0.05) when compared with IL-1β solely group. (D) COX-2 expression of pulp cells after exposure to IL-1β with/without pretreatment and co-incubation by catechol. One representative western blotting result was shown.
Discussion
Catechol is present in a number of food products and cosmetic ingrediens. It can be rapily absorbed by oral, dermal and inhalaton of exposure. Skin application of catechol for 30 min results in absorption of 0.4% of applied dose through human skin and a 24-h application of 4% catechol results in 53% absorption from skin, where it can be metabolized to reactive benzoquinone metabolites or other toxic quinone species by myeloperoxidase [24], [25]. It is also a metabolite of benzene and toluene [4]. In this study, catechol exhibits anti-platelet effects at low concentrations, suggesting its potential role in the prevention of cardiovascular diseases at low levels. Consistently, catechol, resorcinol (1,3-benzenediol) and hydroquinone have been previously shown to suppress AA-induced rabbit platelet aggregation although with varying levels of potency (about µM concentrations) [12]. Catechol also inhibits collagen-induced platelet aggregation at concentration of 100 µM, and a catechol group of phenolics has been suggested to responsible for its anti-platelet effect on collagen-induced platelet aggregation [13], [26]. In the present study, catechol significantly inhibits AA-induced platelet aggregation even at concentrations higher than 5 µM, which is in general agreement with the results described by Kitagawa et al. [12]. It is well known that platelet dysfunction plays a critical role in the pathogenesis of cardiovascular diseases [9]–[11]; therefore, exposure to catechol or consumption of catechol-containing products at suitable amounts may potentially prevent cardiovascular diseases via its anti-platelet effect.
AA-induced platelet aggregation is mediated by COX-1 activation, TXA2 production and subsequent activation of thromboxane receptors and the downstream signaling to induce full platelet aggregation. In this stdy, the anti-platelet effect of catechol may be partly due to its inhibition of COX-1 enzymatic activity given that platelets only express COX-1 and the binding and inhibition of catechol on COX and phospholipase A2 (PLA2) has been reported [27], [28]. Consistently, catechol almost completely inhibited AA-induced platelet production of TXA2, which interacts with platelet thromboxane receptor and induces full platelet aggregation. But catechol showed only partial inhibition on COX activity. This indicates that catechol possibly also affects other critical molecules such as ROS and PLA2 as mentioned previously [28]. As a stable TXA2 metabolite, TXB2 was obviously inhibited by catechol in AA-stimulated platelets. Binding of TXA2 to platelet TXA2 receptors may further induce downstream signaling for platelet aggregation [29]. Platelet TXA2 production is mediated mainly through metabolism of AA by COX-1 to produce TXA2, that may bind and activate thromboxane receptor to induce further full platelet activation. The TXA2 receptor agonist, U46619, was shown to stimulate marked platelet aggregation. However, catechol showed little inhibitory effect on U46619-induced platelet aggregation, suggesting that the anti-platelet effect of catechol is associated with COX-1 inhibition and that it cannot suppress platelet aggregation signaling downstream of thromboxane receptors.
The anti-platelet effect of catechol was not due to cytotoxicity as no obvious changes in platelet extracellular and intracellular LDH levels were noted after exposure to catechol. Catechol is not toxic to glioblastoma cells at concentrations below 0.2 mM [30]; however, it induces a 44% increase in esophageal cell death at 35 µM [4], suggesting that it exhibits differential cytotoxicity that is cell type-dependent.
ROS may regulate many physiological and pathological processes in the pulmonary and cardiovascular systems [31], [32]. ROS and redox changes also critically modulate platelet function [20], [33]. In this study, we found that AA stimulated ROS production in platelets, which was suppressed by catechol. Catechol further inhibited PMA-induced ROS production in PMNs, which is suggestive of a potential role as an anti-oxidant and anti-inflammatory mediator. Thus, the anti-platelet and anti-inflammatory effects of catechol are possibly related to its scavenging ROS or attenuation of ROS production in platelets. Consistently, the catechol group within the B-ring of flavonoids is responsible for their anti-oxidant effects toward PMA-induced oxidative burst [34] and 1,1-diphenyl-2-picrylhydrazyl free radicals [35]). However, at higher concentrations (150–600 µM), catechol is toxic to cultured Muller cells possibly via ROS production [36]. Therefore, more in vitro and in vivo studies are necessary to further clarify the beneficial and detrimental effects of catechol exposure.
ERK and p38 MAPKs play an important role in the regulation of platelet granule secretion, ATP release and platelet aggregation as well as the stimulation of myosin light chain to stimulate blood clot retraction [37], [38]. Some anti-platelet agents, including caffeic acid phenethyl ester, simvastatin and davallialactone, suppress platelet activation via inhibition of p38 and ERK [38]–[40]. Inhibition of both ERK and p38 phosphorylation is required to suppress AA-induced platelet aggregation [41]. In this study, we found that catechol effectively prevented AA-induced ERK and p38 MAPK phosphorylation, which is crucial for platelet AA release and TXA2 production [39]. At comparable concentrations, catechol obviously inhibited AA-induced platelet aggregation and TXA2 production in the present study, suggesting that the inhibitory effect of catechol on platelet TXA2 production is possibly mediated by blocking p38 and ERK signaling or other upstream signaling molecules such as phospholipase C (PLC), protein kinase C, glycoproein αIIbβ3, proline-rich tyrosine kinase 2 [42]–[44], and that it may have therapeutic potential as an anti-platelet agent.
In the present study, intravenous administration of catechol to mice inhibited ex vivo platelet aggregation, indicating that catechol may have anti-platelet effects even in the presence of plasma. Because catechol inhibits both COX-1 and COX-2 activities, we further analyzed the effects of catechol on IL-1β-induced PGE2 production of oral fibroblasts, especially given that it exhibits anti-microbial and anti-inflammatory activities by suppression of leukotriene B4 production in rat peritoneal exudate cells [45]. Therefore, catechol may increase the PGE2/leukotriene B4 ratio in PMNs. These phenols also inhibit PGE2 production in whole blood. Low concentrations of catechol stimulated PGE2 formation in PMNs; however, a repression was noted at high concentrations [46]. Vascular wall inflammation is related to platelet activation and constitutes a major pathogenic event in atherosclerosis [10]. Stimulation of COX-2 expression and PGE2 production is crucial for tissue inflammation, pain response and cardiovascular disease [23], [47], [48]. Dental pulp is a specialized connective tissue popularly suffered from inflammatory and pain response, where IL-1β and PGE2 are involved [22]. Recently, we observed the stimulation of PGE2 by IL-1β and possible activation of pulp cells via prostaglandin EP receptors, and suppression of IL-1β-induced PGE2 production in pulpal fibroblasts by plant phenolic was noted [16], [23]. In this study, catechol also attenuated the IL-1β-induced PGE2 production, but no COX-2 expression of pulp cells. These results support the anti-inflammatory effect of catechol. The inhibition of IL-1β-induced PGE2 production of pulp cells by catechol is possibly due to its direct interaction with COX-2 and inhibition of COX-2 enzyme activities, but not due to its suppression of COX-2 protein expression.
Conclusions
Catechol exhibits anti-platelet and anti-inflammatory effects. The anti-platelet effects of catechol are related to its inhibition of platelet COX activity, TXA2 production, ROS production as well as ERK/p38 signaling. The anti-platelet effect of catechol was confirmed using an ex vivo study. Catechol further suppresses PMA-induced ROS production in PMNs, which is suggestive of possible anti-inflammatory properties. Catechol also exhibits anti-inflammatory effects via inhibition of COX-2 activity and IL-1β-induced prostaglandin production. Catechol showed little cytotoxicity at concentrations below 0.1 mM. Taken together, exposure to catechol or suitable consumption of catechol-containing food/nutritional supplements, such as marine algae tablets or plants, may be beneficial to cardiovascular health. Delineation of its full pharmacological potential requires further investigation.
Data Availability
The authors confirm that all data underlying the findings are fully available without restriction. All relevant data are within the paper and its Supporting Information files
Funding Statement
The authors would like to thank Miss HF Jeng for her technical assistance. This study is supported by grants from National Science Council (NSC102-2314-B-255-003-MY2, NSC102-2628-B-255-001-MY3, NSC101-2320-B-255-002, NSC-100-2314-B-002-094 and NSC-101-2320-B-255-002) and Chang Gung Memorial Hospital (CMRPF1C0011, CMRPF1C0012, NMRPF3C0091, NMRPF3C0061). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. DeCaprio AP (1999) The toxicology of hydroquinone-relevance to occupational and environmental exposure. Crit Rev Toxicol 29: 283–330. [DOI] [PubMed] [Google Scholar]
- 2. McGregor D (2007) Hydroquinone: an evaluation of the human risks from its carcinogenic and mutagenic properties. Crit Rev Toxicol 37: 887–914. [DOI] [PubMed] [Google Scholar]
- 3.IARC (1985) Betel quid and areca nut chewing. Monographs on the evaluation of the carcinogenic risk of chemicals to humans. IARC Scientific Publications No 37, IARC, Lyon, pp. 141–202. [PubMed]
- 4. Myburg RB, Dutton MF, Chuturgoon AA (2002) Cytotoxicity of fumonisin B1, diethylnitrosamine and catechol on the SNO esophageal cancer cell line. Environ Health Perspect 110: 813–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Thavasi V, Bettens RP, Leong LP (2009) Temperature and solvent effects on radical scavenging ability of phenols. J Phys Chem A 113: 3068–3077. [DOI] [PubMed] [Google Scholar]
- 6. Chang MC, Uang BJ, Tsai CY, Wu HL, Lin BR, et al. (2007) Hydroxychavicol, a novel betel leaf component, inhibits platelet aggregation by suppression of cyclooxygenase, thromboxane production and calcium mobilization. Br J Pharmacol 152: 73–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lee SF, Lin JK (1994) Generation of hydrogen peroxide, superoxide anion and hydroxyl free radical from polyphenols and active benzene metabolites: Their possible role in mutagenesis. J Biomed Sci 1: 125–130. [DOI] [PubMed] [Google Scholar]
- 8. Schaefer D, Jurgensmeier JM, Baur G (1995) Catechol interferes with TGF-beta-induced elimination of transformed cells by normal cells: implicatios for the survival of transformed cells during carcinogenesis. Int J Cancer 60: 520–526. [DOI] [PubMed] [Google Scholar]
- 9. Alexandru N, Popov D, Georgescu A (2012) Platelet dysfunction in vascular pathologies and how can it be treated. Thromb Res 129: 116–126. [DOI] [PubMed] [Google Scholar]
- 10. Tsoumani ME, Kalantzi KI, Goudevenos IA, Tselepis AD (2012) Platelet-mediated inflammation in cardiovascular disease. Potential role of platelets-endothelium interaction. Curr Vasc Pharmacol 10: 39–549. [DOI] [PubMed] [Google Scholar]
- 11. Willoughby S, Holmes A, Losalzo J (2002) Platelets and cardiovascular disease. European Journal of Cardiovascular Nursing. 1: 273–288. [DOI] [PubMed] [Google Scholar]
- 12. Kitagawa S, Fujisawa H, Baba S, Kametani F (1991) Inhibitory effect of catechol derivatives on arachidonic acid-induced aggregation of rabbit platelets. Chem Pharm Bull 39: 1062–1064. [DOI] [PubMed] [Google Scholar]
- 13. Ostertag LM, O'Kennedy N, Horgan GW, Kroon PA, Duthie GG, et al. (2011) In vitro antiplatelet effects of simple plant-derived phenolic compounds are only found at high, non-physiological concentrations. Mol Nutr Food Res 55: 1624–1636. [DOI] [PubMed] [Google Scholar]
- 14. Chang MC, Chan CP, Ho YS, Lee JJ, Lin PS, et al. (2009) Signaling pathways for induction of platelet aggregation by SAS tongue cancer cells – a mechanism of hematogenous metastasis. J Oral Pathol Med 38: 434–440. [DOI] [PubMed] [Google Scholar]
- 15. Jeng JH, Chen SY, Liao CH, Tung YY, Lin BR, et al. (2002) Modulation of platelet aggregation by areca nut and betel leaf ingredients: roles of reactive oxygen species and cyclooxygenase. Free Radic Biol Med 32: 860–871. [DOI] [PubMed] [Google Scholar]
- 16. Chang MC, Chang HH, Chan CP, Chou HY, Chang BE, et al. (2012) Antiplatelet effect of phloroglucinol is related to inhibition of cyclooxygenase, reactive oxygen species, ERK/p38 signaling and thromboxane A(2) production. Toxicol Appl Pharmacol 263: 287–295. [DOI] [PubMed] [Google Scholar]
- 17. Born GVR, Cross MJ (1963) The aggregation of blood platelets. J Physiol 168: 178–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Teng CM, Ko FN (1988) Comparison of the platelet aggregation induced by three thrombin-like enzymes of snake venoms and thrombin. Thromb Haemost 59: 304–309. [PubMed] [Google Scholar]
- 19. Wroblewski F, La Due JS (1955) Lactic dehydrogenase activity in blood. Proc Soc Exp Biol Med USA 90: 210–213. [DOI] [PubMed] [Google Scholar]
- 20. Iuliano L, Pedersen JZ, Pratico D, Rotilio G, Violi F (1994) Role of hydroxyl radicals in the activation of human platelets. Eur J Biochem 221: 695–704. [DOI] [PubMed] [Google Scholar]
- 21. Chang MC, Lin HK, Peng HC, Huang TF (1998) Antithrombotic effect of crotalin, a platelet membrane glycoprotein 1b antagonist from venom of Crotalus atrox. Blood 91: 1582–1589. [PubMed] [Google Scholar]
- 22. Martinho FC, Chiesa WM, Leite FR, Cirelli JA, Gomes BP (2012) Correlation between clinical/radiographic features and inflammatory cytokine networks produced by macrophages stimulated with endodontic content. J Endod 38: 740–750. [DOI] [PubMed] [Google Scholar]
- 23. Chang MC, Chen YJ, Tai TF, Tai MR, Li MY, et al. (2006) Cytokine-induced prostaglandin E2 production and cyclooxygenase-2 expression in dental pulp cells: downstream calcium signalling via activation of prostaglandin EP receptor. Int Endod J 39: 819–826. [DOI] [PubMed] [Google Scholar]
- 24. Jung CT, Wickett RR, Desai PB, Bronaugh RL (2003) In vitro and in vivo percutaneous absorption of catechol. Food Chem Toxicol 41: 885–895. [DOI] [PubMed] [Google Scholar]
- 25. Subrahmanyam VV, Kolachana P, Smith MT (1991) Metabolism of hydroquinone by human myeloperoxidase: mechanisms of stimulation by other phenolic compounds. Arch Biochem Biophys 286: 76–84. [DOI] [PubMed] [Google Scholar]
- 26. Wright B, Tindall MJ, Lovegrove JA, Gibbins JM (2013) Investigating flavonoids as molecular templates for the design of small-molecule inhibitors of cell signaling. J Food Science 78: N1921–1928. [DOI] [PubMed] [Google Scholar]
- 27. Laughton MJ, vans PJ, Moroney MA, Hoult J, Halliwell B (1991) Inhibition of mammalian 5-lipoxygenase and cyclooxygenase by flavonoids and phenolic dietary additives: relationship to antioxidant activity and to iron-reducing ability. Biochem Pharmacol 42: 1673–1681. [DOI] [PubMed] [Google Scholar]
- 28. Dileep KV, Tintu I, Mandal PK, Karthe P, Haridas M, et al. (2012) Binding of PLA2 may contribute to the anti-inflammatory activity of catechol. Chem Biol drug Des 79: 143–147. [DOI] [PubMed] [Google Scholar]
- 29. Lopez-Farre A, Sanchez de Miguel L, Monton M, Jimenez A, Lopez-Bloya A, et al. (2001) Angiotensin II AT(I) receptor antagonists and platelet activation. Nephrol Dial Transplant 16 Suppl 145–49. [DOI] [PubMed] [Google Scholar]
- 30. De Oliveira DM, Pitanga BP, Grangeiro MS, Lima RM, Costa MF, et al. (2010) Catechol cytotoxicity in vitro: induction of glioblastoma cell death by apoptosis. Hum Exp Toxicol 29: 199–212. [DOI] [PubMed] [Google Scholar]
- 31. Al Ghouleh I, Khoo NK, Knaus UG, Griendling KK, Touyz RM, et al. (2011) Oxidase and peroxidase in cardiovascular and lung disease: new concept in reactive oxygen species signaling. Free Radic Biol Med 51: 1271–1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zinkevich NS, Gutterman DD (2011) ROS-induced ROS release in vascular biology: redox-redox signaling. Am J Physiol Heart Circ Physiol 301: H647–H653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Essex DW (2009) Redox control of platelet function. Antioxid Redox Signal 11: 1191–1225. [DOI] [PubMed] [Google Scholar]
- 34. Ribeiro D, Freitas M, Tome SM, Silva AM, Porto G, et al. (2013) Modulation of human neutrophils' oxidative burst by flavonoids. Eur J Med Chem 67: 280–292. [DOI] [PubMed] [Google Scholar]
- 35. De Martino L, Mencherini T, Mancini E, Aquino RP, De Almeida LF, et al. (2012) In vitro phytotoxicity and antioxidant activity of selected flavonoids. Int J Mol Sci 13: 5406–5419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mansoor S, Gupta N, Luczy-Bachman G, Limb GA, Kuppermann BD, et al. (2010) Protective effects of memantine and epicatechin on catechol-induced cytotoxicity to Muller cells in vitro. Toxicology 271: 107–114. [DOI] [PubMed] [Google Scholar]
- 37. Flavaris P, Li Z, Zhang G, Zheng Y, Liu J, et al. (2009) Two distinct roles of mitogen-activated protein kinases in platelets and a novel Rac1-MAPK-dependent integrin outside-in retractile signaling pathway. Blood 113: 893–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hsiao G, Lee JJ, Lin KH, Shen CH, Fong TH, et al. (2007) Characterization of a novel and potent collagen antagonist, caffeic acid phenethyl ester in human platelets: in vitro and in vivo studies. Cardiovasc Res 75: 782–792. [DOI] [PubMed] [Google Scholar]
- 39. Lee YM, Chen WF, Chou DS, Jayakumar T, Hou SY, et al. (2010) Cyclic nucleotides and mitogen-activated protein kinases: regulation of simvastatin in platelet activation. J Biomed Sci 17: 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kim SD, Lee IK, Lee WM, Cho JY, Park HJ, et al. (2008) The mechanism of antiplatelet activity of davallialactone: involvement of intracelluar calcium ions, extracellular signal-regulated kinase 2 and p38 mitogen-activated protein kinase. Eur J Pharmacol 584: 367–367. [DOI] [PubMed] [Google Scholar]
- 41. McNicol A, Jackson EC (2003) Inhibition of the MEK/ERK pathway has no effect on agonist-induced aggregation of human platelets. Biochem Pharmacol 65: 1243–1250. [DOI] [PubMed] [Google Scholar]
- 42. Lee YM, Chen WF, Chou DS, Jayakumar T, Hou SY, et al. (2010) Cyclic nucleotides and mitogen-activated protein kinases: regulation of simvastatin in platelet activation. J Biomed Sci 17: 45–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Yacoub D, Theoret JF, Villeneuve L, Abou-Saleh H, Mourad W, et al. (2006) Essential role of protein kinase Cδ in platele signaling, αIIbβ3 activation, and thromboxane A2 release. J Biol Chem 281: 30024–30035. [DOI] [PubMed] [Google Scholar]
- 44. Sun L, Feng S, Resendiz JC, Lu X, Durante W, et al. (2004) Role of the Pyk2-MAP kinase-cPLA2 signaling pathway in shear-dependent platelet aggregation. Ann Biomed Eng 32: 1193–1201. [DOI] [PubMed] [Google Scholar]
- 45. Yamada K, Shoji K, Mori M, Ueyama T, Matsuo N, et al. (1999) Structure-activity relationship of polyphenols on inhibition of chemical mediator release from rat peritoneal exudate cells. In Vitro Cell Dev Biol Anim 35: 169–174. [DOI] [PubMed] [Google Scholar]
- 46. Alanko J, Riutta A, Vapaatalo H (1995) Phenols inhibit prostaglandin E2 synthesis in A23187-stimulated human whole blood and modify the ratio of arachidonic acid metabolites. Prostaglangins Leukot Essent Fatty Acids 52: 299–301. [DOI] [PubMed] [Google Scholar]
- 47. Beck J, Garcia R, Heiss G, Vokonas PS, Offenbacher S (1996) Periodontal disease and cardiovascular disease. J Periodontol 67 10 Suppl: 1123–1137. [DOI] [PubMed] [Google Scholar]
- 48. Wang M, Song WL, Cheng Y, Fitzgerald GA (2008) Microsomal prostaglandin E synthase-1 inhibition in cardiovascular inflammatory disease. J Intern Med 263: 500–505. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The authors confirm that all data underlying the findings are fully available without restriction. All relevant data are within the paper and its Supporting Information files