

Summary
While gender-based differences in heart disease have raised the possibility that estrogen (ES) or progesterone (PG) may have cardioprotective effects, recent controversy regarding hormone replacement therapy has questioned the cardiac effects of these steroids. Using cardiomyocytes, we tested whether ES or PG has protective effects at the cellular level. We found that PG but not ES protects cardiomyocytes from apoptotic cell death induced by doxorubicin (Dox). PG inhibited apoptosis in a dose dependent manner, by 12 ± 4.0% at 1 μM and 60 ± 1.0 % at 10 μM. The anti-apoptotic effect of PG was also time dependent, causing 18 ± 5% or 62 + 2% decrease in caspase-3 activity within 1 or 72 hours of pretreatment. While PG causes nuclear translocation of its receptor within 20 mins, the cytoprotective effect of PG was cancelled by mifepristone (MF), a PG receptor antagonist. Analyses using Affymetrix high-density oligonucleotide array and RT-PCR found that PG induced Bcl-xL, metallothionine, NADPH quinone oxidoreductase 1, glutathione peroxidase-3, and 4 isoforms of glutathione S-transferase. Western blot analyses revealed that PG indeed induced an elevation of Bcl-xL protein in a dose and time dependent manner. Nuclear run-on assay indicated that PG induced Bcl-xL gene transcription. Inhibiting the expression of Bcl-xL using siRNA reduced the cytoprotective effect of PG. Our data suggests that PG induces a cytoprotective effect in cardiomyocytes in association with induction of Bcl-xL gene.
Keywords: Cytoprotection, nuclear receptor, gene expression, antioxidant, oxidative stress, Bcl-xL
Introduction
Gender-based differences in heart disease have raised the possibility that estrogen (ES) or progesterone (PG) have cardioprotective effects. Women tend to develop heart disease later than men (Low et al. 2002). Also, the duration between the onset of symptoms and heart failure is longer for women than men. Many studies including the Framingham Heart Study have shown an increased risk of heart disease in postmenopausal women relative to their age matched cycling counterparts (Murabito 1995; Wexler 1999). While these studies have lead to the assumption that supplementation of ES and PG in postmenopausal women may be beneficial for heart health, recent controversies regarding hormone replacement therapy in postmenopausal women have raised questions about whether ES or PG is indeed cardiac protective. While the effect of ES on the heart and cardiovascular system has been studied extensively, little is known about the cardiovascular effect of PG.
ES or PG binds to its receptor in the cytosol, causing nuclear translocation of the receptor. The PG receptor (PR) is present in cardiac muscle of a variety of mammalian species including the human (Lin et al. 1982; Ingegno et al. 1988; Grohe et al. 1997; Pasanen et al. 1997; Knowlton & Sun 2001; Goldstein et al. 2004). Administration of PG has been shown to induce a number of physiologic effects including increasing cardiac output (Kuebler et al. 2003), decreasing arterial pressure (Pecins-Thompson & Keller-Wood 1997; Roesch & Keller-Wood 1997), dilating coronary arteries (Minshall et al. 2002), induction of angiotensin I (Nickenig et al. 2000), and cardiac muscle protein synthesis (Goldstein et al. 2004). During pregnancy, physiological levels of ES and PG increase significantly along with a concomitant increase in cardiac output in addition to increases in blood volume. Although the PR is expressed in cardiac muscle, the molecular action of PG or PR remains largely undefined in cardiomyocytes.
The PR is encoded by a single gene that produces two isoforms, A and B, from alternative promoters (Graham & Clarke 1997; Conneely et al. 2002). The A isoform is essentially a truncated version of the B isoform and lacks the amino-terminal 128 amino acids of PR-B (Weigel et al. 1995). Both isoforms can bind to PG and change conformation upon ligand binding. The receptors then dimerize (A-A, B-B or A-B) and bind to hormone response elements (HRE) in the promoter regions of target genes. In addition, PRs also interact with co-activators, co-repressors and transcription factors (Weigel et al. 1995; Graham & Clarke 1997; Conneely et al. 2002). The ratio of PR-A versus PR-B differs by tissues and contributes to the difference in downstream genes regulated by PG (Richer et al. 2002; Smid-Koopman et al. 2003).
Many cardiac protective agents act to prevent death of cardiomyocytes from damaging agents. Doxorubicin (Dox) is an anthracycline chemotherapeutic agent currently used for the treatment of many neoplastic conditions such as lymphoma, leukemia, breast, ovarian, gastric, thyroid and bronchogenic carcinomas. The effectiveness of this drug is limited due to its dose dependent cardiac toxicity (Keizer et al. 1990; Singal & Iliskovic 1998; Singal et al. 2000). The administration protocol has been revised to reduce acute cardiac toxicity. However, chronic cardiac toxicity usually develops 2–10 years after drug administration as patients start to show signs of dilated cardiomyopathy. This toxicity is associated with Dox induced oxidative radicals, since Dox can accept electrons from oxoreductive enzymes in the mitochondria to form semiquinone free radicals, which can initiate a chain of redox reactions (Keizer et al. 1990; Singal & Iliskovic 1998; Singal et al. 2000). Dox has been shown to produce superoxide and H2O2 when incubated with the mitochondrial fraction of cardiac tissue extracts (Doroshow & Davies 1986). In addition to inducing oxidative stress, Dox at high concentrations can interact with DNA topoisomerase and cause DNA strand breaks (Gewirtz 1999). At the cellular level, Dox has often been used as a model compound for inducing apoptosis in a number of experimental systems including cardiomyocytes (Kumar et al. 1999; Arola et al. 2000; Kalyanaraman et al. 2002; Ueno et al. 2006; Bruynzeel et al. 2007).
Apoptosis is known to be an important pathological event in cardiac injury and heart failure. Recent studies have shown the presence of apoptosis in biopsy samples of failing human hearts and in the myocardium of experimental animals (Narula et al. 1996; Anversa et al. 1997; Bialik et al. 1997; Condorelli et al. 1999; Webster et al. 1999). There is evidence that inhibiting apoptosis may alleviate certain clinical manifestation of heart failure in experimental animals (Feuerstein et al. 1998; Ma et al. 1999; Kotamraju et al. 2000). Therefore agents protect cells from apoptosis may exhibit therapeutic potential to prevent cardiac injury and heart failure.
Results
Progesterone Inhibits Apoptosis Induced by Dox
Cardiomyocytes are adherent in culture but round up, detach and show Annexin V binding when undergo apoptosis. To test the effect of PG, cardiomyocytes were pretreated with PG for 24 hours before being placed in fresh medium for Dox treatment. With PG pretreatment, inhibition of apoptosis was observed through morphology and annexin V binding (Fig. 1A&B). Under normal culture conditions, cardiomyocytes beat synchronally in clusters at a frequency of 27.2 ± 1.7 beats per minute. PG treatment did not affect the beating frequency. By 24 hrs of Dox treatment, a fraction of cells undergo apoptosis while remaining cells survived but could no longer beat. With PG pretreatment, surviving cells beat at a frequency of 17.6 ± 2.9 beats per minute.
Figure 1. Morphological Evidence of PG Inhibiting Dox Induced Apoptosis.
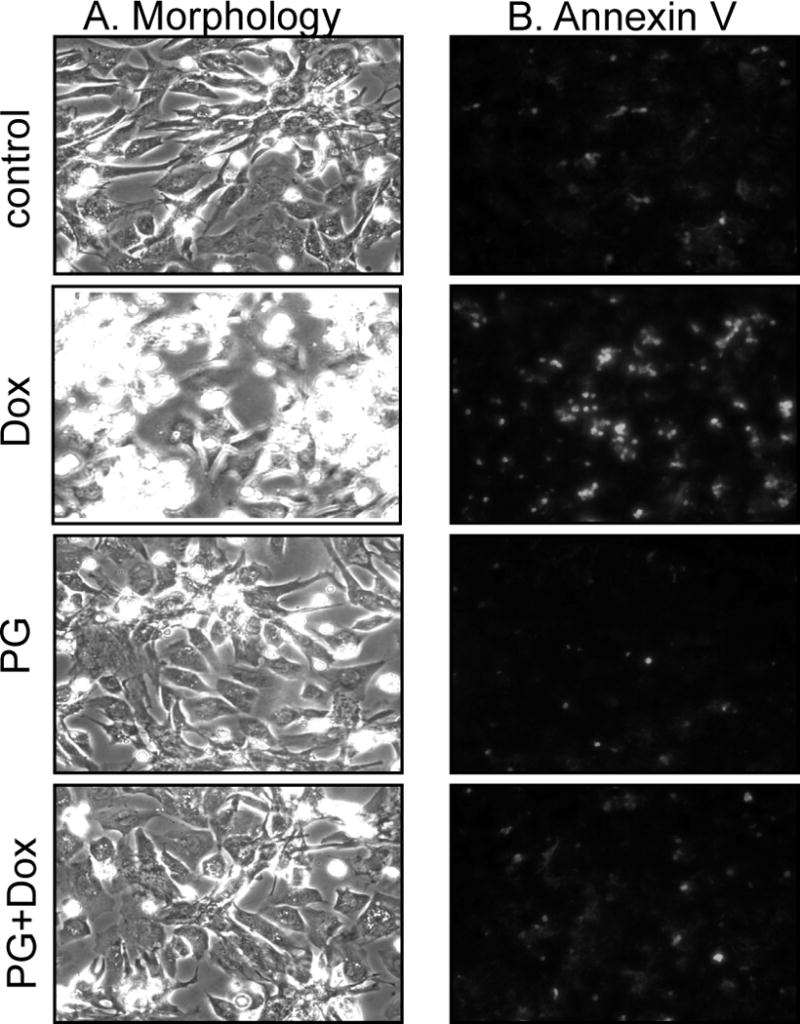
Primary cultured cardiomyocytes were pretreated with 10 μM PG for 24 hrs and then treated with 0.8 μM Dox in the absence or presence of 10 μM PG. At 24 hrs after Dox treatment, the morphology was recorded under a phase contrast microscope. Detached cells were combined with adherent cells for Annexin V-Fluo staining for recording under a fluorescent microscope.
To quantify the inhibitory effect of PG on Dox induced apoptosis, we measured caspase-3 activity. Dox causes a dose dependent increase of caspase-3 activity in primary cardiomyocytes (Chen et al. 2005). When pretreated with PG at various doses before 0.8 μM Dox treatment, PG at 1 or 10 μM reduced caspase activity by 12 ± 4% (p<0.05) or 60 ± 1% (p<0.005) respectively (Fig. 2A). The inhibitory effect was dependent on how long cells being pretreated with PG. A decrease in caspase-3 activity was evident with 1 hr (18 ± 5% inhibition, p<0.05) pretreatment with 10 μM PG and was maximal with 72 hours of PG pretreatment (62 ± 2% inhibition, p<0.005, Fig. 2B). In addition to primary cultured cardiomyocytes, PG showed a protective effect in H9C2 rat cardiomyocyte cell line (data not shown).
Figure 2. PG Inhibits Dox Induced Caspases.
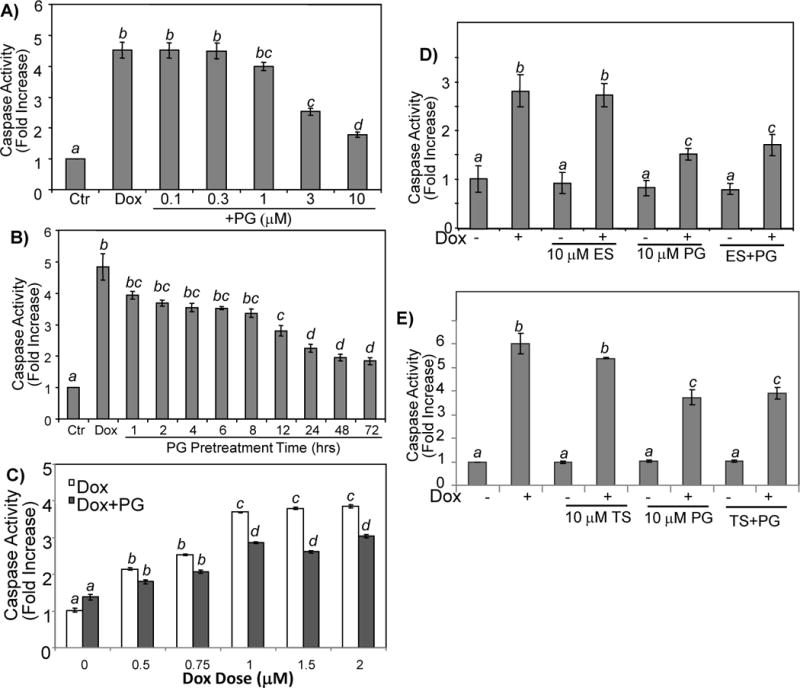
Primary cultured cardiomyocytes were treated with 0.8 μM Dox for 24 hrs following pretreatment of PG at indicated dose for 24 hrs (A) or at 10 μM over indicated time (B). For Dox dose response, cardiomyocytes were treated with 10 μM PG and Dox at indicated doses for 16 hrs (C). Following pretreatment with 10 μM PG in the absence or presence of 10 μM 17β-estradiol (ES) or 10 μM testosterone (TS), cells were treated with 0.8 μM Dox for 24 hrs before harvesting for measurements of caspase activity (D, E). The data are from one experiment representative of three with means ± standard deviations from three samples. A superscript letter indicates significant difference (P<0.05) from means labeled with a different letter as determined by ANOVA analysis.
A number of studies suggest that ES can be cytoprotective. We determined whether ES can prevent Dox from inducing apoptosis of cardiomyocytes. Cardiomyocytes were pretreated for 24 hours with doses of 17β–estradiol varying from 1 to 50 μM before treatment with Dox. No protective effect was observed with any of the dose of 17β-estrodial tested (data not shown). Male sex hormone testosterone (TS) did not show significant protective effect (Fig 2E). To determine if there was a synergistic effect of ES or TS with PG, we pretreated cells with ES or TS with PG. The results show that ES or TS did not potentiate the inhibitory effect of PG (Fig. 2D, E).
Dependence on Receptor of PG Induced Cytoprotection
PG can diffuse through the plasma membrane and binds to its receptor in the cytosol, causing nuclear translocation. To verify that the PG receptor is indeed expressed in cardiomyocytes, hearts from neonatal rats, adult male or female rats, and adult male or female mice were used for Western blot analyses. Rat uterus tissue was included as a positive control. Neonatal rat cardiomyocytes express both A and B isoforms of PR, while adult mice or rat hearts express mainly the A isoform (Fig. 3A). The H9C2 rat cardiomyocyte cell line contains the B isoform of PR (Fig. 3A). Since PG shows a protective effect in H9C2 cardiomyocytes, the B isoform likely plays a critical role in cytoprotection.
Figure 3. Expression of PR in Cardiac Tissue or Cardiomyocytes.
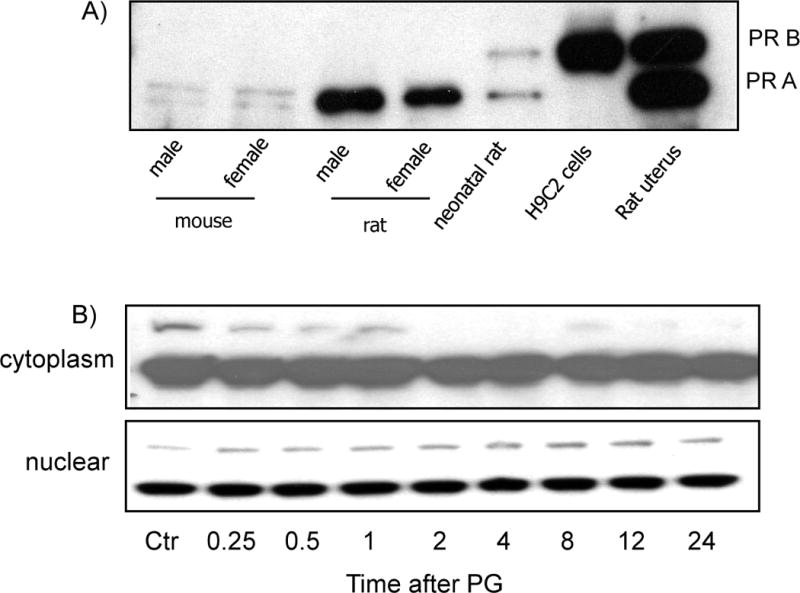
Cardiac tissue was collected from 8 weeks old male or female C57BL6 mice or Sprague Dawley rats by quick frozen in liquid nitrogen. The uterus tissue from a pregnant rat was collected immediately after birth to serve as a positive control. Animal tissues were grinded in liquid nitrogen bath for dissolving in lysis buffer for Western blot analyses (A). Primary cultured cardiomyocytes were treated with 10 μM PG and harvested at indicated time for isolation of cytoplasmic or nuclear fractions (B). An equal amount of proteins (20 μg/lane) was loaded for Western blot analyses.
To determine if PG treatment causes activation of PR in cardiomyocytes, nuclear translocation was measured after cells were incubated with PG for various time points from 15 mins to 72 hours. The cells were harvested for isolation of cytoplasmic and nuclear fractions. The results show that PG indeed induces PR-B translocation in cardiomyocytes detectable 20 mins after PG exposure (Fig. 3B).
The cytoprotective effect of PG was eliminated by mifepristone (MF), a PR antagonist, as measured by caspase-3 activity (Fig. 4A). The ability of MF in canceling the cytoprotective effect of PG was confirmed with measurements of caspase-3 cleavage (Fig. 4B) and mitochondrial cytochrome c release (Fig. 4B).
Figure 4. PR Receptor Antagonist MF Blocks Cytoprotective Effect of PG.
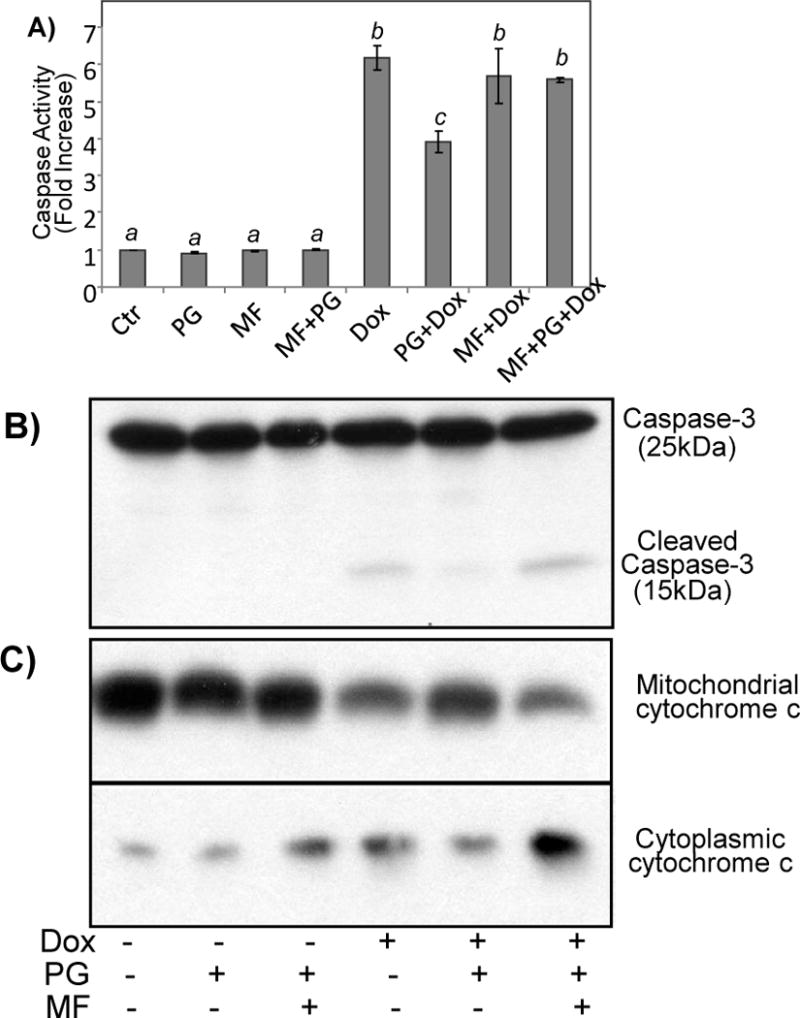
Primary cultured cardiomyocytes were treated with 10 μM PG in the absence or presence of 1 μM MF for 24 hrs before addition of 0.8 μM Dox. The cells were harvested at 24 hrs later for measurements of caspase 3 using DEVD-AMC as a substrate (A), or for Western blot to measure caspase-3 cleavage or mitochondrial release of cytochrome c (B, C). A superscript letter indicates significant difference (P<0.05) from means labeled with a different letter as determined by ANOVA analysis.
Progesterone Induces Antioxidant and Anti-apoptosis Genes
To understand the mechanism of cytoprotection induced by PG, we utilized Affymetrix gene array to screen the expression of 20,000 genes. RNAs were harvested at 24 hrs after incubating cardiomyocytes with 10 μM PG or vehicle. The results show that PG caused 180 genes and 207 ESTs to be upregulated 1.5 fold or higher (p<0.05). Among the long list of genes are the anti-apoptosis gene Bcl-xL and 12 antioxidant/detoxification genes (Table 1 in Supplement). In contrast to these upregulated genes, 73 genes and 123 ESTs showed downregulation at 1.5 fold or lower. The downregulated genes include 8 DNA replication factors and enzymes, 5 cell cycle regulators, 12 cytoskeletal binding or morphogenesis proteins, and 7 cytokines/chemokines or their regulators (Table 2 in Supplement).
To verify the data from microarray, we have chosen 15 genes, 9 of which belong to the category of antioxidant, detoxification and anti-apoptosis genes. With every gene we picked, RT-PCR was able to verify the upregulation in PG treated cells (Fig. 5).
Figure 5. RT-PCR Verification of Microarray Detected Genes Induced by PG.
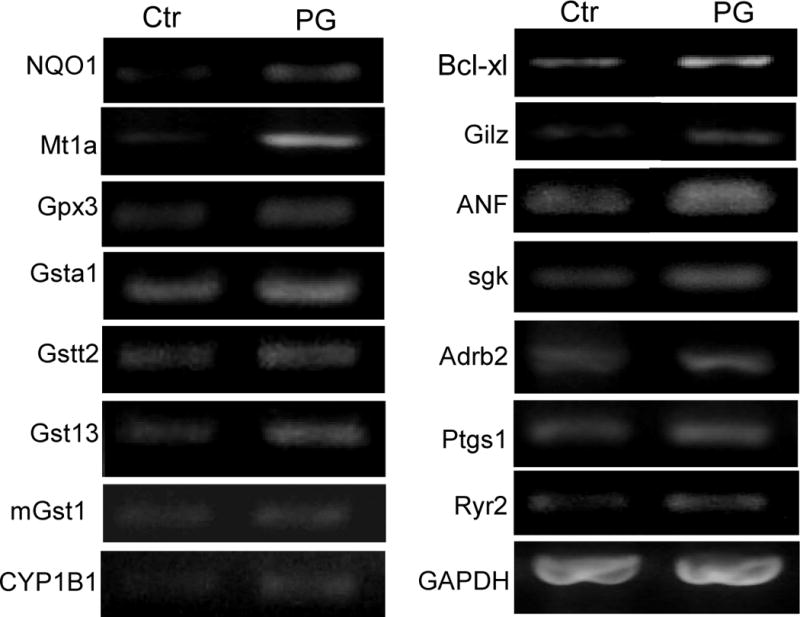
Total RNA was harvested from primary cultured cardiomyocytes 24 hrs after 10 μM PG treatment for RT-PCR analyses as described in the Method. GAPDH was used as a loading control.
Bcl-xL Expression in PG Induced Cytoprotection
Bcl-xL gene is a prosurvival member of bcl-2 family and was identified by microarray as a PG inducible gene in cardiomyocytes. To characterize PG induced expression of Bcl-xL, cells were treated with PG for 4 hrs to 3 days. Bcl-xL elevation was first detectable at 4 hrs, reached the highest level at 24 hrs, and remained elevated for 3 days (Fig. 6A). In primary cultured cardiomyocytes, a dose dependent increase of Bcl-xL was observed (Fig. 6B). In H9C2 cardiomyocytes, the best dose for inducing Bcl-xL was 0.5 μM (data not shown). MF prevented PG from inducing Bcl-xL expression (Fig. 6C).
Figure 6. PG Induces Bcl-xL protein.
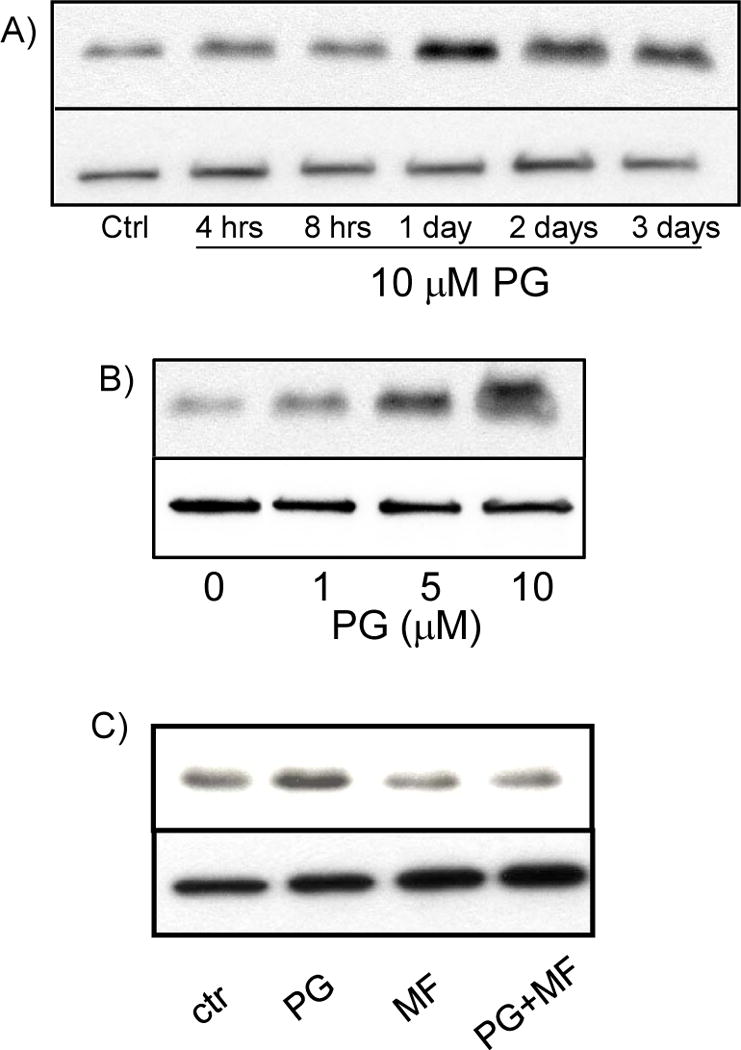
Primary cultured cardiomyocytes were treated with 10 μM PG (A, C) or indicated dose (B) for the time points shown or 24 hrs (B, C). MF (1 μM) was added to cells 1 hr before PG (C). Cell lysates were used for Western blots to measure Bcl-xL protein (20 μg protein/lane, upper panels) with vinculin being used as a loading control (bottom panels). The Data shown is representative of 3 experiments.
To address whether Bcl-xL elevation resulting from transcriptional activation of bcl-x gene, we measured the level of Bcl-xL transcription in vitro using nuclear run-on assay. PG treated cells showed a clearly elevated level of Bcl-xL transcription (Fig. 7A). Unlike corticosterone (CT), which induces transcription of COX-2 gene (Sun et al. 2008a; Sun et al. 2008b), PG did not cause elevation of COX-2 per microarray analyses, allowing COX-2 to serve as a negative control for in vitro transcription assay (Fig. 7A). Measurements of Bcl-xL mRNA decay rate indicate that PG did not cause Bcl-xL mRNA stabilization (Fig. 7B). Our previous studies show that CT induces Bcl-xL and cytoprotection in a Glucocorticoid Receptor (GR) dependent manner (Chen et al. 2005). To differentiate PG effect from CT, we measured the GR transactivity using a luciferase reporter construct under the control of Mammary Tumor Virus (MTV) GR cis-element (GRE). While CT activated GRE, PG did not (data not shown). With CT induced Bcl-xL expression, 905 bp bcl-x promoter is activated (Fig. 8A). In contrast, PG did not activate this promoter fragment (Fig. 8A). With 3.2 kb bcl-x promoter, a minor but significant activation was observed with 10 μM PG treatment (Fig. 8B).
Figure 7. PG Causes An Increase in the Rate of Bcl-xL Transcription.
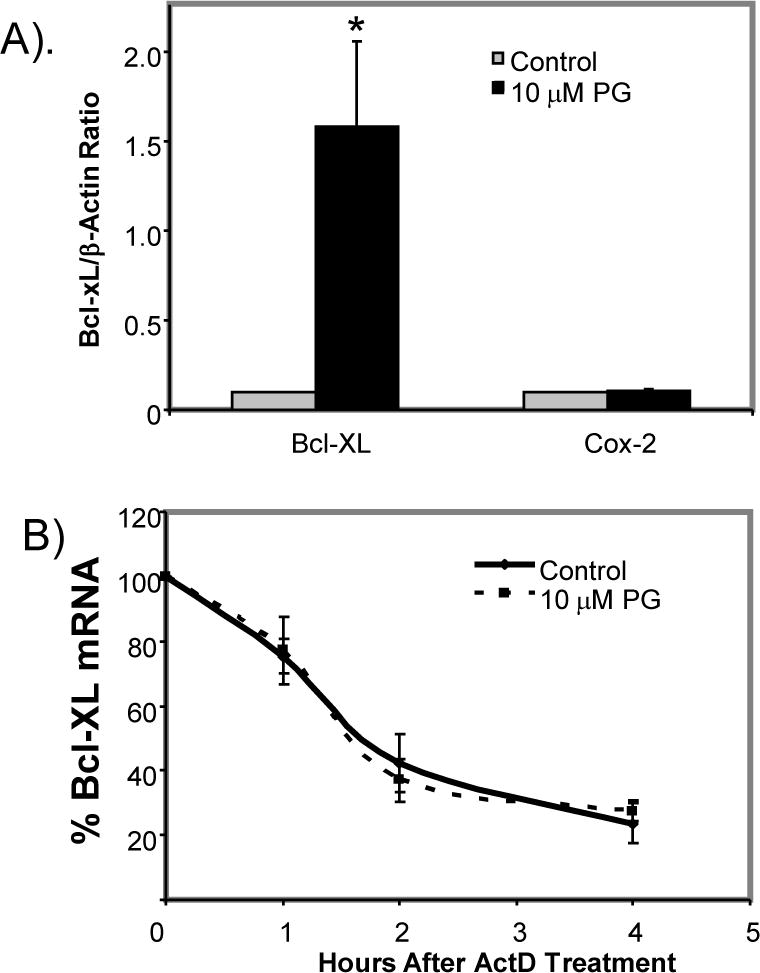
Primary cultured cardiomyocytes were treated with 10 μM PG for 24 hrs before being harvested for nuclear run-on assay (A) or measurements of Bcl-xL mRNA stability by addition of 5 μg/ml actinomycin D (ActD) over indicated time (B). The relative Ct method was used to calculate mRNA levels with normalization to β-Actin (A). The level of Bcl-xL was set to 100 at 0 time point for determining Bcl-xL mRNA stability (B). The data represent average ± standard deviations of three independent experiments. An asterisk (*) indicates p<0.05 when the treated group was compared to control using Student’s t test.
Figure 8. bcl-x Gene Promoter Activity by PG Treatment.
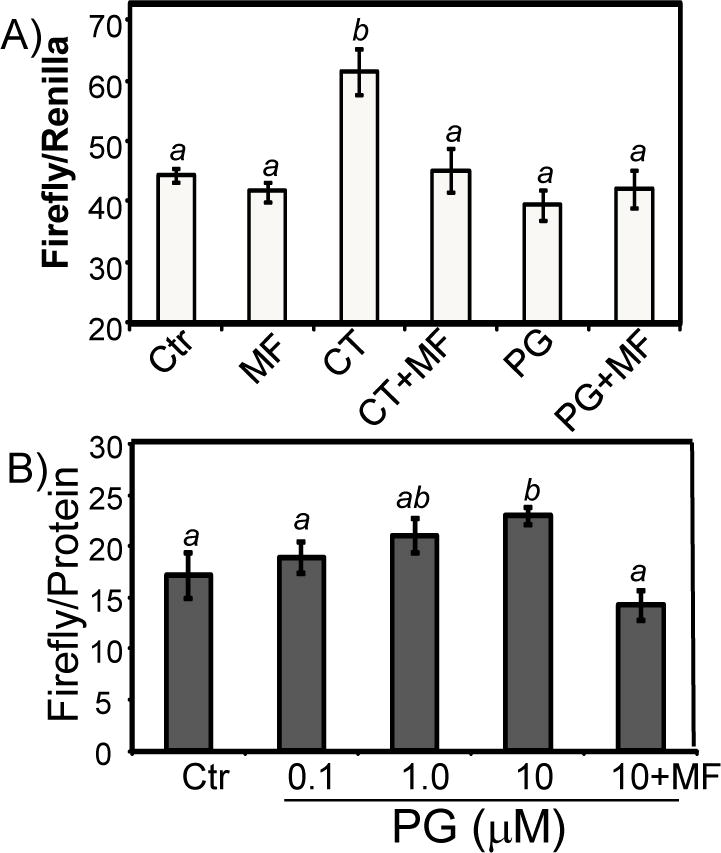
Primary cultured cardiomyocytes were transfected with luciferase constructs under the control of −905 bp human bcl-x promoter (A) or −3.2 kb mouse bcl-x promoter (B) using Fugene6 liposomes. At 48 hrs after transfection, cells were placed in 0.5% FBS for 24 hrs before treatment with 1 μM corticosterone (CT, A), 10 μM PG (A) or PG at indicated doses (B) for 24 hrs before harvesting for measurement of luciferases. A superscript letter indicates significant difference (P<0.05) from means labeled with a different letter as determined by ANOVA analysis.
To determine whether Bcl-xL induction was necessary or sufficient for the cytoprotective effect of PG, we used siRNA to inhibit the expression of Bcl-xL mRNA. Cells were transfected with a combination of two siRNAs against Bcl-xL. Transfected cells were pretreated with PG for 24 hours before Dox treatment to induce apoptosis. Western blot analyses show that the expression of Bcl-xL was indeed decreased in the siRNA treated samples (Fig. 9A). Caspase-3 activity assay indicated that Bcl-xL siRNAs abolished PG-mediated cytoprotection (Fig. 9B).
Figure 9. Cardioprotective Effect of PG Is Dependent on Bcl-xL.
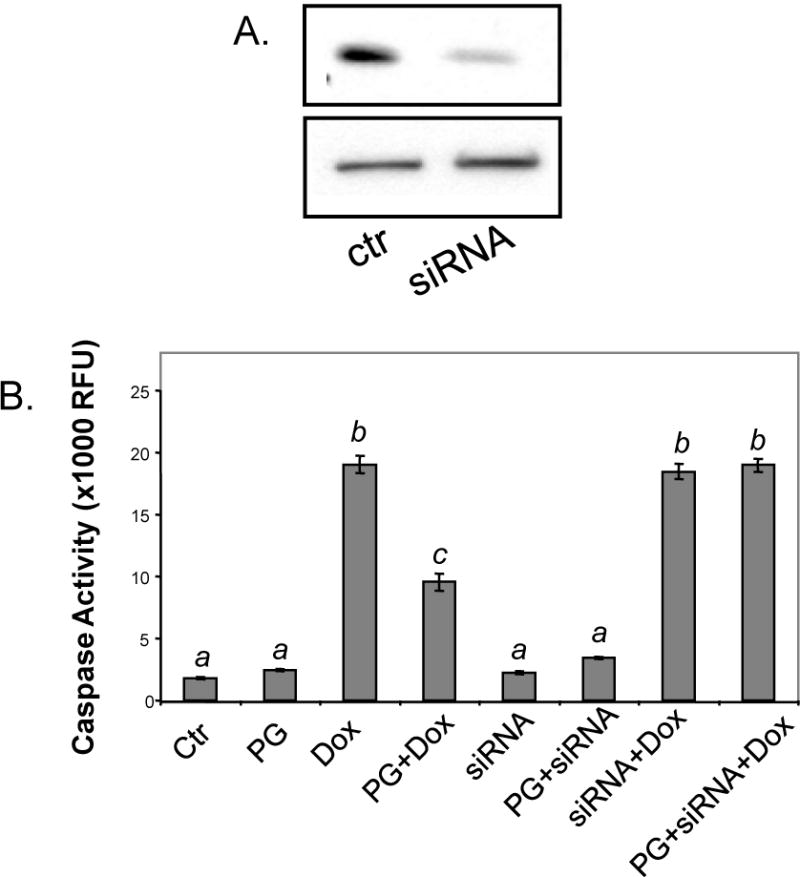
Primary cultured cardiomyocytes were transfected with Bcl-xL siRNA or negative control siRNA as described in the Methods. At 48 hrs after siRNA transfection, cells were collected to verify Bcl-xL down regulation (upper panel) by Western blot with vinculin (bottom panel) as a loading control (A) or were pretreated with 10 μM PG for 24 hrs prior to 16 hrs treatment with 1 μM Dox for measurement of caspase-3 activity (B). The data is from one experiment representative of three. A superscript letter indicates significant difference (P<0.05) from means labeled with a different letter as determined by ANOVA analysis.
Induction of Bcl-xL suggests that PG may inhibit apoptosis from other toxicants. We tested the effect of PG on toxicants such as hydrogen peroxide, rotenone, palmitate, 2-deoxyglucose and glutamate. PG pretreatment significantly decreased caspase-3 activity with any of these toxicants (Fig. 10A&B).
Figure 10. PG Inhibits Caspase-3 Activation by A Variety of Toxicants.
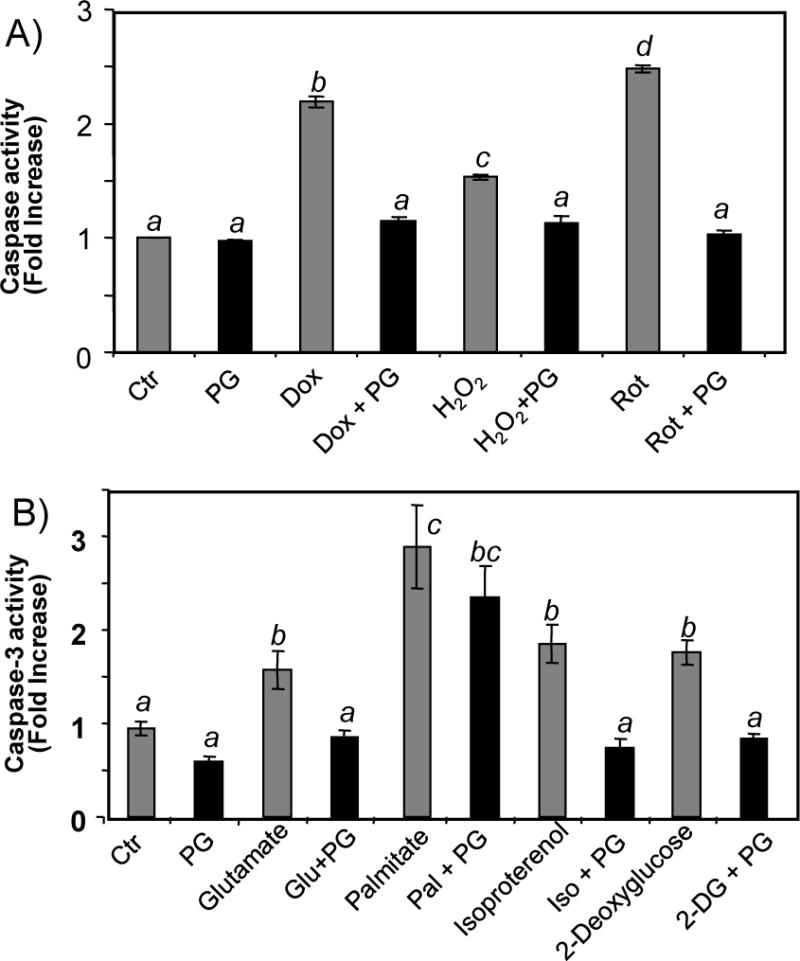
Primary cultured cardiomyocytes were pretreated with 10 μM PG 24 hrs prior to treatment with 100 μM H2O2, 1 μM rotenone (Rot), 0.5 μM palmitate (Palm), 5 μM 2-deoxyglucose (2-DG) or 25 μM glutamate (Glu) in the absence or presence of 10 μM PG. Dox treatment (0.8 μM) was included for comparison. Cells were harvested at 24 hrs after for measurements of caspase-3 activity. The data represents mean ± SEM from triplicate samples of one experiment representative of three. A superscript letter indicates significant difference (P<0.05) from means labeled with a different letter as determined by ANOVA analysis.
Discussion
This study found that PG decreases apoptotic-like cell death induced by Dox and several toxicants in primary cultures of rat cardiomyocytes. Affymetrix gene array analyses revealed that PG induces the expression of anti-apoptotic gene Bcl-xL. This finding was confirmed by measurements of Bcl-xL protein or mRNA using Western blot or RT-PCR. Blocking Bcl-xL induction by siRNAs results in a reduction in the protective effect of PG, indicating that this gene mediates the observed anti-apoptotic effect of PG. Overexpression of Bcl-xL has been shown to inhibit Dox induced apoptosis in cardiomyocytes (Kunisada et al. 2002).
While PG induces apoptosis in various tumor cells, PG has been shown to inhibit apoptosis in several tissue types, including the ovaries (Svensson et al. 2001; Okuda et al. 2004), uterus (Pecci et al. 1997; Kurita et al. 2001), breast (Feng et al. 1995; Moore et al. 2000; Berg et al. 2002), and anterior pituitary (Candolfi et al. 2005). In our study, the doses of PG necessary to inhibit Dox induced apoptosis, i.e. 1 – 10 μM, are comparable to that required to inhibit TNF-α induced apoptosis in rat lactotropes and somamotropes of the anterior pituitary (Candolfi et al. 2005). Cyclical release of PG in females is important for normal cyclical ovulatory function, which involves apoptosis during periods of the endometrial cycle with low PG concentrations (Martin et al. 1970; Terada et al. 1989; Sato et al. 1997). While it is not known why PG induces apoptosis in one cell type while inhibiting apoptosis in others, induction of Bcl-xL correlates with anti-apoptotic effect of PG in endometrial cells (Pecci et al. 1997).
The mechanism of Bcl-xL induction by PG appears to involve transcriptional regulation. The Bcl-xL protein is encoded by bcl-x gene with a complex promoter structure and alternative splicing patterns. The Bcl-xL mRNA is the major transcript of bcl-x gene, which encodes 4 additional mRNA species, bcl-xs, bcl-xβ, bcl-xγ and bcl-xΔTM, due to alternative splicing (Pecci et al. 2001). The mouse bcl-x gene has 5 promoters, P1 – P5, and is predicted to produce five mRNA species sharing the same translational start site with various lengths of 5′-untranslated region (5′UTR). P1 – P5 promoter is located from −151, −802, −1886, −2721 and −3412 bp from the translational start site respectively. In mouse mammary epithelial cells, glucocorticoids induce elevation of Bcl-xL mRNA from P4 promoter (Viegas et al. 2004). Two hormone response element (HRE)-like sequences have been identified at positions −3040 (TGgTgTGTCTGTTCc) and −3001 (aGcTCTCCAGcACA) upstream of P4 promoter. In addition to HRE binding sites, several cis-elements have been identified for binding of transcription factors such as Sp1, AP-1, Oct-1, Ets, Rel/NF-kB, GATA-1 and STATs within −3.2 kb promoter region of mouse bcl-x gene (Grillot et al. 1997). Within −1.2 kb of human bcl-x gene, binding of Rel/NF-kB, Ets, STATs or AP-1 transcription factors mediates transcriptional activation of bcl-x gene encoding Bcl-xL protein (Grad et al. 2000). The Sp1 transcription factor has been shown to mediate PG induced gene expression (Owen et al. 1998; Hewetson & Chilton 2003; Sriraman et al. 2003). PG also upregulates GATA-1 transcription factor and activates AP-1 promoter (Bamberger et al. 1996; da Silva Santos Duarte et al. 2002). While lack of activation of 905 bcl-x promoter excludes the possibility of some of these transcription factors, the discrepancy between a minor activation of 3.2 kb bcl-x promoter and a clearly elevated bcl-x gene transcription suggests the possibility of additional mechanisms such as sequence upstream of 3.2 kb in the promoter regulating PG induced Bcl-xL expression. Therefore it is possible that multiple pathways may mediate PG induced Bcl-xL gene expression in cardiomyocytes.
Although Bcl-xL plays a critical role in cell survival response, other genes identified from the microarray study may also play a role in PG induced cytoprotection. Several genes induced by PG have been shown to have cytoprotective effects, such as metallothionein I, a metal binding protein, which can function as an antioxidant and protect cardiomyocytes from Dox toxicity (Kang 1999). NADPH Quinone-Oxidoreductase I (NQO1) is able to reduce quinones similar to Dox (Lind et al. 1982; Floreani et al. 2002). Since Dox causes oxidative stress, induction of GPx3 may also contribute to the cytoprotective effect of PG. In addition to cytoprotective genes, we observed induction of several muscle contractile genes by PG, consistent with a recent report (Goldstein et al. 2004). Therefore not only PG can prevent apoptosis, it likely enhances the synthesis of muscle proteins and the contractile function of cardiomyocytes.
Rat cardiomyocytes express A and B-isoforms of PR proteins. As a member of the conserved nuclear receptor family, PR contains an N-terminal region with two transcription activator domains (AF1, AF3), a Zinc finger-containing central DNA binding domain (DBD) and a C-terminal ligand binding domain. Both PR isoforms are able to bind to PG and undergo conformational change upon ligand binding. Subsequently, the receptors dimerize (AA, AB or BB), can become phosphorylated, bind to the HRE in the promoter regions of target genes, or interact with co-factors or transcription factors (Weigel et al. 1995; Graham & Clarke 1997; Conneely et al. 2002). PR-A and PR-B may turn on the expression of different sets of genes (Richer et al. 2002). The Bcl-xL has been found to be upregulated by PR-A in human breast cancer cells (Richer et al. 2002). Since H9C2 cells express PR-B and respond to PG by increasing Bcl-xL expression, PR-B may be a key player in Bcl-xL expression in cardiomyocytes. While the myocardium of adult rats or mice express predominant PR-A, it is not known whether the human expresses both isoforms in the myocardium and whether the ratio of the isoforms changes during growth and development. This type of information is important since PG may provide therapeutic option for treatment against Dox induced cardiomyopathy if PR-B is expressed in the human myocardium.
Experimental Procedures
Cell Culture and Treatment of Drugs
Cardiomyocytes were prepared from 1 to 2 days old neonatal Sprague-Dawley rats (Harland, Indianapolis, IN) as previously described (Chen et al. 2000). The myocytes were seeded at a density of 2 × 106 cells per 100 mm dish, 0.3 × 106 cells per well of 6-well plates or 7.5 × 104 cells per well of 24-well plates. Cells were cultured in low glucose DMEM with 1 mM pyruvate, 10% fetal bovine serum, 100 units/ml penicillin and 100 units/ml streptomyocin for 3–4 days before experiments. Using this protocol, over 90% of the cells are myocytes at the time of experiments as judged by sarcomeric myosin content. At 3–4 days after plating, cardiomyocytes were cultured in DMEM containing 0.5% FBS for treatment of PG at 10 μM or indicated doses.
Caspase Activity Assay
Detached cells were collected by centrifugation and were combined with adherent cells harvested from the same well in 6-well plates. The combined cells were dissolved in 250 μl of lysis buffer (0.5% Nonidet P-40, 0.5 mM EDTA, 150 mM NaCl, and 50 mM Tris pH 7.5). Cell lysates (50 μl) were incubated 1 hour at 37°C with 40 μM of N-acetyl-Asp-Glu-Val-Asp-7-amino-4-methylcoumarin (Ac-DEVD-AMC, Alexis Biochemicals, San Diego, CA) in 100 μl reaction buffer (10 mM HEPES, pH 7.5, 0.05 M NaCl and 2.5 mM DTT). The released AMC was measured using a 96-well fluorescence plate reader (Cambridge Bioresearch Model 7620) with an excitation wavelength of 365 nm and an emission wavelength of 450 nm.
Annexin V Binding Assay
Cells were seeded onto coverglasses in 24-well plates. Detached cells in the supernatant were collected by 5 min centrifugation at 1000 rpm. After washing the detached cells and adherent cells with PBS, detached cells were combined with its corresponding group of cells remained adherent to the coverglass. Annexin V-FLUOS (Roche Applied Science, Indianapolis, IN) was diluted 1: 50 with incubation buffer (10 mM HEPES/NaOH, pH 7.4, 140 mM NaCl, 5 mM CaCl2) and cells were incubated with 25 μl/well labeling solution. The cells were examined under a Nikon E800M fluorescent microscope. The images were acquired using a Hamamatsu C5180 digital camera with the Adobe Photoshop software.
Western Blot
Cells in 100 mm dishes were lysed by scraping in EB buffer (1% Triton X-100, 10 mM Tris pH 7.4, 5 mM EDTA pH 8.0, 50 mM NaCl, 50 mM NaF, 2 mM Na3VO3) for PR or Laemini buffer [0.5 M Tris, pH 6.8, 2.4% (w/v) SDS, 50% (v/v) glycerol] for Bcl-xl measurements. Both types of lysate buffer contain freshly added protease inhibitors, 10 μg/ml aprotinin and 1 mM phenylmethanesulfonyl fluoride. Protein concentration was measured by the Bradford method (Bio-Rad, Richmond, CA), the bicichoninic acid (BCA) method (Pierce, Rockford, IL) or the Warburg-Christian method (Layne 1957). Proteins were separated by SDS polyacrylamide gel electrophoresis using a mini-Protean II electrophoresis apparatus (Bio-Rad, Richmond, CA) and transferred to immobilon-P membranes (Millipore, Bedford, MA) by electrophoresis. The membrane was incubated with primary antibodies and then secondary antibodies conjugated with horseradish peroxidase. The bound antibodies were detected via an enhanced chemiluminescent reaction.
Cytosolic and Mitochondrial Fractionation
The mitochondria and cytosolic fractions were prepared using a fractionation kit as instructed by the manufacturer (BioVision, catalog# K256-100). Briefly, 1×107 cells were scraped and washed in PBS, and suspended in 500 μl of cytosol extraction buffer containing 1 mM DTT and 1 tablet of protease inhibitors. After 10 mins incubation on ice, the cells were lysed using an ice cold Dounce homogenizer. The homogenates were examined under a microscope for complete breakage of cells. After 4°C centrifugation at 700 g for 10 mins to remove the nuclei, the supernatant was transferred to a new tube. The cytosolic fraction was collected as the supernatant by centrifugation at 10,000 g for 30 mins at 4°C. The pellet was resuspended in 50 μl of mitochondrial extraction buffer containing DTT and protease inhibitors as mitochondrial fraction.
Microarray
Cardiomyocytes were harvested by Trizol reagent (Sigma, St. Louis, MO) for extraction of total RNA. RNA was cleaned with an RNeasy mini kit (Qiagen). The quality of RNA was examined by agarose gel electrophoresis and the Agilent 2100 bioanalyzer (Agilent Technologies) to ensure the purity and integrity of RNA suitable for microarray. The RNA was converted to cDNA by reverse transcription using a SuperScript Choice kit from Invitrogen with a T7-(dT)24 primer incorporating a T7 RNA polymerase promoter. The cRNA was prepared and labeled with biotin via in vitro transcription using the BioArray High Yield RNA Transcript labeling kit (Enzo Biochemical). Labeled cRNA was fragmented by incubation at 94°C for 35 mins. For hybridization, 15 μg of fragmented cRNA was incubated for 16 hrs at 45°C with a Rat Expression 230A Gene chip. After hybridization, the gene chips were automatically washed and stained with streptavidin-phycoerythrin using a fluidics station (Affymetrix). The probed arrays were scanned at 3 μm resolution using the Genechip System confocal scanner made for Affymetrix by Agilent. Affymetrix Microarray Suite 5.0 was used to scan and analyze the relative abundance of each gene from the average difference of intensities. Analysis parameters used by the software was set to values corresponding to 1.5 fold increase or decrease (Purdom-Dickinson et al. 2007).
RT-PCR
Total RNA extracted using Trizol was used as a template for RT-PCR. Superscript II was used for reverse transcription at 37–50°C for 1 hr. For PCR, the denaturation and primer extension were carried out at 94°C and 72°C. The annealing temperature was calculated based on GC content of the primers as suggested by the oligonucleotide synthesis company (Biosynthesis, Lewisville, TX). The products were detected by agarose gel electrophoresis with ethidium bromide staining or during real-time PCR (BioRad CFX96) using SYBR Green dye.
Nuclear Run-on Assay
A non-radioactive method was used as described (Sun et al. 2008b). Briefly, the nuclei from 5 × 107 trypsinized cells were collected by centrifugation following lysis in a buffer (10 mM Tris, pH 7.4, 3 mM MgCl2, 10 mM NaCl, 150 mM sucrose, and 0.5% NP-40). The nuclei were incubated in 2× transcription buffer (200 mM KCl; 20 mM Tris-HCl, pH 8.0; 5 mM MgCl2; 5 mM dithiothreitol; 4 mM each of ATP, GTP, and CTP; 200 mM sucrose; and 20% glycerol). The reaction was initiated by addition of 4 mM biotin-16-UTP and was carried out with 2 hrs incubation at 30°C. The reaction was terminated by addition of Trizol for extraction of RNA. Streptavidin agarose was used to isolate biotin-labeled newly synthesized RNA. The RNA was reextracted with Trizol for reverse transcription and subsequent real-time PCR (BioRad CFX96) using SYBR Green dye.
Transfection
Primary cultured cardiomyocytes were transfected with 0.5 μg of DNA from pGL3 firefly luciferase under the control of MTV GRE, −905 bp human bcl-x promoter or −3.2 kbp mouse bcl-x promoter using FuGene-6 liposomes on the 3rd day of plating. A TK-Renilla luciferase plasmid (0.05 μg) was cotransfected as a reference for transfection efficiency. For siRNA, the cells in 6 well plates were incubated 6 hrs with 100 nM each 5′-GGCUGGCGAUGAGUUUGAAtt-3′ (abbreviated as SI, Ambion) and 5′-GGUAGUGAAUGAACUCUUUtt-3′ (abbreviated as SII, Ambion) or a silencer negative control siRNA of 19 bases oligonucleotide derived from a scrambled sequence (Ambion) in 1 ml mixture of Opti-MEM (Invitrogen) and DMEM containing 3 μl Oligofectamine (Invitrogen). At 48 hours after transfection, cells were placed in fresh media and treated with 10 μM PG for 24 hours for measurements of luciferases or for treatment with Dox to measure for apoptosis.
Statistics
Student’s t-test was used to compare the means of two groups of samples. Significant differences (p<0.05) were indicated with asterisks (*). ANOVA one-way analysis of variance (p<0.05) followed by Bonferroni correction was used to verify the significant difference using Prism 4.0 software. Means that are not significant different from each other are labeled with a common letter. Therefore, means in the “a” group are significant different from means labeled with “b”, “bc” or “c” and so on. Means labeled with “bc” are not significant different from those with “b” or “c”, although means labeled with “b” is significant different from that labeled “c”.
Supplementary Material
Acknowledgments
This work was supported by NIH R01 HL 076530, T32 ES007091, Arizona Disease Control Research Commission (QMC) and Mark and Mary Anne Fay Investigator Awards (SM and BX). We thank Ms. Yan Lin for technical assistance and Dr. Roger Miesfeld for MTV-GRE luciferase construct.
References
- Anversa P, Olivetti G, Leri A, Liu Y, Kajstura J. Myocyte cell death and ventricular remodeling. Curr Opin Nephrol Hypertens. 1997;6:169–176. doi: 10.1097/00041552-199703000-00011. [DOI] [PubMed] [Google Scholar]
- Arola OJ, Saraste A, Pulkki K, Kallajoki M, Parvinen M, Voipio-Pulkki LM. Acute doxorubicin cardiotoxicity involves cardiomyocyte apoptosis. Cancer Res. 2000;60:1789–1792. [PubMed] [Google Scholar]
- Bamberger AM, Bamberger CM, Gellersen B, Schulte HM. Modulation of AP-1 activity by the human progesterone receptor in endometrial adenocarcinoma cells. Proc Natl Acad Sci USA. 1996;93:6169–6174. doi: 10.1073/pnas.93.12.6169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg MN, Dharmarajan AM, Waddell BJ. Glucocorticoids and progesterone prevent apoptosis in the lactating rat mammary gland. Endocrinology. 2002;143:222–227. doi: 10.1210/endo.143.1.8584. [DOI] [PubMed] [Google Scholar]
- Bialik S, Geenen DL, Sasson IE, Cheng R, Horner JW, Evans SM, Lord EM, Koch CJ, Kitsis RN. Myocyte apoptosis during acute myocardial infarction in the mouse localizes to hypoxic regions but occurs independently of p53. J Clin Invest. 1997;100:1363–1372. doi: 10.1172/JCI119656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruynzeel AM, Abou El, Hassan MA, Torun E, Bast A, van der Vijgh WJ, Kruyt FA. Caspase-dependent and -independent suppression of apoptosis by monoHER in Doxorubicin treated cells. Br J Cancer. 2007;96:450–456. doi: 10.1038/sj.bjc.6603598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Candolfi M, Jaita G, Zaldivar V, Zarate S, Ferrari L, Pisera D, Castro MG, Seilicovich A. Progesterone antagonizes the permissive action of estradiol on tumor necrosis factor-alpha-induced apoptosis of anterior pituitary cells. Endocrinology. 2005;146:736–743. doi: 10.1210/en.2004-1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen QM, Alexander D, Sun H, Xie L, Lin Y, Terrand J, Morrissy S, Purdom S. Corticosteroids inhibit cell death induced by doxorubicin in cardiomyocytes: induction of antiapoptosis, antioxidant, and detoxification genes. Mol Pharmacol. 2005;67:1861–1873. doi: 10.1124/mol.104.003814. [DOI] [PubMed] [Google Scholar]
- Chen QM, Tu VC, Wu Y, Bahl JJ. Hydrogen peroxide dose dependent induction of cell death or hypertrophy in cardiomyocytes. Arch Biochem Biophys. 2000;373:242–248. doi: 10.1006/abbi.1999.1558. [DOI] [PubMed] [Google Scholar]
- Condorelli G, Morisco C, Stassi G, Notte A, Farina F, Sgaramella G, de Rienzo A, Roncarati R, Trimarco B, Lembo G. Increased cardiomyocyte apoptosis and changes in proapoptotic and antiapoptotic genes bax and bcl-2 during left ventricular adaptations to chronic pressure overload in the rat. Circulation. 1999;99:3071–3078. doi: 10.1161/01.cir.99.23.3071. [DOI] [PubMed] [Google Scholar]
- Conneely OM, Mulac-Jericevic B, DeMayo F, Lydon JP, O’Malley BW. Reproductive functions of progesterone receptors. Recent Prog Horm Res. 2002;57:339–355. doi: 10.1210/rp.57.1.339. [DOI] [PubMed] [Google Scholar]
- da Silva Santos, Duarte A, Sales TS, Mengel JO, Costa FF, Saad ST. Progesterone upregulates GATA-1 on erythroid progenitors cells in liquid culture. Blood Cells Mol Diseases. 2002;29:213–224. doi: 10.1006/bcmd.2002.0551. [DOI] [PubMed] [Google Scholar]
- Doroshow JH, Davies KJ. Redox cycling of anthracyclines by cardiac mitochondria. II. Formation of superoxide anion, hydrogen peroxide, and hydroxyl radical. J Biol Chem. 1986;261:3068–3074. [PubMed] [Google Scholar]
- Feng Z, Marti A, Jehn B, Altermatt HJ, Chicaiza G, Jaggi R. Glucocorticoid and progesterone inhibit involution and programmed cell death in the mouse mammary gland. J Cell Biol. 1995;131:1095–1103. doi: 10.1083/jcb.131.4.1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feuerstein G, Yue TL, Ma X, Ruffolo RR. Novel mechanisms in the treatment of heart failure: inhibition of oxygen radicals and apoptosis by carvedilol. Prog Cardiovasc Dis. 1998;41:17–24. doi: 10.1016/s0033-0620(98)80027-3. [DOI] [PubMed] [Google Scholar]
- Floreani M, Napoli E, Palatini P. Role of antioxidant defences in the species-specific response of isolated atria to menadione. Comp Biochem Physiol Toxicol Pharmacol. 2002;132:143–151. doi: 10.1016/s1532-0456(02)00060-1. [DOI] [PubMed] [Google Scholar]
- Gewirtz DA. A critical evaluation of the mechanisms of action proposed for the antitumor effects of the anthracycline antibiotics adriamycin and daunorubicin. Biochem Pharmacol. 1999;57:727–741. doi: 10.1016/s0006-2952(98)00307-4. [DOI] [PubMed] [Google Scholar]
- Goldstein J, Sites CK, Toth MJ. Progesterone stimulates cardiac muscle protein synthesis via receptor-dependent pathway. Fertil Steril. 2004;82:430–436. doi: 10.1016/j.fertnstert.2004.03.018. [DOI] [PubMed] [Google Scholar]
- Grad JM, Zeng XR, Boise LH. Regulation of Bcl-xL: a little bit of this and a little bit of STAT. Curr Opin Oncol. 2000;12:543–549. doi: 10.1097/00001622-200011000-00006. [DOI] [PubMed] [Google Scholar]
- Graham JD, Clarke CL. Physiological action of progesterone in target tissues. Endocr Rev. 1997;18:502–519. doi: 10.1210/edrv.18.4.0308. [DOI] [PubMed] [Google Scholar]
- Grillot DA, Gonzalez-Garcia M, Ekhterae D, Duan L, Inohara N, Ohta S, Seldin MF, Nunez G. Genomic organization, promoter region analysis, and chromosome localization of the mouse bcl-x gene. J Immun. 1997;158:4750–4757. [PubMed] [Google Scholar]
- Grohe C, Kahlert S, Lobbert K, Stimpel M, Karas RH, Vetter H, Neyses L. Cardiac myocytes and fibroblasts contain functional estrogen receptors. FEBS Lett. 1997;416:107–112. doi: 10.1016/s0014-5793(97)01179-4. [DOI] [PubMed] [Google Scholar]
- Hewetson A, Chilton BS. An Sp1-NF-Y/progesterone receptor DNA binding-dependent mechanism regulates progesterone-induced transcriptional activation of the rabbit RUSH/SMARCA3 gene. J Biol Chem. 2003;278:40177–40185. doi: 10.1074/jbc.M303921200. [DOI] [PubMed] [Google Scholar]
- Ingegno MD, Money SR, Thelmo W, Greene GL, Davidian M, Jaffe BM, Pertschuk LP. Progesterone receptors in the human heart and great vessels. Lab Invest. 1988;59:353–356. [PubMed] [Google Scholar]
- Kalyanaraman B, Joseph J, Kalivendi S, Wang S, Konorev E, Kotamraju S. Doxorubicin-induced apoptosis: implications in cardiotoxicity. Mol Cell Biochem. 2002:234–235. [PubMed] [Google Scholar]
- Kang YJ. The antioxidant function of metallothionein in the heart. Proc Soc Exp Biol Med. 1999;222:263–273. doi: 10.1046/j.1525-1373.1999.d01-143.x. [DOI] [PubMed] [Google Scholar]
- Keizer HG, Pinedo HM, Schuurhuis GJ, Joenje H. Doxorubicin (adriamycin): a critical review of free radical-dependent mechanisms of cytotoxicity. Pharmacol Therap. 1990;47:219–231. doi: 10.1016/0163-7258(90)90088-j. [DOI] [PubMed] [Google Scholar]
- Knowlton AA, Sun L. Heat-shock factor-1, steroid hormones, and regulation of heat-shock protein expression in the heart. Am J Physiol Heart Circ Physiol. 2001;280:H455–464. doi: 10.1152/ajpheart.2001.280.1.H455. [DOI] [PubMed] [Google Scholar]
- Kotamraju S, Konorev EA, Joseph J, Kalyanaraman B. Doxorubicin-induced apoptosis in endothelial cells and cardiomyocytes is ameliorated by nitrone spin traps and ebselen. Role of reactive oxygen and nitrogen species. J Biol Chem. 2000;275:33585–33592. doi: 10.1074/jbc.M003890200. [DOI] [PubMed] [Google Scholar]
- Kuebler JF, Jarrar D, Bland KI, Rue L, 3rd, Wang P, Chaudry IH. Progesterone administration after trauma and hemorrhagic shock improves cardiovascular responses. Crit Care Med. 2003;31:1786–1793. doi: 10.1097/01.CCM.0000063441.41446.23. [DOI] [PubMed] [Google Scholar]
- Kumar D, Kirshenbaum L, Li T, Danelisen I, Singal P. Apoptosis in isolated adult cardiomyocytes exposed to adriamycin. Ann N Y Acad Sci. 1999;874:156–168. doi: 10.1111/j.1749-6632.1999.tb09233.x. [DOI] [PubMed] [Google Scholar]
- Kunisada K, Tone E, Negoro S, Nakaoka Y, Oshima Y, Osugi T, Funamoto M, Izumi M, Fujio Y, Hirota H, Yamauchi-Takihara K. Bcl-xl reduces doxorubicin-induced myocardial damage but fails to control cardiac gene downregulation. Cardiovasc Res. 2002;53:936–943. doi: 10.1016/s0008-6363(01)00506-5. [DOI] [PubMed] [Google Scholar]
- Kurita T, Wang YZ, Donjacour AA, Zhao C, Lydon JP, O’Malley BW, Isaacs JT, Dahiya R, Cunha GR. Paracrine regulation of apoptosis by steroid hormones in the male and female reproductive system. Cell Death Differ. 2001;8:192–200. doi: 10.1038/sj.cdd.4400797. [DOI] [PubMed] [Google Scholar]
- Layne E. Spectrophotometric and turbidimetric methods for measuring proteins. Meth Enzymol. 1957;3:447–454. [Google Scholar]
- Lin AL, McGill HC, Jr, Shain SA. Hormone receptors of the baboon cardiovascular system. Biochemical characterization of aortic and myocardial cytoplasmic progesterone receptors. Circ Res. 1982;50:610–616. doi: 10.1161/01.res.50.5.610. [DOI] [PubMed] [Google Scholar]
- Lind C, Hochstein P, Ernster L. DT-diaphorase as a quinone reductase: a cellular control device against semiquinone and superoxide radical formation. Arch Biochem Biophys. 1982;216:178–185. doi: 10.1016/0003-9861(82)90202-8. [DOI] [PubMed] [Google Scholar]
- Low AK, Russell LD, Holman HE, Shepherd JM, Hicks GS, Brown CA. Hormone replacement therapy and coronary heart disease in women: a review of the evidence. Am J Med Sci. 2002;324:180–184. doi: 10.1097/00000441-200210000-00003. [DOI] [PubMed] [Google Scholar]
- Ma XL, Kumar S, Gao F, Louden CS, Lopez BL, Christopher TA, Wang C, Lee JC, Feuerstein GZ, Yue TL. Inhibition of p38 mitogen-activated protein kinase decreases cardiomyocyte apoptosis and improves cardiac function after myocardial ischemia and reperfusion. Circulation. 1999;99:1685–1691. doi: 10.1161/01.cir.99.13.1685. [DOI] [PubMed] [Google Scholar]
- Martin L, Finn CA, Carter J. Effects of progesterone and oestradiol-17 beta on the luminal epithelium of the mouse uterus. J Reprod Fertil. 1970;21:461–469. doi: 10.1530/jrf.0.0210461. [DOI] [PubMed] [Google Scholar]
- Minshall RD, Pavcnik D, Browne DL, Hermsmeyer K. Nongenomic vasodilator action of progesterone on primate coronary arteries. J Appl Physiol. 2002;92:701–708. doi: 10.1152/japplphysiol.00689.2001. [DOI] [PubMed] [Google Scholar]
- Moore MR, Conover JL, Franks KM. Progestin effects on long-term growth, death, and Bcl-xL in breast cancer cells. Biochem Biophys Res Commun. 2000;277:650–654. doi: 10.1006/bbrc.2000.3728. [DOI] [PubMed] [Google Scholar]
- Murabito JM. Women and cardiovascular disease: contributions from the Framingham Heart Study. J Am Med Womens Assoc. 1995;50:35–39. 55. [PubMed] [Google Scholar]
- Narula J, Haider N, Virmani R, DiSalvo TG, Kolodgie FD, Hajjar RJ, Schmidt U, Semigran MJ, Dec GW, Khaw BA. Apoptosis in myocytes in end-stage heart failure. N Engl J Med. 1996;335:1182–1189. doi: 10.1056/NEJM199610173351603. [DOI] [PubMed] [Google Scholar]
- Nickenig G, Strehlow K, Wassmann S, Baumer AT, Albory K, Sauer H, Bohm M. Differential effects of estrogen and progesterone on AT(1) receptor gene expression in vascular smooth muscle cells. Circulation. 2000;102:1828–1833. doi: 10.1161/01.cir.102.15.1828. [DOI] [PubMed] [Google Scholar]
- Okuda K, Korzekwa A, Shibaya M, Murakami S, Nishimura R, Tsubouchi M, Woclawek-Potocka I, Skarzynski DJ. Progesterone is a suppressor of apoptosis in bovine luteal cells. Biol Reprod. 2004;71:2065–2071. doi: 10.1095/biolreprod.104.028076. [DOI] [PubMed] [Google Scholar]
- Owen GI, Richer JK, Tung L, Takimoto G, Horwitz KB. Progesterone regulates transcription of the p21(WAF1) cyclin- dependent kinase inhibitor gene through Sp1 and CBP/p300. J Biol Chem. 1998;273:10696–10701. doi: 10.1074/jbc.273.17.10696. [DOI] [PubMed] [Google Scholar]
- Pasanen S, Ylikomi T, Syvala H, Tuohimaa P. Distribution of progesterone receptor in chicken: novel target organs for progesterone and estrogen action. Mol Cell Endocrinol. 1997;135:79–91. doi: 10.1016/s0303-7207(97)00192-5. [DOI] [PubMed] [Google Scholar]
- Pecci A, Scholz A, Pelster D, Beato M. Progestins prevent apoptosis in a rat endometrial cell line and increase the ratio of bcl-XL to bcl-XS. J Biol Chem. 1997;272:11791–11798. doi: 10.1074/jbc.272.18.11791. [DOI] [PubMed] [Google Scholar]
- Pecci A, Viegas LR, Baranao JL, Beato M. Promoter choice influences alternative splicing and determines the balance of isoforms expressed from the mouse bcl-X gene. J Biol Chem. 2001;276:21062–21069. doi: 10.1074/jbc.M008665200. [DOI] [PubMed] [Google Scholar]
- Pecins-Thompson M, Keller-Wood M. Effects of progesterone on blood pressure, plasma volume, and responses to hypotension. Am J Physiol. 1997;272:R377–385. doi: 10.1152/ajpregu.1997.272.1.R377. [DOI] [PubMed] [Google Scholar]
- Purdom-Dickinson S, Lin Y, Dedek M, Johnson J, Chen Q. Induction of Antioxidant and Detoxification Response by Oxidants in Cardiomyocytes: Evidence from Gene Expression Profiling and Activation of the Nrf2 Transcription Factor. J Mol Cell Cardiol. 2007;42:159–176. doi: 10.1016/j.yjmcc.2006.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richer JK, Jacobsen BM, Manning NG, Abel MG, Wolf DM, Horwitz KB. Differential gene regulation by the two progesterone receptor isoforms in human breast cancer cells. J Biol Chem. 2002;277:5209–5218. doi: 10.1074/jbc.M110090200. [DOI] [PubMed] [Google Scholar]
- Roesch DM, Keller-Wood M. Progesterone rapidly reduces arterial pressure in ewes. Am J Physiol. 1997;272:H386–391. doi: 10.1152/ajpheart.1997.272.1.H386. [DOI] [PubMed] [Google Scholar]
- Sato T, Fukazawa Y, Kojima H, Enari M, Iguchi T, Ohta Y. Apoptotic cell death during the estrous cycle in the rat uterus and vagina. Anat Rec. 1997;248:76–83. doi: 10.1002/(SICI)1097-0185(199705)248:1<76::AID-AR9>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- Singal PK, Iliskovic N. Doxorubicin-induced cardiomyopathy. N Engl J Med. 1998;339:900–905. doi: 10.1056/NEJM199809243391307. [DOI] [PubMed] [Google Scholar]
- Singal PK, Li T, Kumar D, Danelisen I, Iliskovic N. Adriamycin-induced heart failure: mechanism and modulation. Mol Cell Biochem. 2000;207:77–86. doi: 10.1023/a:1007094214460. [DOI] [PubMed] [Google Scholar]
- Smid-Koopman E, Blok LJ, Kuhne LC, Burger CW, Helmerhorst TJ, Brinkmann AO, Huikeshoven FJ. Distinct functional differences of human progesterone receptors A and B on gene expression and growth regulation in two endometrial carcinoma cell lines. J Soc Gynecol Investig. 2003;10:49–57. [PubMed] [Google Scholar]
- Sriraman V, Sharma SC, Richards JS. Transactivation of the progesterone receptor gene in granulosa cells: evidence that Sp1/Sp3 binding sites in the proximal promoter play a key role in luteinizing hormone inducibility. Mol Endocrin. 2003;17:436–449. doi: 10.1210/me.2002-0252. [DOI] [PubMed] [Google Scholar]
- Sun H, Sheveleva E, Xu B, Inoue H, Bowden TG, Chen QM. Corticosteroids induce COX-2 expression in cardiomyocytes: role of glucocorticoid receptor and C/EBP-beta. Am J Physiol Cell Physiol. 2008a;295:915–922. doi: 10.1152/ajpcell.90646.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun H, Xu B, Inoue H, Chen QM. P38 MAPK mediates COX-2 gene expression by corticosterone in cardiomyocytes. Cell Signal. 2008b;20:1952–1959. doi: 10.1016/j.cellsig.2008.07.003. [DOI] [PubMed] [Google Scholar]
- Svensson EC, Markstrom E, Shao R, Andersson M, Billig H. Progesterone receptor antagonists Org 31710 and RU 486 increase apoptosis in human periovulatory granulosa cells. Fertil Steril. 2001;76:1225–1231. doi: 10.1016/s0015-0282(01)02891-6. [DOI] [PubMed] [Google Scholar]
- Terada N, Yamamoto R, Takada T, Miyake T, Terakawa N, Wakimoto H, Taniguchi H, Li W, Kitamura Y, Matsumoto K. Inhibitory effect of progesterone on cell death of mouse uterine epithelium. J Steroid Biochem. 1989;33:1091–1096. doi: 10.1016/0022-4731(89)90414-7. [DOI] [PubMed] [Google Scholar]
- Ueno M, Kakinuma Y, Yuhki K, Murakoshi N, Iemitsu M, Miyauchi T, Yamaguchi I. Doxorubicin induces apoptosis by activation of caspase-3 in cultured cardiomyocytes in vitro and rat cardiac ventricles in vivo. J Pharmacol Sci. 2006;101:151–158. doi: 10.1254/jphs.fp0050980. [DOI] [PubMed] [Google Scholar]
- Viegas LR, Vicent GP, Baranao JL, Beato M, Pecci A. Steroid hormones induce bcl-X gene expression through direct activation of distal promoter P4. J Biol Chem. 2004;279:9831–9839. doi: 10.1074/jbc.M312402200. [DOI] [PubMed] [Google Scholar]
- Webster KA, Discher DJ, Kaiser S, Hernandez O, Sato B, Bishopric NH. Hypoxia-activated apoptosis of cardiac myocytes requires reoxygenation or a pH shift and is independent of p53. J Clin Invest. 1999;104:239–252. doi: 10.1172/JCI5871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weigel NL, Bai W, Zhang Y, Beck CA, Edwards DP, Poletti A. Phosphorylation and progesterone receptor function. J Steroid Biochem Mol Biol. 1995;53:509–514. doi: 10.1016/0960-0760(95)00098-k. [DOI] [PubMed] [Google Scholar]
- Wexler LF. Studies of acute coronary syndromes in women–lessons for everyone. N Engl J Med. 1999;341:275–276. doi: 10.1056/NEJM199907223410409. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.