

Abstract
Rheumatoid arthritis (RA) is an autoimmune disease, resulting in a chronic, systemic inflammatory disorder. It may affect many tissues and organs, but it primarily affects the flexible joints. In clinical practice patient care generates many questions about diagnosis, prognosis, and treatment. It is challenging for health care specialists to keep up to date with the medical literature. This review summarizes the pathogenesis, the polymorphisms of interleukin and interleukin genes and the standard available and possible future immunologic targets for RA treatment. The identification of disease-associated interleukin and interleukin receptor genes can provide precious insight into the genetic variations prior to disease onset in order to identify the pathways important for RA pathogenesis. The knowledge of the complex genetic background may prove useful for developing novel therapies and making personalized medicine based on the individual’s genetics.
Keywords: Rheumatoid arthritis, Interleukins, Polymorphisms, Immunologic targets, Therapy
Core tip: Rheumatoid arthritis (RA) is an autoimmune disease, resulting in a chronic, systemic inflammatory disorder. It may affect many tissues and organs, but mainly attacks the flexible joints. This review provides a comprehensive overview about the genetic background, especially with regard to inflammatory cytokines to understand the pathogenesis of the disease. Furthermore it summarizes the current therapy and the future therapeutic agents for RA.
INTRODUCTION
Rheumatoid arthritis
Rheumatoid arthritis (RA) is one of the most common systemic autoimmune diseases which affects approximately 1% of the population[1-3], which can lead to significant morbidity and mortality rates, and can shorten the lifespan by 10 years[4]. RA affects people all over the world, but it is more uncommon in Africa[5]. In contrast, its largest prevalence is registered among North-American Chippewa- and Pima-tribes[5]. Like in other autoimmune diseases females are more often affected than males.
RA is a severely disabling chronic inflammatory disease characterized by inflammation, persistent synovitis, progressive joint destruction, and systemic, extraarticular manifestations (e.g., pericarditis, episcleritis/scleritis, secondary Sjögren syndrome, Felty syndrome, cervical myelopathy, neuropathy, interstitial lung disease, rheumatoid nodules, and vasculitis)[5,6]. Accelerated atherosclerosis is the leading cause of death among patients with RA. The incidence of lymphoma increases twofold in RA patients which is thought to be caused by the underlying severity of the inflammatory process, and not a consequence of the medical treatment[7].
Patients with RA typically present with pain, stiffness in multiple joints, swollen peripheric joints, regional osteoporosis, narrowing of the synovial space and fibrous ankylosis. The wrists, proximal interphalangeal joints, and metacarpophalangeal joints are most commonly affected, however, the clinical appearance can be heterogenous[5,6]. In 2010 the American College of Rheumatology (ACR) and European League Against Rheumatism (EULAR) developed a new classification criteria, aiming to support diagnosis and facilitate the early introduction of effective RA therapy[8]. The 2010 criteria include tender and swollen joint count, acute phase reactants [C-reactive protein levels (CRP) and erythrocyte sedimentation rate (ESR)], anti-citrullinated protein antibodies (ACPA) or rheumatoid factor (RF), and symptom duration[8]. RF is not specific for RA and it may be present in patients with other diseases, such as hepatitis C and in also healthy older persons. However, patients seropositive for RF are characterized by rapid and more severe course of the disease, and it is frequently coupled with extraarticular manifestations. ACPA is more specific to RA (97.1%)[9]. It is produced against citrullinated proteins which derive from an RA-specific dysregulation of the humoral immune response[10,11]. Likewise RF, the presence of ACPA contributes to a more severe and extended type of RA[10]. The simultaneous presence of these two serological factors predestinates the onset of RA at a 28.9 times higher risk, compared to the population negative for both autoantibodies[10].
RA is an inflammatory arthritis that results from a systemic autoimmune response stimulated by an as yet unidentified antigen. It is commonly believed that the generation of autoantibodies through interactions of the innate immune system (antigen-presenting cells) with the adaptive immune system (CD4+ T cells and B cells) is central to the pathogenesis[12]. Although the clear mechanisms of RA pathogenesis still remain to be defined, cytokines are considered to play an important role in the disease. The imbalance between pro- and anti-inflammatory cytokines promotes the induction of autoimmunity, inflammation and joint destruction. The synovial membrane in patients with RA is characterized by hyperplasia, proliferation, angiogenesis and an infiltrate of predominantly CD4+ T helper (Th) cells. The pro-inflammatory cytokines, especially tumor necrosis factor alpha (TNF-α), and two interleukins (ILs), IL-1B and IL-6 are the key cytokines which drive inflammation and the destructive process[13]. However, it is likely that other cytokines such as IL-23, IL-17A and interferon gamma (IFN-γ) also play crucial roles in the pathogenesis of RA. IL-4 and IL-10, on the other hand, have been suggested to improve arthritis[14]. Joint damage results from the degradation of connective tissue by tissue-destroying matrix metalloproteinases (MMP) and the stimulation of osteoclastogenesis through the receptor activator of nuclear factor-kB ligand (RANKL). Activated CD4+ T cells also stimulate B cells to produce immunoglobulins, including RF[15].
Similarly to other autoimmune disorders, RA is a disease of multifactorial etiology. The genetic predisposition is responsible for approximately 60% of the whole disease risk, while environmental factors, such as infections by microbial agents[16], smoking[17,18], obesity, or schizophrenia of first-degree relatives[19,20], and abnormalities of the autoimmune processes also play a role. The association with the human leukocyte antigen (HLA)-DRB1 locus was the first to be described to confer risk for RA (50% of the overall genetic predisposition)[21]. Linkage and genome-wide association studies identified over 30 validated additional genetic loci associated with RA, example HLA-DRB1, protein tyrosine phosphatase N22 (PTPN22), tumor necrosis factor alpha-induced protein 3 (TNFAIP3), TNF receptor-associated factor 1 (TRAF1), signal transducer and activator of transcription 4 (STAT4), chemokine (C-C motif) receptor 6 (CCR6), PX domain containing serine/threonine kinase (PXK)[22-24]. Further studies have revealed the importance of numerous other predisposing genes and their variants, including several pro-inflammatory and anti-inflammatory cytokine genes, especially interleukins (Figure 1).
Figure 1.
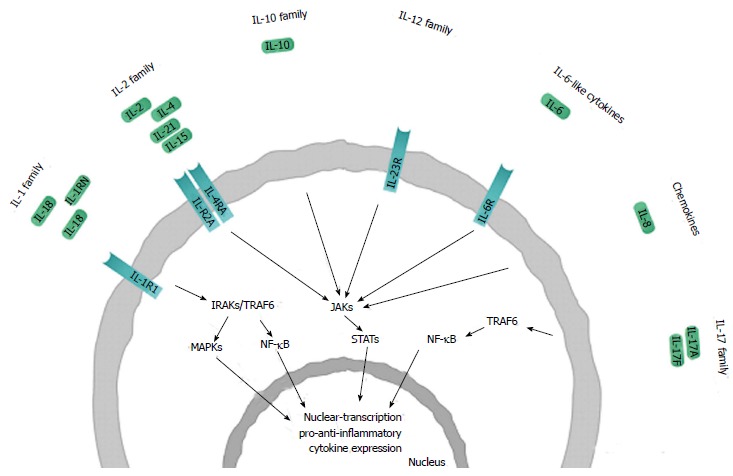
Schematic representation of the interleukin families and receptors involved in the pathogenesis of rheumatoid arthritis. Only those interleukins and interleukin (IL) receptors are shown where studies have demonstrated positive association between genes/SNPs and disease phenotype. Interleukins are assigned to each family based on sequence homology and receptor chain similarities or functional properties, considerable overlap between these families exists. Polymorphisms in genes encoding ILs and ILRs have been found to be involved in rheumatoid arthritis. Ligand binding initiates intracellular phosphorylation cascades that are mediated by kinases [i.e., interleukin 1 receptor associated kinase (IRAK); mitogen-activated protein kinase (MAPK); Janus kinase (JAK) and tumor necrosis factor (TNF) receptor associated factor, TRAF], resulting in signal transduction through certain transcription factors [including signal transducers and activators of transcription (STAT); nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB)]. These transcription factors stimulate the expression of a number of pro-inflammatory and anti-inflammatory cytokine genes involved in the pathogenesis of rheumatoid arthritis.
RESEARCH
We conducted a systematic review of the literature of the last 10 years on the polymorphisms of interleukin and interleukin genes associated with RA, and also standard available and possible future therapeutic possibilities of RA. PubMed was searched for papers and abstracts published in English-language journals, using the following terms and/or text words alone and in combination “rheumatoid arthritis”, “interleukins”, “interleukin receptors”, “polymorphisms” and “therapy”. No restrictions were placed on race, ethnicity, or geographic area. Extraction from each study was conducted independently by all authors, and consensus was achieved for all data.
INTERLEUKIN AND INTERLEUKIN RECEPTOR GENE POLYMORPHISMS
ILs are a large group of cytokines which are especially important in stimulating immune responses, such as inflammation. Once an IL has been produced, it proceeds to its target cell and binds to it via a receptor molecule on the cell surface. This interaction triggers a cascade of signals within the target cell which finally alters the behaviour of the cell. Different types of ILs are known. The nomenclature is based on sequence homology and receptor chain similarities or functional nature (www.genenames.org/genefamilies/IL) (Table 1)[25]. Within the interleukin families several IL and ILR gene polymorphisms have been investigated that are associated with RA (Table 2).
Table 1.
Characteristics of cytokines in rheumatoid arthritis
| Family | Cytokine | Cytogenetic location | Molecular weight | Receptor | Cell source |
| IL-1 | IL-1B | 2q14 | 17 kD | IL-1R1 | Macrophages, monocytes, lymphocytes, keratinocytes, microglia, megakaryocytes, neutrophils, fibroblasts and synovial lining cells |
| (IL-1F2) | IL-1R2 | ||||
| IL-1RN | 2q14.2 | 16.1-20 kD | IL-1R1 | Monocytes, macrophages, fibroblasts, neutrophils, epithelial cells and keratinocytes | |
| (IL-1F3) | IL-1R2 | ||||
| IL-18 | 11q22.2-q22.3 | 22.3 kD | IL-18R1 | Macrophages, Kupffer cells, keratinocytes, osteoblasts, astrocytes, and DCs | |
| (IL-1F4) | IL-18RAP | ||||
| IL-2 | IL-2 | 4q26-q27 | 15.5 kD | IL-2R | CD4+, CD8+ activated T cells, DCs, NK and NKT cells |
| IL-2RA | 10p15-p14 | 30.8 kD | IL-2R | Activated T and B cells, thymocytes, myeloid precursors and olygodendrocytes | |
| IL-4 | 5q23-q31 | 15 kD | IL-4R1 | Th2 cells, basophils, eosinophils, mast cells, NKT and γ/δ T cells | |
| IL-4R2 | |||||
| IL-15 | 4q31 | 14-15 kD | IL-15R | Monocytes, activated CD4+ T cells, keratinocytes, skeletal muscle cells | |
| IL-21 | 4q26-q27 | 15 kD | IL-21R | T and NKT cells | |
| IL-10 | IL-10 | 1q31-q32 | 18.6 kD | IL-10RA/IL-10RB | T and B cells, monocytes, macrophages and DCs |
| IL-12 | IL-23 | 12q13.13 | 19 kD | IL-12RB1/IL-23R | Macrophages and activated DCs |
| IL-6-like cytokines | IL-6 | 7p21-p15 | 19-26 kD | IL-6R/IL-6ST | Endothelial cells, fibroblasts, monocytes/ macrophages |
| IL-17 | IL-17A | 6p12 | 35 kD | IL-17RA/IL-17RC | Th17, CD8+ T cells, NK cells, NKT cells, γ/δ T cells and neutrophils |
| IL-17F | 6p12 | 44 kD | IL-17RA/IL-17RC | Th17, CD8+ T cells, NK cells, NKT cells, γ/δ T ells and neutrophils | |
| Chemokines | IL-8 | 4q13-q21 | 16 kD | IL-8RA/IL-8RB | Monocytes, macrophages, neutrophils, lymphocytes, endothelial cells, epithelial cells, fibroblasts, keratinocytes, chondrocytes, synovial cells, and hepatocytes |
NK: Natural killer cells, NKT: Natural killer T cells, DCs: Dendritic cells; IL: Interleukin.
Table 2.
The examined interleukin and interleukin receptor gene polymorphisms that are associated with rheumatoid arthritis
| Gene | Polymorphism | Population | Ref. |
| IL-1B | rs16944 | Algerian, British, Turkish | [34,35,40] |
| rs1143623 | British | [35] | |
| rs1143634 | Algerian, Asian, Turkish | [34,40-42] | |
| IL1-RN | rs419598 | Black South Africans | [36] |
| IL-2/IL-21 | rs907715 | Australasian | [72] |
| rs6822844 | Australasian, Dutch | [65,72] | |
| rs17388568 | Australasian | [72] | |
| IL-2RA | rs2104286 | Dutch | [64] |
| IL-4 | rs2243250 | Egyptian, Polish | [51,52] |
| IL-4R | rs1801275 | African American, Egyptian | [54,55] |
| rs1805010 | African American, Egyptian | [54,55] | |
| IL-6 | rs1800795 | Iranian, United Kingdom, Spain, Spanish, Turkish | [34,114,116, 117,122] |
| rs1800796 | Han Chinese, Taiwan, Turkish | [34,43,47,118] | |
| IL-8 | rs112664 | Taiwan | [118] |
| rs2227306 | Caucasian | [137] | |
| IL-10 | rs1800871 | Malaysian, Polish | [80,81] |
| rs1800872 | Chinese, Malaysian, Polish | [80-82] | |
| rs1800896 | Malaysian, Polish | [80,81] | |
| IL-15 | rs2322182 | North European | [59] |
| rs4371699 | North European | [59] | |
| rs6821171 | North European | [59] | |
| rs7665842 | North European | [59] | |
| rs7667746 | North European | [59] | |
| IL-17A | rs1974226 | Japanese | [136] |
| rs2275913 | Norwegian | [135] | |
| rs3748067 | Japanese | [136] | |
| rs3804513 | Japanese | [136] | |
| IL-17F | rs763780 | Polish | [134] |
| rs2397084 | Polish | [134] | |
| IL-18 | rs187238 | Chinese, Egyptian | [46,47] |
| rs549908 | Taiwan | [48] | |
| rs360718 | Japanese | [49] | |
| rs360722 | Japanese | [49] | |
| rs1946518 | Chinese, Egyptian, Japanese | [46,47,49] | |
| IL-23R | rs1004819 | European, New Zealand, Spanish | [97,100,106] |
| rs1343151 | European, New Zealand, Spanish | [97,100,106] | |
| rs1495965 | Spanish | [97] | |
| rs2201841 | European, New Zealand, Hungarian | [96,97,100,106] | |
| rs7517847 | European, New Zealand, Spanish | [97,100,106] | |
| rs7530511 | Caucasian | [98] | |
| rs10889677 | Hungarian | [96] | |
| rs11209026 | European, Caucasian, New Zealand | [98,100,106] | |
| rs11209032 | Spanish | [97] | |
| rs10489629 | Europan, New Zealand, Spanish | [97,100,106] |
The IL-1 family
Members of the IL-1 family (IL-1A, IL-1B, IL-1RN, IL-18, IL-33, IL-36A, IL-36B, IL-36G, IL-36RN, IL-37 and IL-38) have similar gene structures and induce a complex network of pro-inflammatory cytokines. IL-1 family has also expanded to 9 distinct genes including coreceptors, decoy receptors, binding proteins and inhibitory receptors[26].
IL-1: IL-1 is a key mediator of inflammation which has an effect on cell proliferation and differentiation. It mediates many inflammatory diseases by initiating and potentiating immune and inflammatory responses[25]. IL-1 is involved in several systemic autoinflammatory syndromes and in juvenile RA. It also plays a pathogenic role in inflammation and tissue destruction[27,28]. The most studied IL-1 proteins are IL-1α and IL-1β. IL-1A (OMIM 147760) encodes IL-1α which is cell-bound, and IL-1B (OMIM 147720) encodes IL-1β, a secreted cytokine. The IL-1 receptor antagonist (IL-1RN) (OMIM 147679) is an anti-inflammatory protein which binds to IL-1 receptor type 1 (IL-1R1, OMIM 147810) without transducing signal[29,30].
Two SNPs, the -511 C/T (rs16944) in IL-1B gene promoter and +3953 C/T (rs1143634) in the exon 5 of IL-1B are thought to influence IL-1 expression[31,32]. Both loci may influence erosive damage in RA[33]. The rarer (-511) C-allele is associated with milder erosive disease in patients with a disease duration in excess of 20 years. In a Turkish cohort it was found that patients carrying the 2/2 (T/T) genotype of IL-1B +3953 gene are susceptible to RA. However, the 1/2 (C/T) genotype of IL-1B -511 has a protective role against RA[34]. In British Caucasian RA patients the IL-1B -1464 C/G (rs1143623) G allele showed the possibility to have protective effect in RA. Meta-analysis revealed that IL-1B -511 SNP is associated with increased susceptibility to RA[35].
Variable number of tandem repeat (VNTR) + 2018 (rs419598) SNP of IL-1RN gene were investigated in Black South Africans where no significant differences were found in genotype and allele frequencies between RA group and healthy controls. Within the RA group, the IL1RN*2 (two repeats of an 86bp tandem repeat) at the VNTR locus was independently associated with higher Larsen radiologic damage scores (LDS), corrected for disease duration. Furthermore the IL1RN*2 and + 2018 C allele defined haplotype was associated with significantly higher LDS on average 15 points higher, compared to the base haplotype of IL1RN*long (three or more repeats) and + 2018 T allele. The authors concluded that IL1RN*2 is a marker of erosive joint damage in Black South Africans RA patients[36]. With respect to IL1R1 loci, negative findings were reported in Indian, Swedish and Chinese RA patients[37-39]. In an Algerian population IL-1B (-511), IL-1 (+3953), and IL-1RN VNTR polymorphisms were examined, where no significant differences were observed in the three polymorphisms in genotype, allele and haplotype frequencies between the RA group and the healthy controls. However, the TT genotype of IL-1B-511 is more frequent in the patients’ cohort with positive ACPA compared with negative ACPA group. The IL-1RN*1/IL-1B-511T/IL-1B + 3953C haplotype was more frequent in the positive ACPA group. The association between IL-1RN allele 1 of VNTR, IL-1B-511 T allele and IL-1B + 3953 C allele seems to predispose to the synthesis of ACPA and therefore to the occurrence of ACPA positive RA[40].
Meta-analysis of 10 European, 7 Asian, and 1 Latin American RA population showed that the IL-1B + 3953 polymorphism was associated with the development of RA only in the Asian RA cohort[41]. Similarly to these results the genotype and allele distributions of IL-1B + 3953 showed significantly increased risks in a RA cohort from Northwest China compared to controls[42].
IL-18: The main function of IL-18 (OMIM 600953) is to promote the production of IFN-γ from T and natural killer (NK) cells, in particular the presence of IL12p70. IL-18 binds to its ligand binding chain, to the interleukin 18 receptor 1 (IL-18R1, OMIM 604494). It also recruits its co-receptor, the IL-18 receptor accessory protein (IL-18RAP, OMIM 604509). With these reactions IL-18 virtually initiates the activation of nuclear factor kappa-light-chain-enhancer of activated B cells/mitogen activated protein 8 (NF-κB/MAPK8)[43]. IL-18 is an important proinflammatory cytokine and plays a potential pathological role in RA[44]. It is highly expressed in sera, synovial fluids and synovial tissues of RA patients; furthermore, elevated IL-18 levels are correlated with RA disease activity, indicating an important role of IL-18 in the pathogenesis of RA[45].
Several studies examined the association of IL-18 gene polymorphisms with RA, but these studies showed inconclusive and controversial results. In an Egyptian population the -607 C/A (rs1946518) and -137 G/C (rs187238) were analysed in the promoter region. The frequency of -137CC genotype was significantly lower in RA patients compared to controls. As IL-18 -137CC and IL-18 -607 were negatively associated with RA, they may not be risk factors for RA in the Egyptian patients[46].The same polymorphisms were analysed in a Chinese Han population. The genotype and allele frequency of -607 of the IL-18 gene showed significant differences between RA patients and controls. There was no statistical significance in the distribution of genotype frequencies of -137. Significance was found in the data on statistical basis only on allele frequency levels[47]. The controls had significantly higher AA genotype frequency in the Chinese population at position-607 compared to RA patients. At position-137 no significant differences were observed in the distribution of either allelic or genotypic frequencies. Furthermore there was no association between the examined genotypes and the presence of rheumatoid factors. In the Chinese population only the AA genotype at position-607 is associated with a protective effect against development of RA. Meta-analysis was conducted on the associations between these promoter polymorphisms and RA in the Asian population. They found significant differences in genotype and allele frequencies only in the Chinese population which the previous study has also demonstrated[44].
Another polymorphism, the 105 A/C (rs549908) was analysed in a Chinese population living in Taiwan. There were significant differences in the genotype distribution of this polymorphism between patients and controls. The distribution of the AA homozygote in the RA patients was higher compared to the control group. The allele frequency also differed significantly between RA patients and controls[48].
In a Japanese study three haplotype tag SNP, rs1946518 A/C, rs360718 T/G, and rs360722 T/C, spanning from the 5’UTR region to intron 1 were genotyped using allelic discrimination with use of specific TaqMan probes, and three haplotypes (ATT, CTC and AGC). Among these polymorphisms, the T allele frequency of rs360722 which tags the ATT haplotype, was significantly lower in the RA cohort compared with the normal subjects. Having the TT genotype further increased the significance. The presence of the T allele and TT genotype at rs360722 reduces the susceptibility of Japanese people to RA[49].
The IL-2 family
The members of the IL-2 cytokine family are: IL-2, IL-4, IL-7, IL-9, IL-15 and IL-21. This family includes a group of ILs which share a common receptor subunit, the “common γ chain”. This acts in unison with a subtype specific α-chain to initiate the signaling cascade. These ILs act mainly as growth and proliferation factors for progenitors and mature cells, and they also have a role in lineage-specific cell differentiation[25].
IL-4: IL-4 (OMIM 147780) is a major stimulus of Th2-cell development which regulates allergic conditions and the protective immune response against helminthes and other extracellular parasites[50].
The -590 C/T (rs2243250) promoter polymorphism of the IL-4 gene was tested in an Egyptian population with RA. In RA patients, the frequencies of TT genotype and T allele were significantly increased compared to controls. Subjects with TT genotype and carriers of T allele were significantly more likely to develop RA. In non-erosive RA patients, the frequencies of TT genotype were significantly increased compared to controls. CT and TT genotypes were significantly increased in erosive RA patients compared to control group. Carriers of T allele had significantly increased risk to develop erosive RA as compared to the control group. The frequencies of -590 CT and TT genotypes were significantly increased in erosive RA patients. RA patients carrying CT, TT genotypes were significantly more likely to have erosive arthropathy. In RA patients with positive anti-CPP, the frequencies of CT and TT genotypes were significantly increased compared to anti-CCP group. Carriers of T allele were significantly more likely to have positive anti-CCP[51]. The same polymorphism was tested in a Polish RA population, where no significant differences were observed in the genotype and allele frequencies of controls vs RA patients[52]. Meta-analysis showed that the -590 C/T polymorphism is a risk factor for RA among Europeans[53].
Two functional polymorphisms in the IL-4 receptor gene rs1801275 (Q551R) and rs1805010 (I50V) were analysed in African American RA patients. They found that patients positive for HLA-DRB1 shared epitope (SE) and autoantibodies had a higher risk of developing rheumatoid nodules in the presence of rs1801275 AA and AG alleles, while patients positive for the HLA-DRB1 SE and RF alone had a higher risk of developing rheumatoid nodules in presence of rs1801275 AA and AG alleles and rs1805010 AA allele[54]. These variants were examined in an Egyptian population as well. In RA patients, the IV genotype frequency was significantly increased compared to controls. Subjects with IV genotype were significantly more potential to have the disease. In patients with erosive RA, the VV genotype frequency was significantly increased compared to patients with non-erosive RA. Subjects with VV genotype were significantly more susceptible to erosive arthropathy. The QR genotype frequency was significantly decreased in patients with erosive RA compared to patients with non-erosive RA. Carriers of V allele and Q allele were significantly more potential to be RF-positive, respectively and consequently develop severe RA[55].
IL-15: IL-15 (OMIM 600554) is a T cell activating factor which has pleiotropic and physiological activities in both the innate and acquired immune responses[56]. It plays an essential role in the differentiation, survival and activation of NK cells[56]. These functions are mediated through IL -15 receptor (IL-15R)[57]. IL-15 exerts pro-inflammatory effect in several diseases like allergy, transplant rejection and autoimmune disorders. Abnormalities in IL-15 expression may be involved in the pathogenesis of inflammatory autoimmune disorders like RA[56]. IL-15 may be implicated in the perpetuation of synovial inflammation in RA by generating positive-feedback loop, in which IL-15 synthesis by activated synovial macrophages or fibroblasts could induce a continuous T-cell recruitment[58].
Five SNPs in IL-15 gene (rs7667746, rs7665842, rs2322182, rs6821171 and rs4371699) were significantly associated with rate of joint destruction in North European RA patients[59]. Thirteen SNPs were screened within the IL-15 regulatory regions [promoter, 5’ and 3’ untranslated region (UTR) regions]. In addition, an association study of these SNPs was conducted in three independent case-control cohorts with Spanish Caucasian origin. The presence of the 13 selected IL-15 SNPs was confirmed and no new genetic variants were found. The distribution of the IL-15 selected SNPs in RA patients and controls showed no statistically significant difference in any studied populations. The haplotype analysis revealed that the three IL-15 haplotype blocks were not associated with RA susceptibility or severity in the analysed cohorts. It was suggested that the IL-15 gene polymorphisms may not play major role in RA genetic predisposition, and probably other molecules which are implicated in the IL-15 pathway might possibly be implicated in RA susceptibility[60]. These results are in accordance with two previous studies which analysed the contribution of IL-15 gene to the genetics of immunity-related diseases. There was no significant association observed in two different Caucasian populations for a number of IL-15 SNPs and allergic disorders[61].
IL-2/21: IL-2 (OMIM 147680) promotes proliferation and expansion of both antigen-specific clones of CD4+ and CD8+ T cells. The IL-2 receptor (CD25) susceptibility locus has recently been reported to be associated with RA[62]. IL-21 is involved in cell-mediated and humoral responses. It has pleiotropic effect on a variety of immune and nonimmune cells. In RA, the synovial fluid and tissue have enhanced inflammatory responses to IL-21 and elevated IL-21 receptor expression[63]. In Dutch RA patients was found that the IL-2RA locus may predispose to less destructive course of RA. The minor C allele of IL-2RA (rs2104286) was associated with less progression of joint destruction. The IL-2RA (rs2104286) protective genotype was associated with lower circulating levels of soluble interleukin-2 receptor A (sIL2RA). Lower sIL2RA levels were associated with a lower rate of joint destruction[64]. The SNP rs6822844 for IL-2/IL-21 was investigated for associations with the disease and for associations with autoantibody status in a Dutch RA cohort. IL-2/IL-21 rs6822844 showed a clear trend toward association with RA[65].
The KIAA1109-TENR-IL2-IL21 region has been associated with a wide variety of autoimmune diseases like type 1 diabetes (T1D)[66], ulcerative colitis[67], Crohn’s disease[68], celiac disease, Graves’ disease (GD)[66], systemic lupus erythematosus (SLE)[69], psoriatic arthritis[70], and juvenile idiopathic arthritis[71]. The rs6822844 within the KIAA1109-TENR-IL2-IL21 gene cluster has been linked to RA. Other variants within this cluster, for example rs17388568 is not in linkage disequilibrium (LD), but rs907715 is in moderate LD with rs6822844. The rs17388568 has been described to be in association with a number of autoimmune phenotypes, including T1D. Australasian RA patients and controls of European Caucasian ancestry were genotyped for rs6822844, rs17388568 and rs907715. No statistically significant difference was observed in the Australasian sample set for rs6822844or rs17388568 or rs907715. In a meta-analysis there was a genome-wide level of significance supporting association of rs6822844 with RA. Meta-analysis of rs17388568 showed no significant association with RA, while the meta-analysis of rs907715 only a trend towards association, but this was not independent of the association at rs6822844. The analysis of the KIAA1109-TENR-IL2-IL-21 gene cluster supported its association with RA and rs6822844 is the dominant association in this locus[72].
The IL-10 family
IL-10 cytokine family members (IL-10, IL-19, IL-20, IL-22, IL-24, IL-26, IL-28 and IL-29) are mainly linked through their similar intron-exon structure[73].
IL-10: IL-10 (OMIM 124092) is an anti-inflammatory cytokine produced by B cells, T cells, NK cells, monocytes, macrophages, and dendritic cells (DCs). It inhibits both antigen presentation and subsequent release of pro-inflammatory cytokines, so it attenuates the activated immune system[74]. IL-10 has been shown to suppress the inflammatory cytokines IL-1, IL-6, IL-8, IL-12, TNF-α, hematopoietic growth factors and inhibit the synthesis of nitric oxide, gelatinase, and collagenase[75].
Three promoter polymorphisms of IL-10 rs1800896, rs1800871 and rs1800872 have been studied in some populations with controversial results. The rs1800896 polymorphism is localized within a putative E-twenty six (Ets) transcription factor binding site, while rs1800871 is located within the putative positive regulatory region[76-78]. The rs1800872 is localized within the putative STAT3 binding site and negative regulatory region. These polymorphisms are in strong LD and appear in three potential haplotypes: GCC, ACC and ATA. The production of IL-10 depends on the genotypes. The ACC/ACC, ACC/ATA, and ATA/ATA are correlated with low, ACC/GCC and ATA/GCC with intermediate, whereas GCC/GCC with high IL-10 production[79]. In a Malaysian population the distribution of the IL-10 genotypes did not differ significantly between RA patients and healthy controls. However, significant difference was found in the allele frequencies of rs1800896CT, rs1800871TT, rs1800872CA and AA between the RA patients and healthy subjects[80].
A Polish population study could not show association between IL-10 genotypes and age at disease diagnosis, disease activity in a physician’s global assessment, joint and extra-articular involvement. They found also no correlation between IL-10 polymorphisms and disease activity parameters (ESR and CRP), number of swollen and tender joints, and duration of morning stiffness. The frequency of GCC, ACC, and ATA haplotypes in RA patients did not differ from that in the control group. These results suggest that IL-10 promoter polymorphisms are not risk factors for RA activity[81].
In a Chinese population only rs1800872 was studied. The allele and genotype frequencies were significantly different between the RA patients and controls. In addition, significant differences of allelic and genotypic frequencies were also detected between the patients with or without anti-CCP. The CA genotype is the most frequently observed genotype in both patients and controls. However, the distributions of CC and AA genotype in patients and control were reverse. The AA genotype frequency was higher, whereas the CC genotype frequency was lower in patients than in controls. The A allele frequency showed increased level comparing the results to the controls. It showed decreased A allele frequency, but increased C allele frequency level[82].
In a meta-analysis related to rs1800896, 10 case–control studies were carried out with the result that G allele carriers (GG + GA) had 25% decreased risk of RA, compared to the homozygote AA. In the analysis of Europeans, significantly decreased risks were associated with G allele carriers. The results of this meta-analysis provided evidence for the association between rs1800896 polymorphism and the risk of RA[83].
The IL-12 family
The IL-12 family consists of IL-12, IL-23, IL-27, IL-30 and IL-35, which are important mediators of inflammatory disorders. The common part is the heterodimeric complex composed of two subunits, the expression of which is regulated independently and have very different biological activities[84].
IL-23: IL-23 (OMIM 605580) was first described as a member of the IL-6/IL-12 superfamily[85]. It is a heterodimeric cytokine composed of 2 subunits[86-90]. The α-subunit is homologous to type I cytokines and β-subunit is related to the extracellular domain of other hematopoietin receptor family members[91]. IL-23 is expressed by activated monocytes, macrophages, DCs, T cells, B cells and endothelial cells[85,92] which strongly correlate with the cellular responsiveness to IL-23. IL-23 binds to a heterodimeric receptor complex composed of IL12RB1 (OMIM 601604) and IL23R (OMIM 607562) subunits[93]. IL12RB1 is also part of the IL-12 receptor, while IL-23R is unique to the IL-23 receptor complex.
The genetic variants of IL-23 and its receptor (IL-23R) were first examined in the context of inflammatory bowel diseases (IBDs) in non-Jewish subjects[94]. Previous studies using genetically deficient animals showed experimental evidence that a strong association exists not only between the carriage of certain IL-23R gene variants and IBDs, but the correlation also stands for collagen-induced arthritis as inactivation of the IL-23R gene resulting in disease resistance[95].
On the basis of these findings, the possible associations between the functional variants of the IL-23R gene and RA were studied in the Hungarian population, and it was reported that some allelic variants represent an elevated risk for the disease, particularly in the RF- and/or anti-CCP-seropositive subsets of patients[96]. An increased prevalence of the homozygous rs10889677 AA genotype of the exon-3’-UTR 2370 C/A variant and the homozygous rs2201841 CC genotypes of the intronic SNP could be observed not only in Crohn’s disease, but also in the RA groups compared to the controls.
In a Spanish population the examinations were expanded with genotyping of rs1004819, rs7517847, rs10489629, rs1343151, rs11209032 and rs1495965 SNPs, but none of the examined allelic variants and genotypes showed an increased prevalence in RA patients, not even when patients were stratified according to their clinical and demographic features (gender, age at disease onset, presence of shared epitope, RF, rheumatic nodules and extra-articular disease)[97].
These findings were supported by a large cohort genotyping of Caucasian subjects for rs7530511 and rs11209026 variants of the IL-23R gene, but none of these SNPs proved to contribute to the predisposition to RA[98]. A Spanish study proved that the minor allele of the rs7517847 variant is responsible for a slightly elevated risk to RA[99] which is in accordance with the former findings[98]. Also the exonic rs11209026 contributes to the onset of the disease as well. The same variant was reported to be significantly less frequent in RA patients compared to controls[94].
Analyses of six IL-23R SNPs (rs11209026, rs1004819, rs7517847, rs10489629, rs2201841 and rs1343151) were analysed in a New Zealand Caucasian set of RA patients and extended by the reanalysis of the Wellcome Trust Case Control Consortium[62] and the previously published Spanish data set[97]. Unfortunately, the results emphasized the lack of association of the exonic rs11209026 with RA, but provided evidence for a weak allelic association of the rs1343151 variant with the disease. The study also tested LD relationships between 11 IL-23R markers. Results showed that several SNPs which were reported to confer risk for RA (rs1343151 in the same paper, and rs10889677 and rs2201841 in the Hungarian cohort) seemed to be independent risk factors[96,100], while others, such as rs7530511 in the North America Rheumatoid Arthritis Consortium (NARAC) and the Swedish Epidemiological Investigation of Rheumatoid Arthritis EIRA genome wide scan[101] seemed to confer risk only due to their weak LD with the markers associated in the Hungarian cohort[96]. Additionally, the NARAC plus EIRA data did not support the predisposing nature of the rs1343151 genetic variant which contradicted the results of a New Zealand study and a meta-analysis performed by a Chinese medical research group, who were able to replicate the association in a data set consisting of four European Caucasian populations[102].
As several studies have examined the association between IL-23R polymorphisms and RA but the results were contradictory. There was a high need for a carefully designed meta-analysis which can clarify the issue. In 2012, two papers summarized the observations published before February 2012[103,104]. In a meta-analyis, all relevant original papers gathered from electronic databases were selected by strict inclusion criteria[104]. Six studies involving more than 5000 European patients and controls were assessed. The C allele of the rs10489629 and the G allele of the rs7517847 proved susceptibility to RA. Interestingly, these alleles are protective factors in ankylosing spondylitis, but the same genetic variants may not share a common mechanism in different autoimmune diseases[105].
A meta-analysis evaluated the possible role of four other IL-23R polymorphisms in the etiology of RA (rs10489629, rs11209026, rs1004819 and rs2201841). It revealed significant association between the A allele of the rs134151 variant and RA in European subjects, and also confirmed the results of the previous meta-analysis on the predisposing feature of the rs10489629. A allele in the overall population[106]. Interestingly, no association was found between the rs7517847 polymorphism and RA, although this variant is in moderate LD with rs1343151. None of the other SNPs in the focus of this meta-analysis (rs11209026, rs1004819, rs2201841) showed association with the disease. Although three, possibly predisposing IL-23R variants after the meta-analyses have been convincingly identified; the exact functional significance of these polymorphisms remains unclear.
The IL-6-like cytokines
Members of this family (IL-6, IL-11, IL-27 and IL-31) signal through receptors containing gp130 which are commonly referred to as the IL-6-like or gp130 utilizing cytokines family[107].
IL-6: IL-6 (OMIM 147620) is one of those pro-inflammatory cytokines which are involved in the pathogenesis of RA. It acts as a major mediator of the acute phase response[108]. IL-6 signals through a cell-surface type I cytokine receptor complex consisting of the ligand-binding IL-6R chain (OMIM 147880) and the shared signal-transducing component IL6ST (also called gp130; OMIM 600694)[109].
Conventional radiography is considered a well-established imaging technique for identifying progressive joint damage[110]. The IL-6 gene polymorphism-174 G/C (rs1800795) have been associated with RA susceptibility and radiographic severity of bone-erosive damage[111-115]. Significant association was observed between IL-6 -174 C allele and early disease onset of RA[116]. However no relationship was found between disease susceptibility and IL-6 -174 C allele in a Spanish study[117]. Another SNP, the -572 G/C (rs1800796) of IL-6 is associated with RA in a Chinese Han population[47] but with no association with RA in Taiwan[118]. Similarly to these studies no association was found between IL-6 -174, -572, -597 genotype distributions and allele frequencies in Turkish RA patients[34].
Elevated levels of IL-6 and soluble IL-6R (sIL6R) were found both in the serum and also in synovial fluid of joints in Han Chinese patients with RA[119]. IL-6R can be released in vivo in sIL6R through differential mRNA splicing and proteolytic rupture controlled by a disintegrin and metalloprotease domain (ADAM17, also called tumor necrosis factor-α-converting enzyme, TACE). This process is influenced by the SNP rs8192284 resulting in an aspartic acid to alanine substitution (D358A) at the proteolytic cleavage site. Trans-signalling extends the IL-6 range of action to cells lacking constitutive IL-6R[120]. It plays a key role in the pathophysiology of RA[121], where synoviocytes and chondrocytes react to IL-6 through this pathway. In a Spanish RA study was found that rs8192284 polymorphism determines the sIL6R plasma level. Furthermore, increased sIL6R plasma levels and expression of spliced isoform generating sIL6R are genotype dependent.
Meta-analysis of different studies with different ethnicities found association between RA and IL-6-174 G/C polymorphism in the European population. An Asian study also revealed significant association between the same IL-6 polymorphism and RA. Regarding to the IL-6-572 G/C polymorphism, the ethnicity-specific analysis in the Asian study revealed an association between RA and the IL-6-572 G/C. However no association was found between the IL-6-174 G/C polymorphism and RA in the Iranian study[122].
The IL-17 family
This recently discovered interleukin family contains six cytokines (IL-17A, IL-17B, IL-17C, IL-17D, IL-17E and IL-17F). IL-17A was the first member and the others were discovered shortly after the first one by large-scale sequencing of the human genome[123-126]. Members of this family share the highest amino acid sequence homology and perform distinct biological functions[127].
IL-17: IL-17A (OMIM 603149) is a pro-inflammatory cytokine which was the first discovered member of this family in 1993[128]. It acts on a variety of cells involving the development of autoimmunity, inflammation, and tumors. IL-17A and IL-17F genes share the highest degree of homology of about 50%, while the others have only 16%-30% of identity at the primary sequence level[126,129-132]. IL-17F (OMIM 606496) is a novel pro-inflammatory cytokine which induces the expression of cytokines and chemokines. Furthermore, it may play a role in skeletal tissue destruction and inflammatory processes in RA. In arthritis, IL-17A and IL-17F induce significant cartilage matrix release, inhibit new cartilage matrix synthesis and directly regulate cartilage matrix turnover[130]. The IL-17 receptor (IL17R) family includes five members: IL-17RA (OMIM 605461), IL-17RB (OMIM 605458), IL-17RC (OMIM 610925), IL-17RD (OMIM 606807), and IL-17RE (OMIM 614995)[133].
The IL-17F gene polymorphisms 7488 A⁄G (rs763780) and 7383 A⁄G (rs2397084) were investigated in Polish RA patients. The examined polymorphisms were not correlated with susceptibility to RA, but the 7488A⁄G (His161Arg) variant was associated with parameters of disease activity (number of tender joints), Health Assessment Questionnaire (HAQ) score or Disease Activity Score (DAS-28)-CRP. The authors supposed that the 7383 A⁄G (Glu126Gly) polymorphism may be correlated with longer disease duration in patients with RA. Probably these two SNPs directly regulate the IL-17F expression. They hypothesized that polymorphisms in IL-17 gene may cause redundant production of IL-1 and TNF-α which can mediate inflammatory pathology in many autoimmune diseases, including RA[134].
In Norwegian patients with RA, five IL-17A SNPs were analysed. They found a weak but significant RA correlation with the IL-17A promoter polymorphism rs2275913[135]. A Japanese study concerning early RA examined the association between age at RA onset, radiographic progression and three SNPs (rs3804513, rs3748067, rs1974226) in the IL-17A gene. They described a weak association between the intronic rs3804513 and joint destruction (Larsen score), but found no association with the risk of developing RA[136].
Chemokines
This group contains only two ILs, IL-8 and IL-16.
IL-8: In a Taiwan population the 2767 A/G (rs112664) polymorphism in the 3’-UTR of the IL-8 gene were investigated and no significant differences were found in the genotype and allele frequencies between RA patients and controls. Clinical characteristics such as age at onset, RF positivity, joint erosion and extra-articular manifestations were compared among patients, and it was found that patients with IL-8 3’-UTR 2767AA genotype had a significantly younger age of onset of RA than patients without that genotype[118].
In caucasian RA patients and healthy controls the 781 C/T (2227306) SNP of IL-8 gene was examined with the result that CC genotype is associated with the early onset of RA[137].
CLINICAL IMPLICATIONS, TREATMENTS
Till the 1950s, aspirin and non-steroid anti-inflammatory drugs (NSAIDs) were the mainstay of RA therapy. Oral, intramuscular or intra-articular corticosteroids are recommended for controlling pain and inflammation. Ideally, NSAIDs and corticosteroids are used only for short-term management of RA. The development of disease-modifying anti-rheumatic drugs (DMARDs) has revolutionized the long-term therapeutic approach of the chronic inflammatory rheumatic diseases, particularly in patients resistant to standard treatment.
In respect of the new nomenclature for DMARDs[138], the term conventional, synthetic DMARDs (csDMARDs) is used to subsume chemical agents such as methotrexate (MTX), sulfasalazine, leflunomide, hydroxychloroquine, minocycline (less commonly used: gold sodium, thiomalate, penicillamine, cyclophosphamide, cyclosporine, azathioprine); whereas tofacitinib, a new sDMARD specifically designed to target janus kinases (JAKs), will be designated as a targeted sDMARD (tsDMARD). Biologic agents (bDMARDs) include monoclonal antibodies (mAbs), soluble recombinant cytokine receptors and natural antagonists to block cytokines which promote the inflammatory cascade responsible for RA[139]. Biological originator (bo) DMARDs encompass the five currently available TNF-α inhibitors (infliximab, etanercept, adalimumab, certolizumab pegol and golimumab), the T cell costimulation inhibitor abatacept, the anti-B cell agent rituximab, and the IL6R-blocking monoclonal antibody tocilizumab, as well as the IL-1 inhibitor anakinra. While biosimilars (bs), such as bs-infliximab, approved newly in the United States and/or Europe, will be named bsDMARDs[140].
The last set of ACR recommendations for the treatment of RA were published in 2008[141] with an update in 2012 for the use of DMARDs[142]. The 2010 EULAR guideline was renewed in 2013[140]. Goals of RA therapy include reaching a target of remission or low disease activity in every patient. The commonly used indices to depict clinical response to therapy in RA include the ACR response[143], HAQ score[144] and DAS[145]. DAS28 is derived by the number of swollen joints and tender joints using the 28-joint count, and measures the CRP and and the patient’s own assessment on a visual analogue scale[145].
Therapy with DMARDs should be started as soon as the diagnosis of RA is made[140]. Methotrexate is recommended as the first-line treatment in patients with active RA, unless contraindicated or not tolerated[138,141]. Leflunomide may be used as an alternative to methotrexate, although gastrointestinal adverse effects are more common. Sulfasalazine or hydroxychloroquine is recommended as monotherapy in patients with low disease activity or without poor prognostic features (e.g., seronegative, non-erosive RA)[141,146]. Low-dose glucocorticoids should be considered as part of the initial treatment strategy (in combination with one or more csDMARDs) for up to 6 mo, but should be tapered as rapidly as clinically feasible. Combination therapy with two or more DMARDs is more effective than monotherapy; however, adverse effects may also be greater[15]. If RA is not well controlled with a csDMARD, a biologic DMARD should be initiated with MTX[140,141,146]. Current practice would be to start a TNFα inhibitor. If TNF inhibitors have failed, other biologic therapies can be considered (e.g., abatacept, rituximab or tocilizumab). Simultaneous use of more than one biological therapy (e.g., adalimumab with abatacept) is not recommended because of an unacceptable rate of adverse effects[141]. Tofacitinib, a new tsDMARD, may be considered after biological treatment has failed[138].
Although the precise etiology of RA still remains unknown, improved understanding of the pathogenesis of the disease DMARD treatment has undergone dramatic changes during the past decade. Biological DMARDs have the potential to inhibit the behaviour of cytokine, cellular activation, and inflammatory gene transcription by various means. Cardiovascular disease due to accelerated atherosclerosis is the major cause of excessive mortality in RA. Cytokine antagonists have shown a favourable response in endothelial cell dysfunction in these patients[147].
Because of the elevated levels of TNFα, IL-1 and IL-6 in the synovial fluid of patients with RA, these three cytokines have been targeted at the beginning of RA therapy. Numerous other biological therapies are in various stages of development. We now review the main biological drugs classifing according to the targeted mechanism of action.
Tumor necrosis factor inhibitors
TNFα is a pleiotropic, pro-inflammatory cytokine which plays a pivotal role in the origin and progression of RA. TNF is a 17-kD trimeric protein cytokine that is produced mainly by monocytes and macrophages. Newly synthesized TNFα is inserted into the cell membrane. Subsequently, the TNFα converting enzyme (TACE) cleaves this cell-bound TNFα to release it into circulation[148]. Both soluble (sTNF) and membrane TNF (mTNF) are biologically active when interacting with either of two distinct receptors, TNF receptor 1 (TNFR1, p55) and TNFR2 (p75), expressed on a wide variety of target cells[149]. TNF receptor signalling occurs through two pathways: one arm has death-domain proteins which lead to apoptosis, the second and dominant signalling pathway goes through a series of kinases, leading to the activation of nuclear-factor kappa B (NF-κB).
The mechanism of action of TNF-α antagonists is based on the neutralization of both sTNF and mTNF and has a more global effect on inflammation than the blockade of other cytokines. The interruption of the signal pathways mediated by TNF has numerous consequences, reflecting the pleiotropic effect of the cytokine: apoptosis, inhibition of pro-inflammatory cytokine and chemokine release, but also of chondrocyte, osteoclast, and endothelial cell activation, reduction of leukocyte accumulation and angiogenesis, increase of T reg cell number.
Currently, 5-TNF inhibitors are approved for use by the United States Food and Drug Administration. Infliximab (Remicade®) is a chimeric (75% human + 25% mouse) monoclonal full-length, bivalent IgG1 mAb[150]. Certolizumab Pegol (Cimzia®) is a humanized protein containing amino acid sequences derived from a mouse anti-TNF mAb and inserted into human domains; adalimumab (Humira®) and golimumab (Simponi®) are fully human mAbs[151]. Etanercept (Enbrel®) is a dimeric fusion protein consisting of soluble p75-TNFR2 and the Fc portion of human IgG1. The primary action of etanercept is to bind and inactivate soluble and cell-bound TNF-α and lymphotoxin-α.
Another way to block TNF-α in biological fluids is to inhibit TACE, up to 95% reduction of the TNF production is attainable. TACE inhibitors are under development, but, even after more than a decade no single TACE inhibitor has passed the phase 2 clinical trials[152].
IL-1 antagonism
IL-1 is implicated in the pathogenesis of RA, its level correlates with RA disease activity[153]. IL-1 type 2 receptor (IL-1R2) (OMIM 147811) is a decoy receptor which binds to circulating IL-1[154] and is not involved in signal transduction. An antagonist of these receptors has also been identified (IL1RN) which neutralizes the effects of IL-1, consequently, IL1RN acts as a physiological inhibitor of IL-1. Complete inhibition of IL-1 requires 10-fold to 100-fold molar excess of IL1RN over IL-1. The balance between IL-1 and IL1RN is important in maintaining the normal physiology of the joints and homeostasis of the immune system.
Anakinra (Kinaret®) is a recombinant form of the naturally occurring IL1RN[155], approved in 2001 for the treatment of patients affected by RA. It should also be mentioned that anakinra, while effective in individual patients with RA, did not show a high level of clinical efficacy in clinical trials[156] and therefore has not been recommended as a major biological agent for use in RA[140].
Many IL-1 inhibiting agents are being developed and tested. These include a recombinant form IL-1R2, rilonacept, also known as IL1Trap (recombinant molecule consisting of IL1R1 and IL1RAP fused to human IgG1 Fc portion which acts as a soluble decoy receptor, trapping both IL-1A and IL-1B[155]). Canakinumab, a human anti-IL-1B mAb (currently investigated in phase 3 studies[157]), and an inhibitor of IL-1 converting enzyme[154,158].
IL-6 antagonism
Evidence has indicated that blocking the effects of IL-6 in RA is effective and safe, especially with the IL-6R inhibitor, tocilizumab[159]. Tocilizumab (Actemra®) is a humanized mAb of IgG1 class against IL-6R which prevents the formation of the IL-6/IL-6R complex and the activation of signal transduction cascade through JAKs and STATs. Over the next few years, new biological agents targeting the IL-6 receptor (sarilumab) or IL-6R (clazakizumab, sirukumab) may become available[160].
Co-stimulation signal blockade
There are several sets of T cell co-stimulatory molecules like CD40-CD40 ligand (CD40L) and CD28-CTLA4-B7. Blockade of some of these are under various stages of development[139]. Abatacept (Orencia®) is a biologic agent which blocks T cell activation through the inhibition of CD28-B7 mediated costimulation of the T cell. It has been approved for the treatment of RA[161]. Structurally, abatacept is a recombinant dimeric fusion protein consisting of the extracellular domain of CTLA-4 fused with the modified Fc portion of a human IgG1. Blocking anti-CD40 ligand antibody and anti-CD11a monoclonal antibody (efalizumab) could be also beneficial for the treatment of RA.
B-cell-depleting therapy
B cells behave as antigen presenting cells, stimulating the activation and proliferation of T cells. In addition, the synovium of patients with RA contains a large number of plasma cells producing RF. The easiest method to obtain a reduction in the number of B cells is to use mAbs directed against surface markers such as CD19, CD20, and CD22.
Rituximab (Rituxan®) is a chimeric mouse/human mAb which selectively depletes B cells bearing the CD20 surface marker. Widely used in the treatment of B-cell lymphomas, it has been shown to be surprisingly effective in RA. The rituximab/MTX combination represents a potential therapeutic option for moderate/severe RA patients, resistant or intolerant to at least one TNF antagonist[140]. Epratuzumab is a humanized mAb formed by an IgG1 directed against CD22.
Tumor necrosis family proteins (e.g., death receptors, anti-B lymphocyte stimulator antibodies) are the molecules of the immune system which take part in the negative feedback regulation to eliminate autoimmune cells. Excessive levels of the TNF family ligand B-lymphocyte stimulator (BLyS) have been demonstrated in RA synovial fluid. Approaches targeting the BLyS and other systems (like APRIL) to selectively eliminate the activated autoimmune lymphocytes in RA are under development[162]. Belimumab is a human recombinant IgG mAb which acts by binding BLyS protein and prevents the interaction with the B cell activating factor receptor.
Atacicept is a recombinant fusion protein comprising the extracellular domain of the TACI (Transmembrane Activator and CAML Interactor) receptor joined to a human IgG1 Fc domain. Atacicept also inhibits the survival of long-lived plasmacells directly involved in the pathogenesis of RA and SLE.
Kinase inhibitors
In the case of RA, kinases play a central role in the aberrant immune system activation and hence have been targeted using small molecule inhibitors. Mitogen-activated phosphokinase p38 (MAPK), spleen tyrosine kinase (Syk), and JAKs have been studied extensively in clinical trials in RA[163]. Several p38 MAPK inhibitors proved inefficient in treating rheumatoid arthritis.
The Syk inhibitor, fostamatinib, proved superior to placebo in Phase 2 trials and is currently under phase 3 investigation. Tofacitinib (Xeljanz®), a JAK1/3 inhibitor, was approved for the treatment of RA in the United States, Japan and Russia in April 2013[164]. This new tsDMARD may be considered for use after biological treatment has failed[138]. Ruxolitinib and baricitinib (JAK1/2 inhibitors) and pan-JAK inhibitors (JAKinibs) have also been studied in RA where preliminary results were promising in terms of efficacy and safety in a Phase 2a trial.
Upcoming therapies
IL-1 superfamily: IL-18 could be an interesting target in the treatment of RA and one opportunity for antibody-based biological therapies in RA. Blocking of IL-18 by the administration of a recombinant IL-18 binding protein (IL18BP, OMIM 604113) which has the ability to prevent binding of IL-18 to its receptor, or anti-IL-18 in mice with collagen-induced arthritis resulted in a clear reduction of the disease severity compared with placebo-treated mice[165].
IL-33 (OMIM 608678), a newly identified IL-1 family member cytokine, is a chemoattractant for Th2 cells and facilitates the production of Th2 cytokines. IL-33 binds to its receptor consisting of the orphan receptor ST2 (IL1RL1, OMIM 601203) and IL-1 receptor accessory protein (IL1RAP). The soluble ST2 (sST) acts as a decoy receptor of IL-33 and is a natural inhibitor of IL-33. Increased serum and synovial fluid levels of sST2 in RA patients reflect an active inflammatory state[166,167]. Furthermore, the inhibition of IL-33 receptor signaling with anti-ST2 antibodies or sST2-Fc fusion protein resulted in reduced severity of collagen-induced arthritis[166,168].
IL-2 superfamily: The role of IL-2 in the immunopathogenesis of RA is debated. IL-2 is hardly detectable in the synovial fluid, and only a low percentage of the intra-articular T cells express T cell activation markers (Tac antigen)[169]. IL2-directed therapy may have beneficial effects in RA patients[170]. The humanized monoclonal antibody daclizumab (Zenapax®) against the α-chain of the IL-2R (CD25) caused significant reduction of joint-inflammation and joint-erosion in collagen-induced arthritis in rhesus monkeys[171]. Antagonistic IL2RA mAbs (anti-Tac/daclizumab, basiliximab) are effective in preventing rejections of organ transplants.
IL-15 is a pro-inflammatory, innate response cytokine. In patients with RA, innate response cytokine IL-15 is expressed in the synovial tissue and the serum levels of IL-15 have been reported to correlate with disease severity[172]. Anti-IL15 monoclonal antibodies are being examined for their anti-arthritic activity. Baslund and colleagues conducted a phase 1/2 clinical trial of a human IgG1 anti-IL15 monoclonal antibody, HuMax-IL15. This antibody could neutralize various biological effects of IL-15 in synovial tissue in vitro, and it caused significant improvement in disease activity at 12 wk after treatment initiation[56,173,174]. Clinical trials are underway evaluating the safety and efficacy of monoclonal antibody IL-15 (HuMax-IL15) and CD2 receptor (Alefacept).
IL-21 contributes to joint inflammation and synovial cellular infiltration in RA, as expected for a Th17-related cytokine[25,175]. Increased level of IL-21 has been reported in RA sera, and the concentration of IL-21 in serum and synovial fluid was higher in RA than osteoarthritis[175]. Treatment with IL21RFc chimeric protein in animal models of RA resulted in significantly reduced disease severity[176-178].
The IL-12 family: IL-12 and IL-23 bind to the IL12RB1 of T cells and NK cells via their shared p40 subunit. Evidence shows that IL-23 plays a key role in the development of pathogenic Th17 cells producing IL-17, which further induces the production of several pro-inflammatory cytokines, such as TNFα and IL-6, chemokines, which cause the aggravation of synovial inflammation and osteoclast differentiation leading to joint destruction in patients with RA[179,180]. In addition, the serum level of IL-23 in patients with RA correlates with the number of swollen joints, the DAS28 joints. Studies have shown that IL-23 induces receptor activator of RANKL expression on CD4+ T cells and promotes osteoclastogenesis in an autoimmune arthritis[181]. Thus anti-IL23 therapy could be a therapeutic target not only of inflammation but also bone erosion in RA. The level of interest in this target can be seen from the fact that 15 different IL23R antagonists are now reported to be in clinical or pre-clinical development[182] .
Recent clinical studies associated with IL-23 inhibition in arthritis include the use of apilimod mesylate, an orally administered inhibitor of IL-12/IL-23 in RA[183]. Ustekinumab and briakinumab, fully human mAbs directed against the p40, are currently in phase 2 trials. However, due to the common p40 subunit and IL12RB1 chain, the major drawback of anti-IL23 treatment may be the simultaneous inhibition of IL-12 and a possible shutdown of the immune system. Nevertheless, it would be much more useful to design drugs that target the IL23p19 or IL23RA itself, thus inhibiting IL-23 without modifying the effects of IL-12 (e.g., MP-196, FM-303, IL-23 Adnectin)[182].
The IL-17 family: Increased levels of IL-17A have been found in sera, synovial fluid, and in the T cell-rich area of the synovium in patients with RA[184,185] and these levels are predictive of a more severe joint damage progression[186]. Besides the enhancement of inflammation commonly observed in arthritis, IL-17A also mediates bone and cartilage destruction through the stimulation of fibroblast-like synoviocytes to produce pro-inflammatory cytokines, IL-6 and IL-8, as well as matrix-degrading enzymes, matrix metalloproteinases. In addition, IL-17 upregulates the receptor activator of RANK on osteoclast precursors causing increased sensitivity to RANK signaling.
Treatment of RA patients with a humanized anti–IL-17 antibody (LY2439821) given intravenously is shown to improve the signs and symptoms of the disease[187]. In another study on RA, treatment with AIN457 (anti-IL17) induced clinically relevant responses, although of variable magnitude[188]. The IL-17 blockers secukinumab (anti-IL-17A), ixekizumab (anti-IL17A), and brodalumab (anti-IL17RA) have shown efficacy in phase 2 trials in RA. The results of the ongoing phase 3 trials should help to shed light on whether IL-17A is truly a viable therapeutic target in RA. The effect of blocking other IL-17 family members including IL-17F has yet to be evaluated in human diseases.
Agents blocking the chemokines and adhesion molecules: Agents blocking the chemokines and adhesion molecules are also under trial. These include antibodies to IL-18[165], humanized anti-integrin avb3 monoclonal antibody (MEDI-522) and anti-VCAM antibodies. Suppression of new wessel formation could also be an interesting target in the future treatment of RA.
Anti-inflammatory cytokines in RA: IL-10 and IL-4 are cytokines with counter-regulatory mechanism that down-regulates pro-inflammatory responses. Some anti-inflammatory effects are also naturally provided by the presence of IL-1RN, IL-1R2 decoy receptor and soluble TNF receptor. In vitro, IL-10 and IL-4 inhibit the production of inflammatory cytokines including IL-1, IL-6 and TNF-α RA[189,190], furthermore IL-10 has been shown to reverse the cartilage degradation seen in RA[189]. In vivo, however, they are inherently weak and proved inadequate.
IL-27 (OMIM 605816) is an IL-12 superfamily cytokine that plays a role in the immune effector responses in autoimmune diseases, including arthritis. The role of IL-27 as a pro-versus an anti-inflammatory cytokine has not yet been fully resolved. In collagen-induced arthritis, treatment of mice with IL-27 reduced the severity of arthritis, as well as the levels of IL-6, IL-17[87]. Another mechanism of IL-27-mediated protection against arthritis involves the inhibition of osteoclastogenesis[191].
It has recently been shown that human Tregs express IL-35 and require this cytokine for their optimal suppressive effect[192]. These findings reinforce a potential mechanism (e.g., suppression of Th17 response) that Tregs can be used to control pathogenic T cell responses in RA. Treatment of mice with IL-35 reduced disease severity which was associated with reduction in IL-17, IFN-γ, and an increase in IL-10 production[193].
Small molecular inhibitors of intracellular signalling: Small molecular inhibitors of intracellular signalling (e.g., NF-κB and associated activator molecules) are in focus of numerous clinical and preclinical research and have shown promising results in animal models[194,195].
RANKL inhibition : Denosumab, a human anti-RANKL mAb is approved in the United States for the treatment of postmenopausal osteoporosis but is not currently indicated for the treatment of RA (phase 2).
CONCLUSION
RA is the most common chronic inflammatory disease of the joints and is characterized by a complex genetic architecture. In our review, we discussed the pathogenesis, the polymorphisms of IL and IL genes and also the standard available and possible future immunologic targets for RA treatment. The identification of disease-associated interleukin and interleukin receptor genes could provide precious insight into the genetic variations prior to disease onset in order to identify the pathways important for RA pathogenesis. From the discussed interleukins the IL-1, the IL-6, and the IL-23 were the most investigated.
IL-1 is very important, because it mediates many inflammatory diseases by initiating and potentiating immune and inflammatory responses. Several studies have dealed with the association of IL-1 gene polymorphisms with RA. The IL-1 expression is influenced by two variants of the IL-1B (-511 T/C and +3953 C/T). Carrying the TT genotype of IL-1B +3953 gene is susceptible to RA in a Turkish cohort, while the CT genotype of IL-1B -511 has a protective role against RA. In a British Caucasian RA patients the G allele of IL-1B -1464 C/G was found to possibly have protective effect in RA. In the Asian populations +3953 C/T SNP of the IL-1B polymorphism was associated with the development of RA.
IL-6 is the main pro-inflammatory cytokine which is involved in the pathogenesis of RA. Several SNPs of the IL-6 gene were investigated (promoter polymorphisms: -174, 572, -597, exonic polymorphisms: 869), but with controversial results. In a Spanish, Turkish and Iranian cohort the IL-6-174 G/C is not a risk factor for RA, but meta-analyses with different ethnicities showed an association between RA and these SNP in other European population. An Asian study also revealed a significant association between the same SNP and RA. The -572 G/C SNP is associated with RA in a Han Chinese population, while no correlation could be detected in a Taiwan population. Ethnicity-specific analysis in an Asian study revealed an association between RA and IL-6 -572 G/C polymorphism.
IL-23 is the most extensively studied cytokine. IL-23 is very important in innate and adaptive immunity. The IL-23R gene was identified first as a CD susceptibility gene in North American non-Jewish subjects but the studied were extended to RA. Association between independent functional SNPs in the gene and its neighboring region and RA were investigated (rs10889677, rs11209032, rs1495965, rs2201841, rs1004819, rs11209026, rs7517847, rs10489629, rs1343151) in numerous studies. Several SNPs are susceptible, others are protective to the disease but the predisposition was population dependent.
In the lack of knowing genetic variants that influences the development of RA and in default of adequate therapy, the patient’s way of life continuously declines, even permanent disability might arise. However, conventional DMARD therapy of RA has several limitations like slow onset of action and induction of partial remission. By targeting molecules that are directly involved in pathogenesis pathways, blocking biologic activity of pro-inflammatory cytokines and their receptors may be more specific, more efficacious, and less toxic in the short-term than current treatment modalities. Because biologics are relatively new, evidence is insufficient to determine their long-term benefits and risks, including the risk of lymphoma and malignancies.
Evidence suggests that although biologics are relative expensive, they remain cost-effective because of the major clinical benefits that patients may experience. Currently, data is available for one biosimilar product (infliximab) which shows similar efficiancy and safety profiles to the original biological agent. It has been estimated that the price for biosimilar products will be 65%-85% of their originators. Nevertheless, combinations of biological agents targeting different disease processes may allow more promising results in future. The knowledge of the complex genetic background may prove to be greatly useful for developing novel therapies and producing personalized medicine based on the individual’s genetics.
Footnotes
P- Reviewer: Andonopoulos AP, Olama SM, Sokolove J S- Editor: Ji FF L- Editor: A E- Editor: Wu HL
Supported by The Grant of the Hungarian Science Foundation, No. OTKA K103983
References
- 1.Kiss CG, Lövei C, Sütö G, Varjú C, Nagy Z, Füzesi Z, Illés T, Czirják L. Prevalence of rheumatoid arthritis in the South-Transdanubian region of Hungary based on a representative survey of 10,000 inhabitants. J Rheumatol. 2005;32:1688–1690. [PubMed] [Google Scholar]
- 2.Worthington J. Investigating the genetic basis of susceptibility to rheumatoid arthritis. J Autoimmun. 2005;25 Suppl:16–20. doi: 10.1016/j.jaut.2005.09.011. [DOI] [PubMed] [Google Scholar]
- 3.Lee DM, Weinblatt ME. Rheumatoid arthritis. Lancet. 2001;358:903–911. doi: 10.1016/S0140-6736(01)06075-5. [DOI] [PubMed] [Google Scholar]
- 4.Pincus T, Brooks RH, Callahan LF. Prediction of long-term mortality in patients with rheumatoid arthritis according to simple questionnaire and joint count measures. Ann Intern Med. 1994;120:26–34. doi: 10.7326/0003-4819-120-1-199401010-00005. [DOI] [PubMed] [Google Scholar]
- 5.Järvinen P, Aho K. Twin studies in rheumatic diseases. Semin Arthritis Rheum. 1994;24:19–28. doi: 10.1016/0049-0172(94)90096-5. [DOI] [PubMed] [Google Scholar]
- 6.Scutellari PN, Orzincolo C. Rheumatoid arthritis: sequences. Eur J Radiol. 1998;27 Suppl 1:S31–S38. doi: 10.1016/s0720-048x(98)00040-0. [DOI] [PubMed] [Google Scholar]
- 7.Baecklund E, Iliadou A, Askling J, Ekbom A, Backlin C, Granath F, Catrina AI, Rosenquist R, Feltelius N, Sundström C, et al. Association of chronic inflammation, not its treatment, with increased lymphoma risk in rheumatoid arthritis. Arthritis Rheum. 2006;54:692–701. doi: 10.1002/art.21675. [DOI] [PubMed] [Google Scholar]
- 8.Aletaha D, Neogi T, Silman AJ, Funovits J, Felson DT, Bingham CO, Birnbaum NS, Burmester GR, Bykerk VP, Cohen MD, et al. 2010 rheumatoid arthritis classification criteria: an American College of Rheumatology/European League Against Rheumatism collaborative initiative. Ann Rheum Dis. 2010;69:1580–1588. doi: 10.1136/ard.2010.138461. [DOI] [PubMed] [Google Scholar]
- 9.Balsa A, Cabezón A, Orozco G, Cobo T, Miranda-Carus E, López-Nevot MA, Vicario JL, Martín-Mola E, Martín J, Pascual-Salcedo D. Influence of HLA DRB1 alleles in the susceptibility of rheumatoid arthritis and the regulation of antibodies against citrullinated proteins and rheumatoid factor. Arthritis Res Ther. 2010;12:R62. doi: 10.1186/ar2975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chaiamnuay S, Bridges SL. The role of B cells and autoantibodies in rheumatoid arthritis. Pathophysiology. 2005;12:203–216. doi: 10.1016/j.pathophys.2005.07.007. [DOI] [PubMed] [Google Scholar]
- 11.Baeten D, Peene I, Union A, Meheus L, Sebbag M, Serre G, Veys EM, De Keyser F. Specific presence of intracellular citrullinated proteins in rheumatoid arthritis synovium: relevance to antifilaggrin autoantibodies. Arthritis Rheum. 2001;44:2255–2262. doi: 10.1002/1529-0131(200110)44:10<2255::aid-art388>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 12.Harris ED. Rheumatoid arthritis. Pathophysiology and implications for therapy. N Engl J Med. 1990;322:1277–1289. doi: 10.1056/NEJM199005033221805. [DOI] [PubMed] [Google Scholar]
- 13.Scott DL, Wolfe F, Huizinga TW. Rheumatoid arthritis. Lancet. 2010;376:1094–1108. doi: 10.1016/S0140-6736(10)60826-4. [DOI] [PubMed] [Google Scholar]
- 14.Joosten LA, Radstake TR, Lubberts E, van den Bersselaar LA, van Riel PL, van Lent PL, Barrera P, van den Berg WB. Association of interleukin-18 expression with enhanced levels of both interleukin-1beta and tumor necrosis factor alpha in knee synovial tissue of patients with rheumatoid arthritis. Arthritis Rheum. 2003;48:339–347. doi: 10.1002/art.10814. [DOI] [PubMed] [Google Scholar]
- 15.Choy EH, Smith C, Doré CJ, Scott DL. A meta-analysis of the efficacy and toxicity of combining disease-modifying anti-rheumatic drugs in rheumatoid arthritis based on patient withdrawal. Rheumatology (Oxford) 2005;44:1414–1421. doi: 10.1093/rheumatology/kei031. [DOI] [PubMed] [Google Scholar]
- 16.Czirják L, Kiss CG, Lövei C, Süto G, Varjú C, Füzesi Z, Illés T, Nagy Z. Survey of Raynaud’s phenomenon and systemic sclerosis based on a representative study of 10,000 south-Transdanubian Hungarian inhabitants. Clin Exp Rheumatol. 2005;23:801–808. [PubMed] [Google Scholar]
- 17.Linn-Rasker SP, van der Helm-van Mil AH, van Gaalen FA, Kloppenburg M, de Vries RR, le Cessie S, Breedveld FC, Toes RE, Huizinga TW. Smoking is a risk factor for anti-CCP antibodies only in rheumatoid arthritis patients who carry HLA-DRB1 shared epitope alleles. Ann Rheum Dis. 2006;65:366–371. doi: 10.1136/ard.2005.041079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Klareskog L, Padyukov L, Rönnelid J, Alfredsson L. Genes, environment and immunity in the development of rheumatoid arthritis. Curr Opin Immunol. 2006;18:650–655. doi: 10.1016/j.coi.2006.06.004. [DOI] [PubMed] [Google Scholar]
- 19.Eaton WW, Byrne M, Ewald H, Mors O, Chen CY, Agerbo E, Mortensen PB. Association of schizophrenia and autoimmune diseases: linkage of Danish national registers. Am J Psychiatry. 2006;163:521–528. doi: 10.1176/appi.ajp.163.3.521. [DOI] [PubMed] [Google Scholar]
- 20.Mors O, Mortensen PB, Ewald H. A population-based register study of the association between schizophrenia and rheumatoid arthritis. Schizophr Res. 1999;40:67–74. doi: 10.1016/s0920-9964(99)00030-4. [DOI] [PubMed] [Google Scholar]
- 21.Gibert M, Balandraud N, Touinssi M, Mercier P, Roudier J, Reviron D. Functional categorization of HLA-DRB1 alleles in rheumatoid arthritis: the protective effect. Hum Immunol. 2003;64:930–935. doi: 10.1016/s0198-8859(03)00186-1. [DOI] [PubMed] [Google Scholar]
- 22.Tamiya G, Shinya M, Imanishi T, Ikuta T, Makino S, Okamoto K, Furugaki K, Matsumoto T, Mano S, Ando S, et al. Whole genome association study of rheumatoid arthritis using 27 039 microsatellites. Hum Mol Genet. 2005;14:2305–2321. doi: 10.1093/hmg/ddi234. [DOI] [PubMed] [Google Scholar]
- 23.Barton A, Thomson W, Ke X, Eyre S, Hinks A, Bowes J, Gibbons L, Plant D, Wilson AG, Marinou I, et al. Re-evaluation of putative rheumatoid arthritis susceptibility genes in the post-genome wide association study era and hypothesis of a key pathway underlying susceptibility. Hum Mol Genet. 2008;17:2274–2279. doi: 10.1093/hmg/ddn128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stahl EA, Raychaudhuri S, Remmers EF, Xie G, Eyre S, Thomson BP, Li Y, Kurreeman FA, Zhernakova A, Hinks A, et al. Genome-wide association study meta-analysis identifies seven new rheumatoid arthritis risk loci. Nat Genet. 2010;42:508–514. doi: 10.1038/ng.582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Akdis M, Burgler S, Crameri R, Eiwegger T, Fujita H, Gomez E, Klunker S, Meyer N, O’Mahony L, Palomares O, et al. Interleukins, from 1 to 37, and interferon-γ: receptors, functions, and roles in diseases. J Allergy Clin Immunol. 2011;127:701–721.e1-70. doi: 10.1016/j.jaci.2010.11.050. [DOI] [PubMed] [Google Scholar]
- 26.Dinarello CA. Interleukin-1 in the pathogenesis and treatment of inflammatory diseases. Blood. 2011;117:3720–3732. doi: 10.1182/blood-2010-07-273417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Goldring SR. Pathogenesis of bone and cartilage destruction in rheumatoid arthritis. Rheumatology (Oxford) 2003;42 Suppl 2:ii11–ii16. doi: 10.1093/rheumatology/keg327. [DOI] [PubMed] [Google Scholar]
- 28.Dayer JM. The pivotal role of interleukin-1 in the clinical manifestations of rheumatoid arthritis. Rheumatology (Oxford) 2003;42 Suppl 2:ii3–10. doi: 10.1093/rheumatology/keg326. [DOI] [PubMed] [Google Scholar]
- 29.Arend WP, Guthridge CJ. Biological role of interleukin 1 receptor antagonist isoforms. Ann Rheum Dis. 2000;59 Suppl 1:i60–i64. doi: 10.1136/ard.59.suppl_1.i60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dinarello CA. Biologic basis for interleukin-1 in disease. Blood. 1996;87:2095–2147. [PubMed] [Google Scholar]
- 31.di Giovine FS, Takhsh E, Blakemore AI, Duff GW. Single base polymorphism at -511 in the human interleukin-1 beta gene (IL1 beta) Hum Mol Genet. 1992;1:450. doi: 10.1093/hmg/1.6.450. [DOI] [PubMed] [Google Scholar]
- 32.Pociot F, Mølvig J, Wogensen L, Worsaae H, Nerup J. A TaqI polymorphism in the human interleukin-1 beta (IL-1 beta) gene correlates with IL-1 beta secretion in vitro. Eur J Clin Invest. 1992;22:396–402. doi: 10.1111/j.1365-2362.1992.tb01480.x. [DOI] [PubMed] [Google Scholar]
- 33.Cox A, Camp NJ, Cannings C, di Giovine FS, Dale M, Worthington J, John S, Ollier WE, Silman AJ, Duff GW. Combined sib-TDT and TDT provide evidence for linkage of the interleukin-1 gene cluster to erosive rheumatoid arthritis. Hum Mol Genet. 1999;8:1707–1713. doi: 10.1093/hmg/8.9.1707. [DOI] [PubMed] [Google Scholar]
- 34.Arman A, Yilmaz B, Coker A, Inanc N, Direskeneli H. Interleukin-1 receptor antagonist (IL-1RN) and interleukin-1B gene polymorphisms in Turkish patients with rheumatoid arthritis. Clin Exp Rheumatol. 2006;24:643–648. [PubMed] [Google Scholar]
- 35.Harrison P, Pointon JJ, Chapman K, Roddam A, Wordsworth BP. Interleukin-1 promoter region polymorphism role in rheumatoid arthritis: a meta-analysis of IL-1B-511A/G variant reveals association with rheumatoid arthritis. Rheumatology (Oxford) 2008;47:1768–1770. doi: 10.1093/rheumatology/ken374. [DOI] [PubMed] [Google Scholar]
- 36.Lubbe S, Tikly M, van der Merwe L, Hodkinson B, Ramsay M. Interleukin-1 receptor antagonist gene polymorphisms are associated with disease severity in Black South Africans with rheumatoid arthritis. Joint Bone Spine. 2008;75:422–425. doi: 10.1016/j.jbspin.2007.09.015. [DOI] [PubMed] [Google Scholar]
- 37.Cvetkovic JT, Wallberg-Jonsson S, Stegmayr B, Rantapaa-Dahlqvist S, Lefvert AK. Susceptibility for and clinical manifestations of rheumatoid arthritis are associated with polymorphisms of the TNF-alpha, IL-1beta, and IL-1Ra genes. J Rheumatol. 2002;29:212–219. [PubMed] [Google Scholar]
- 38.Huang CM, Tsai FJ, Wu JY, Wu MC. Interleukin-1beta and interleukin-1 receptor antagonist gene polymorphisms in rheumatoid arthritis. Scand J Rheumatol. 2001;30:225–228. doi: 10.1080/030097401316909576. [DOI] [PubMed] [Google Scholar]
- 39.Grover S, Tandon S, Misra R, Aggarwal A. Interleukin-1 receptor antagonist gene polymorphism in patients with rheumatoid arthritis in India. Indian J Med Res. 2006;123:815–820. [PubMed] [Google Scholar]
- 40.Allam I, Djidjik R, Ouikhlef N, Louahchi S, Raaf N, Behaz N, Abdessemed A, Khaldoun N, Tahiat A, Bayou M, et al. Interleukin-1 and the interleukin-1 receptor antagonist gene polymorphisms study in patients with rheumatoid arthritis. Pathol Biol (Paris) 2013;61:264–268. doi: 10.1016/j.patbio.2013.05.005. [DOI] [PubMed] [Google Scholar]
- 41.Lee YH, Ji JD, Song GG. Association between interleukin 1 polymorphisms and rheumatoid arthritis susceptibility: a metaanalysis. J Rheumatol. 2009;36:12–15. doi: 10.3899/jrheum.080450. [DOI] [PubMed] [Google Scholar]
- 42.You CG, Li JF, Xie XD, Zhu Y, Li PQ, Chen YR. Association of interleukin-1 genetic polymorphisms with the risk of rheumatoid arthritis in Chinese population. Clin Chem Lab Med. 2007;45:968–971. doi: 10.1515/CCLM.2007.156. [DOI] [PubMed] [Google Scholar]
- 43.Arend WP, Palmer G, Gabay C. IL-1, IL-18, and IL-33 families of cytokines. Immunol Rev. 2008;223:20–38. doi: 10.1111/j.1600-065X.2008.00624.x. [DOI] [PubMed] [Google Scholar]
- 44.Chen S, Jiang F, Ren J, Liu J, Meng W. Association of IL-18 polymorphisms with rheumatoid arthritis and systemic lupus erythematosus in Asian populations: a meta-analysis. BMC Med Genet. 2012;13:107. doi: 10.1186/1471-2350-13-107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ji JD, Lee WJ. Interleukin-18 gene polymorphisms and rheumatoid arthritis: a meta-analysis. Gene. 2013;523:27–32. doi: 10.1016/j.gene.2013.03.132. [DOI] [PubMed] [Google Scholar]
- 46.Hashaad NI, El-Din MS, Moustafa EF, Abo Elazem AA. Interleukin-18 promoter polymorphisms in Egyptian patients with rheumatoid arthritis. Egypt J Immunol. 2012;19:13–24. [PubMed] [Google Scholar]
- 47.Huang XZ, Zhuang JH, Ren YG, Zhou LJ, Zhou Q. [Association of interleukin-6 and interleukin-18 gene polymorphism with rheumatoid arthritis in Guangdong Han population] Nanfang Yike Daxue Xuebao. 2007;27:1661–1664. [PubMed] [Google Scholar]
- 48.Lee CC, Lin WY, Wan L, Tsai Y, Lin YJ, Tsai CH, Huang CM, Tsai FJ. Interleukin-18 gene polymorphism, but not interleukin-2 gene polymorphism, is associated with rheumatoid arthritis. Immunogenetics. 2007;59:433–439. doi: 10.1007/s00251-007-0212-z. [DOI] [PubMed] [Google Scholar]
- 49.Sugiura T, Kawaguchi Y, Ikari K, Ichida H, Kawamoto M, Momohara S, Hara M, Yamanaka H. Interleukin-18 promoter polymorphisms in Japanese patients with rheumatoid arthritis: protective effect of the T allele and T/T genotype at rs360722. Mod Rheumatol. 2011;21:359–364. doi: 10.1007/s10165-010-0399-y. [DOI] [PubMed] [Google Scholar]
- 50.Howard M, Farrar J, Hilfiker M, Johnson B, Takatsu K, Hamaoka T, Paul WE. Identification of a T cell-derived b cell growth factor distinct from interleukin 2. J Exp Med. 1982;155:914–923. doi: 10.1084/jem.155.3.914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hussein YM, El-Shal AS, Rezk NA, Abdel Galil SM, Alzahrani SS. Influence of interleukin-4 gene polymorphisms and interleukin-4 serum level on susceptibility and severity of rheumatoid arthritis in Egyptian population. Cytokine. 2013;61:849–855. doi: 10.1016/j.cyto.2013.01.001. [DOI] [PubMed] [Google Scholar]
- 52.Pawlik A, Wrzesniewska J, Florczak M, Gawronska-Szklarz B, Herczynska M. The -590 IL-4 promoter polymorphism in patients with rheumatoid arthritis. Rheumatol Int. 2005;26:48–51. doi: 10.1007/s00296-004-0539-9. [DOI] [PubMed] [Google Scholar]
- 53.Song GG, Bae SC, Kim JH, Lee YH. Interleukin-4, interleukin-4 receptor, and interleukin-18 polymorphisms and rheumatoid arthritis: a meta-analysis. Immunol Invest. 2013;42:455–469. doi: 10.3109/08820139.2013.804084. [DOI] [PubMed] [Google Scholar]
- 54.Burgos PI, Causey ZL, Tamhane A, Kelley JM, Brown EE, Hughes LB, Danila MI, van Everdingen A, Conn DL, Jonas BL, et al. Association of IL4R single-nucleotide polymorphisms with rheumatoid nodules in African Americans with rheumatoid arthritis. Arthritis Res Ther. 2010;12:R75. doi: 10.1186/ar2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hussein Y, El-Tarhouny S, Mohamed R, Pasha H, Abul-Saoud A. Association of interleukin-4 receptor gene polymorphisms with rheumatoid arthritis in Egyptian female patients. Joint Bone Spine. 2012;79:38–42. doi: 10.1016/j.jbspin.2011.02.017. [DOI] [PubMed] [Google Scholar]
- 56.Fehniger TA, Caligiuri MA. Interleukin 15: biology and relevance to human disease. Blood. 2001;97:14–32. doi: 10.1182/blood.v97.1.14. [DOI] [PubMed] [Google Scholar]
- 57.Waldmann TA, Tagaya Y. The multifaceted regulation of interleukin-15 expression and the role of this cytokine in NK cell differentiation and host response to intracellular pathogens. Annu Rev Immunol. 1999;17:19–49. doi: 10.1146/annurev.immunol.17.1.19. [DOI] [PubMed] [Google Scholar]
- 58.McInnes IB, Liew FY. Interleukin 15: a proinflammatory role in rheumatoid arthritis synovitis. Immunol Today. 1998;19:75–79. doi: 10.1016/s0167-5699(97)01205-x. [DOI] [PubMed] [Google Scholar]
- 59.Knevel R, Krabben A, Brouwer E, Posthumus MD, Wilson AG, Lindqvist E, Saxne T, de Rooy D, Daha N, van der Linden MP, et al. Genetic variants in IL15 associate with progression of joint destruction in rheumatoid arthritis: a multicohort study. Ann Rheum Dis. 2012;71:1651–1657. doi: 10.1136/annrheumdis-2011-200724. [DOI] [PubMed] [Google Scholar]
- 60.Rueda B, López-Nevot MA, González-Gay MA, Balsa A, Pascual-Salcedo D, Garcia A, Gonzalez A, Martin J. Molecular screening and association study of IL15 gene polymorphisms in rheumatoid arthritis. Cytokine. 2007;38:84–89. doi: 10.1016/j.cyto.2007.05.005. [DOI] [PubMed] [Google Scholar]
- 61.Christensen U, Haagerup A, Binderup HG, Vestbo J, Kruse TA, Børglum AD. Family based association analysis of the IL2 and IL15 genes in allergic disorders. Eur J Hum Genet. 2006;14:227–235. doi: 10.1038/sj.ejhg.5201541. [DOI] [PubMed] [Google Scholar]
- 62.Wellcome Trust Case Control Consortium. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447:661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Spolski R, Leonard WJ. The Yin and Yang of interleukin-21 in allergy, autoimmunity and cancer. Curr Opin Immunol. 2008;20:295–301. doi: 10.1016/j.coi.2008.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Knevel R, de Rooy DP, Zhernakova A, Gröndal G, Krabben A, Steinsson K, Wijmenga C, Cavet G, Toes RE, Huizinga TW, et al. Association of variants in IL2RA with progression of joint destruction in rheumatoid arthritis. Arthritis Rheum. 2013;65:1684–1693. doi: 10.1002/art.37938. [DOI] [PubMed] [Google Scholar]
- 65.Daha NA, Kurreeman FA, Marques RB, Stoeken-Rijsbergen G, Verduijn W, Huizinga TW, Toes RE. Confirmation of STAT4, IL2/IL21, and CTLA4 polymorphisms in rheumatoid arthritis. Arthritis Rheum. 2009;60:1255–1260. doi: 10.1002/art.24503. [DOI] [PubMed] [Google Scholar]
- 66.Todd JA, Walker NM, Cooper JD, Smyth DJ, Downes K, Plagnol V, Bailey R, Nejentsev S, Field SF, Payne F, et al. Robust associations of four new chromosome regions from genome-wide analyses of type 1 diabetes. Nat Genet. 2007;39:857–864. doi: 10.1038/ng2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Festen EA, Goyette P, Scott R, Annese V, Zhernakova A, Lian J, Lefèbvre C, Brant SR, Cho JH, Silverberg MS, et al. Genetic variants in the region harbouring IL2/IL21 associated with ulcerative colitis. Gut. 2009;58:799–804. doi: 10.1136/gut.2008.166918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Márquez A, Orozco G, Martínez A, Palomino-Morales R, Fernández-Arquero M, Mendoza JL, Taxonera C, Díaz-Rubio M, Gómez-García M, Nieto A, et al. Novel association of the interleukin 2-interleukin 21 region with inflammatory bowel disease. Am J Gastroenterol. 2009;104:1968–1975. doi: 10.1038/ajg.2009.224. [DOI] [PubMed] [Google Scholar]
- 69.Sawalha AH, Kaufman KM, Kelly JA, Adler AJ, Aberle T, Kilpatrick J, Wakeland EK, Li QZ, Wandstrat AE, Karp DR, et al. Genetic association of interleukin-21 polymorphisms with systemic lupus erythematosus. Ann Rheum Dis. 2008;67:458–461. doi: 10.1136/ard.2007.075424. [DOI] [PubMed] [Google Scholar]
- 70.Liu Y, Helms C, Liao W, Zaba LC, Duan S, Gardner J, Wise C, Miner A, Malloy MJ, Pullinger CR, et al. A genome-wide association study of psoriasis and psoriatic arthritis identifies new disease loci. PLoS Genet. 2008;4:e1000041. doi: 10.1371/journal.pgen.1000041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Albers HM, Kurreeman FA, Stoeken-Rijsbergen G, Brinkman DM, Kamphuis SS, van Rossum MA, Girschick HJ, Wouters C, Saurenmann RK, Hoppenreijs E, et al. Association of the autoimmunity locus 4q27 with juvenile idiopathic arthritis. Arthritis Rheum. 2009;60:901–904. doi: 10.1002/art.24296. [DOI] [PubMed] [Google Scholar]
- 72.Hollis-Moffatt JE, Chen-Xu M, Topless R, Dalbeth N, Gow PJ, Harrison AA, Highton J, Jones PB, Nissen M, Smith MD, van Rij A, Jones GT, Stamp LK, Merriman TR: Only one independent genetic association with rheumatoid arthritis within the KIAA1109-TENR-IL2-IL21 locus in Caucasian sample sets: confirmation of association of rs6822844 with rheumatoid arthritis at a genome-wide level of significance. Arthritis Res Ther. 2010;12:R116. doi: 10.1186/ar3053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zdanov A. Structural features of the interleukin-10 family of cytokines. Curr Pharm Des. 2004;10:3873–3884. doi: 10.2174/1381612043382602. [DOI] [PubMed] [Google Scholar]
- 74.Deniz G, Erten G, Kücüksezer UC, Kocacik D, Karagiannidis C, Aktas E, Akdis CA, Akdis M. Regulatory NK cells suppress antigen-specific T cell responses. J Immunol. 2008;180:850–857. doi: 10.4049/jimmunol.180.2.850. [DOI] [PubMed] [Google Scholar]
- 75.Chernoff AE, Granowitz EV, Shapiro L, Vannier E, Lonnemann G, Angel JB, Kennedy JS, Rabson AR, Wolff SM, Dinarello CA. A randomized, controlled trial of IL-10 in humans. Inhibition of inflammatory cytokine production and immune responses. J Immunol. 1995;154:5492–5499. [PubMed] [Google Scholar]
- 76.Turner DM, Williams DM, Sankaran D, Lazarus M, Sinnott PJ, Hutchinson IV. An investigation of polymorphism in the interleukin-10 gene promoter. Eur J Immunogenet. 1997;24:1–8. doi: 10.1111/j.1365-2370.1997.tb00001.x. [DOI] [PubMed] [Google Scholar]
- 77.Guzowski D, Chandrasekaran A, Gawel C, Palma J, Koenig J, Wang XP, Dosik M, Kaplan M, Chu CC, Chavan S, et al. Analysis of single nucleotide polymorphisms in the promoter region of interleukin-10 by denaturing high-performance liquid chromatography. J Biomol Tech. 2005;16:154–166. [PMC free article] [PubMed] [Google Scholar]
- 78.Lee YH, Bae SC, Choi SJ, Ji JD, Song GG. Associations between interleukin-10 polymorphisms and susceptibility to rheumatoid arthritis: a meta-analysis. Mol Biol Rep. 2012;39:81–87. doi: 10.1007/s11033-011-0712-7. [DOI] [PubMed] [Google Scholar]
- 79.Hoffmann SC, Stanley EM, Darrin Cox E, Craighead N, DiMercurio BS, Koziol DE, Harlan DM, Kirk AD, Blair PJ. Association of cytokine polymorphic inheritance and in vitro cytokine production in anti-CD3/CD28-stimulated peripheral blood lymphocytes. Transplantation. 2001;72:1444–1450. doi: 10.1097/00007890-200110270-00019. [DOI] [PubMed] [Google Scholar]
- 80.Hee CS, Gun SC, Naidu R, Gupta E, Somnath SD, Radhakrishnan AK. Comparison of single nucleotide polymorphisms in the human interleukin-10 gene promoter between rheumatoid arthritis patients and normal subjects in Malaysia. Mod Rheumatol. 2007;17:429–435. doi: 10.1007/s10165-007-0612-9. [DOI] [PubMed] [Google Scholar]
- 81.Pawlik A, Kurzawski M, Szklarz BG, Herczynska M, Drozdzik M. Interleukin-10 promoter polymorphism in patients with rheumatoid arthritis. Clin Rheumatol. 2005;24:480–484. doi: 10.1007/s10067-004-1066-5. [DOI] [PubMed] [Google Scholar]
- 82.Ying B, Shi Y, Pan X, Song X, Huang Z, Niu Q, Cai B, Wang L. Association of polymorphisms in the human IL-10 and IL-18 genes with rheumatoid arthritis. Mol Biol Rep. 2011;38:379–385. doi: 10.1007/s11033-010-0119-x. [DOI] [PubMed] [Google Scholar]
- 83.Zhang J, Zhang Y, Jin J, Li M, Xie K, Wen C, Cheng R, Chen C, Lu J. The -1082A/G polymorphism in the Interleukin-10 gene and the risk of rheumatoid arthritis: a meta-analysis. Cytokine. 2011;56:351–355. doi: 10.1016/j.cyto.2011.05.022. [DOI] [PubMed] [Google Scholar]
- 84.Vignali DA, Kuchroo VK. IL-12 family cytokines: immunological playmakers. Nat Immunol. 2012;13:722–728. doi: 10.1038/ni.2366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Oppmann B, Lesley R, Blom B, Timans JC, Xu Y, Hunte B, Vega F, Yu N, Wang J, Singh K, et al. Novel p19 protein engages IL-12p40 to form a cytokine, IL-23, with biological activities similar as well as distinct from IL-12. Immunity. 2000;13:715–725. doi: 10.1016/s1074-7613(00)00070-4. [DOI] [PubMed] [Google Scholar]
- 86.Boulay JL, O’Shea JJ, Paul WE. Molecular phylogeny within type I cytokines and their cognate receptors. Immunity. 2003;19:159–163. doi: 10.1016/s1074-7613(03)00211-5. [DOI] [PubMed] [Google Scholar]
- 87.Niedbala W, Cai B, Wei X, Patakas A, Leung BP, McInnes IB, Liew FY. Interleukin 27 attenuates collagen-induced arthritis. Ann Rheum Dis. 2008;67:1474–1479. doi: 10.1136/ard.2007.083360. [DOI] [PubMed] [Google Scholar]
- 88.Del Vecchio M, Bajetta E, Canova S, Lotze MT, Wesa A, Parmiani G, Anichini A. Interleukin-12: biological properties and clinical application. Clin Cancer Res. 2007;13:4677–4685. doi: 10.1158/1078-0432.CCR-07-0776. [DOI] [PubMed] [Google Scholar]
- 89.Villarino AV, Hunter CA. Biology of recently discovered cytokines: discerning the pro- and anti-inflammatory properties of interleukin-27. Arthritis Res Ther. 2004;6:225–233. doi: 10.1186/ar1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Goriely S, Goldman M. The interleukin-12 family: new players in transplantation immunity? Am J Transplant. 2007;7:278–284. doi: 10.1111/j.1600-6143.2006.01651.x. [DOI] [PubMed] [Google Scholar]
- 91.Goriely S, Goldman M. Interleukin-12 family members and the balance between rejection and tolerance. Curr Opin Organ Transplant. 2008;13:4–9. doi: 10.1097/MOT.0b013e3282f406c4. [DOI] [PubMed] [Google Scholar]
- 92.Pirhonen J, Matikainen S, Julkunen I. Regulation of virus-induced IL-12 and IL-23 expression in human macrophages. J Immunol. 2002;169:5673–5678. doi: 10.4049/jimmunol.169.10.5673. [DOI] [PubMed] [Google Scholar]
- 93.Parham C, Chirica M, Timans J, Vaisberg E, Travis M, Cheung J, Pflanz S, Zhang R, Singh KP, Vega F, et al. A receptor for the heterodimeric cytokine IL-23 is composed of IL-12Rbeta1 and a novel cytokine receptor subunit, IL-23R. J Immunol. 2002;168:5699–5708. doi: 10.4049/jimmunol.168.11.5699. [DOI] [PubMed] [Google Scholar]
- 94.Duerr RH, Taylor KD, Brant SR, Rioux JD, Silverberg MS, Daly MJ, Steinhart AH, Abraham C, Regueiro M, Griffiths A, et al. A genome-wide association study identifies IL23R as an inflammatory bowel disease gene. Science. 2006;314:1461–1463. doi: 10.1126/science.1135245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Murphy CA, Langrish CL, Chen Y, Blumenschein W, McClanahan T, Kastelein RA, Sedgwick JD, Cua DJ. Divergent pro- and antiinflammatory roles for IL-23 and IL-12 in joint autoimmune inflammation. J Exp Med. 2003;198:1951–1957. doi: 10.1084/jem.20030896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Farago B, Magyari L, Safrany E, Csongei V, Jaromi L, Horvatovich K, Sipeky C, Maasz A, Radics J, Gyetvai A, Szekanecz Z, Czirjak L, Melegh B: Functional variants of interleukin-23 receptor gene confer risk for rheumatoid arthritis but not for systemic sclerosis. Ann Rheum Dis. 2008;67:248–250. doi: 10.1136/ard.2007.072819. [DOI] [PubMed] [Google Scholar]
- 97.Orozco G, Rueda B, Robledo G, García A, Martín J. Investigation of the IL23R gene in a Spanish rheumatoid arthritis cohort. Hum Immunol. 2007;68:681–684. doi: 10.1016/j.humimm.2007.05.008. [DOI] [PubMed] [Google Scholar]
- 98.Chang M, Saiki RK, Cantanese JJ, Lew D, van der Helm-van Mil AH, Toes RE, Huizinga TW, Ardlie KG, Criswell LA, Seldin MF, et al. The inflammatory disease-associated variants in IL12B and IL23R are not associated with rheumatoid arthritis. Arthritis Rheum. 2008;58:1877–1881. doi: 10.1002/art.23492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Varade J, Ramón Lamas J, Rodríguez L, Fernández-Arquero M, Loza-Santamaría E, Jover JA, de la Concha EG, Fernández-Gutierrez B, Urcelay E, Martínez A. IL23R and IL12B genes: susceptibility analysis in rheumatoid arthritis. Ann Rheum Dis. 2009;68:1230–1232. doi: 10.1136/ard.2008.099275. [DOI] [PubMed] [Google Scholar]
- 100.Hollis-Moffatt JE, Merriman ME, Rodger RA, Rowley KA, Chapman PT, Dalbeth N, Gow PJ, Harrison AA, Highton J, Jones PB, et al. Evidence for association of an interleukin 23 receptor variant independent of the R381Q variant with rheumatoid arthritis. Ann Rheum Dis. 2009;68:1340–1344. doi: 10.1136/ard.2008.090142. [DOI] [PubMed] [Google Scholar]
- 101.Plenge RM, Seielstad M, Padyukov L, Lee AT, Remmers EF, Ding B, Liew A, Khalili H, Chandrasekaran A, Davies LR, et al. TRAF1-C5 as a risk locus for rheumatoid arthritis--a genomewide study. N Engl J Med. 2007;357:1199–1209. doi: 10.1056/NEJMoa073491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Chen-Xu M, Topless R, McKinney C, Merriman ME, Phipps-Green A, Dalbeth N, Gow PJ, Harrison AA, Highton J, Jones PB, et al. Replication of association of the interleukin 23 receptor rs1343151 variant with rheumatoid arthritis in Caucasian sample sets. Ann Rheum Dis. 2012;71:155–157. doi: 10.1136/annrheumdis-2011-200591. [DOI] [PubMed] [Google Scholar]
- 103.Song GG, Bae SC, Choi SJ, Ji JD, Lee YH. Associations between interleukin-23 receptor polymorphisms and susceptibility to rheumatoid arthritis: a meta-analysis. Mol Biol Rep. 2012;39:10655–10663. doi: 10.1007/s11033-012-1955-7. [DOI] [PubMed] [Google Scholar]
- 104.Zhai Y, Xu K, Huang F, Peng H, Feng CC, Zhu KK, Leng RX, Pan HF, Ye DQ. Association of interleukin 23 receptor gene polymorphisms (rs10489629, rs7517847) with rheumatoid arthritis in European population: a meta-analysis. Mol Biol Rep. 2012;39:8987–8994. doi: 10.1007/s11033-012-1768-8. [DOI] [PubMed] [Google Scholar]
- 105.Lee YH, Choi SJ, Ji JD, Song GG. Associations between interleukin-23R polymorphisms and ankylosing spondylitis susceptibility: a meta-analysis. Inflamm Res. 2012;61:143–149. doi: 10.1007/s00011-011-0398-2. [DOI] [PubMed] [Google Scholar]
- 106.Park JH, Kim YJ, Park BL, Bae JS, Shin HD, Bae SC. Lack of association between interleukin 23 receptor gene polymorphisms and rheumatoid arthritis susceptibility. Rheumatol Int. 2009;29:781–786. doi: 10.1007/s00296-008-0770-x. [DOI] [PubMed] [Google Scholar]
- 107.Heinrich PC, Behrmann I, Haan S, Hermanns HM, Müller-Newen G, Schaper F. Principles of interleukin (IL)-6-type cytokine signalling and its regulation. Biochem J. 2003;374:1–20. doi: 10.1042/BJ20030407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Guerne PA, Terkeltaub R, Zuraw B, Lotz M. Inflammatory microcrystals stimulate interleukin-6 production and secretion by human monocytes and synoviocytes. Arthritis Rheum. 1989;32:1443–1452. doi: 10.1002/anr.1780321114. [DOI] [PubMed] [Google Scholar]
- 109.Boulanger MJ, Chow DC, Brevnova EE, Garcia KC. Hexameric structure and assembly of the interleukin-6/IL-6 alpha-receptor/gp130 complex. Science. 2003;300:2101–2104. doi: 10.1126/science.1083901. [DOI] [PubMed] [Google Scholar]
- 110.Filippucci E, Iagnocco A, Meenagh G, Riente L, Delle Sedie A, Bombardieri S, Valesini G, Grassi W. Ultrasound imaging for the rheumatologist. Clin Exp Rheumatol. 2006;24:1–5. [PubMed] [Google Scholar]
- 111.Newton JL, Harney SM, Wordsworth BP, Brown MA. A review of the MHC genetics of rheumatoid arthritis. Genes Immun. 2004;5:151–157. doi: 10.1038/sj.gene.6364045. [DOI] [PubMed] [Google Scholar]
- 112.Kilding R, Iles MM, Timms JM, Worthington J, Wilson AG. Additional genetic susceptibility for rheumatoid arthritis telomeric of the DRB1 locus. Arthritis Rheum. 2004;50:763–769. doi: 10.1002/art.20043. [DOI] [PubMed] [Google Scholar]
- 113.Chang WW, Su H, He L, Zhao KF, Wu JL, Xu ZW: Association between transforming growth factor-beta1 T869C polymorphism and rheumatoid arthritis: a meta-analysis. Rheumatology (Oxford) 2010;49:652–656. doi: 10.1093/rheumatology/kep417. [DOI] [PubMed] [Google Scholar]
- 114.Marinou I, Healy J, Mewar D, Moore DJ, Dickson MC, Binks MH, Montgomery DS, Walters K, Wilson AG. Association of interleukin-6 and interleukin-10 genotypes with radiographic damage in rheumatoid arthritis is dependent on autoantibody status. Arthritis Rheum. 2007;56:2549–2556. doi: 10.1002/art.22814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Mattey DL, Nixon N, Dawes PT, Kerr J. Association of polymorphism in the transforming growth factor {beta}1 gene with disease outcome and mortality in rheumatoid arthritis. Ann Rheum Dis. 2005;64:1190–1194. doi: 10.1136/ard.2004.031674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Pascual M, Nieto A, Matarán L, Balsa A, Pascual-Salcedo D, Martín J. IL-6 promoter polymorphisms in rheumatoid arthritis. Genes Immun. 2000;1:338–340. doi: 10.1038/sj.gene.6363677. [DOI] [PubMed] [Google Scholar]
- 117.Palomino-Morales R, Gonzalez-Juanatey C, Vazquez-Rodriguez TR, Miranda-Filloy JA, Llorca J, Martin J, Gonzalez-Gay MA. Interleukin-6 gene -174 promoter polymorphism is associated with endothelial dysfunction but not with disease susceptibility in patients with rheumatoid arthritis. Clin Exp Rheumatol. 2009;27:964–970. [PubMed] [Google Scholar]
- 118.Lo SF, Huang CM, Lin HC, Chen WC, Tsai CH, Tsai FJ. Cytokine (IL-6) and chemokine (IL-8) gene polymorphisms among rheumatoid arthritis patients in Taiwan. Clin Exp Rheumatol. 2008;26:632–637. [PubMed] [Google Scholar]
- 119.Zhang S, Zhang P, Huang QY. Association of the D358A polymorphism of IL6R gene with type 2 diabetes in Hubei Han Chinese. Zhonghua Yixue Yichuan Xue Zazhi. 2009;26:452–456. [PubMed] [Google Scholar]
- 120.Jones SA, Rose-John S. The role of soluble receptors in cytokine biology: the agonistic properties of the sIL-6R/IL-6 complex. Biochim Biophys Acta. 2002;1592:251–263. doi: 10.1016/s0167-4889(02)00319-1. [DOI] [PubMed] [Google Scholar]
- 121.Rose-John S, Scheller J, Elson G, Jones SA. Interleukin-6 biology is coordinated by membrane-bound and soluble receptors: role in inflammation and cancer. J Leukoc Biol. 2006;80:227–236. doi: 10.1189/jlb.1105674. [DOI] [PubMed] [Google Scholar]
- 122.Lee YH, Bae SC, Choi SJ, Ji JD, Song GG. The association between interleukin-6 polymorphisms and rheumatoid arthritis: a meta-analysis. Inflamm Res. 2012;61:665–671. doi: 10.1007/s00011-012-0459-1. [DOI] [PubMed] [Google Scholar]
- 123.Li H, Chen J, Huang A, Stinson J, Heldens S, Foster J, Dowd P, Gurney AL, Wood WI. Cloning and characterization of IL-17B and IL-17C, two new members of the IL-17 cytokine family. Proc Natl Acad Sci USA. 2000;97:773–778. doi: 10.1073/pnas.97.2.773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Shi Y, Ullrich SJ, Zhang J, Connolly K, Grzegorzewski KJ, Barber MC, Wang W, Wathen K, Hodge V, Fisher CL, et al. A novel cytokine receptor-ligand pair. Identification, molecular characterization, and in vivo immunomodulatory activity. J Biol Chem. 2000;275:19167–19176. doi: 10.1074/jbc.M910228199. [DOI] [PubMed] [Google Scholar]
- 125.Lee J, Ho WH, Maruoka M, Corpuz RT, Baldwin DT, Foster JS, Goddard AD, Yansura DG, Vandlen RL, Wood WI, et al. IL-17E, a novel proinflammatory ligand for the IL-17 receptor homolog IL-17Rh1. J Biol Chem. 2001;276:1660–1664. doi: 10.1074/jbc.M008289200. [DOI] [PubMed] [Google Scholar]
- 126.Starnes T, Broxmeyer HE, Robertson MJ, Hromas R. Cutting edge: IL-17D, a novel member of the IL-17 family, stimulates cytokine production and inhibits hemopoiesis. J Immunol. 2002;169:642–646. doi: 10.4049/jimmunol.169.2.642. [DOI] [PubMed] [Google Scholar]
- 127.Kawaguchi M, Adachi M, Oda N, Kokubu F, Huang SK. IL-17 cytokine family. J Allergy Clin Immunol. 2004;114:1265–173; quiz 1274. doi: 10.1016/j.jaci.2004.10.019. [DOI] [PubMed] [Google Scholar]
- 128.Rouvier E, Luciani MF, Mattéi MG, Denizot F, Golstein P. CTLA-8, cloned from an activated T cell, bearing AU-rich messenger RNA instability sequences, and homologous to a herpesvirus saimiri gene. J Immunol. 1993;150:5445–5456. [PubMed] [Google Scholar]
- 129.Paradowska A, Maślińiski W, Grzybowska-Kowalczyk A, Łacki J. The function of interleukin 17 in the pathogenesis of rheumatoid arthritis. Arch Immunol Ther Exp (Warsz) 2007;55:329–334. doi: 10.1007/s00005-007-0032-8. [DOI] [PubMed] [Google Scholar]
- 130.Hymowitz SG, Filvaroff EH, Yin JP, Lee J, Cai L, Risser P, Maruoka M, Mao W, Foster J, Kelley RF, et al. IL-17s adopt a cystine knot fold: structure and activity of a novel cytokine, IL-17F, and implications for receptor binding. EMBO J. 2001;20:5332–5341. doi: 10.1093/emboj/20.19.5332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Kolls JK, Lindén A. Interleukin-17 family members and inflammation. Immunity. 2004;21:467–476. doi: 10.1016/j.immuni.2004.08.018. [DOI] [PubMed] [Google Scholar]
- 132.Moseley TA, Haudenschild DR, Rose L, Reddi AH. Interleukin-17 family and IL-17 receptors. Cytokine Growth Factor Rev. 2003;14:155–174. doi: 10.1016/s1359-6101(03)00002-9. [DOI] [PubMed] [Google Scholar]
- 133.Gaffen SL. Structure and signalling in the IL-17 receptor family. Nat Rev Immunol. 2009;9:556–567. doi: 10.1038/nri2586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Paradowska-Gorycka A, Wojtecka-Lukasik E, Trefler J, Wojciechowska B, Lacki JK, Maslinski S. Association between IL-17F gene polymorphisms and susceptibility to and severity of rheumatoid arthritis (RA) Scand J Immunol. 2010;72:134–141. doi: 10.1111/j.1365-3083.2010.02411.x. [DOI] [PubMed] [Google Scholar]
- 135.Nordang GB, Viken MK, Hollis-Moffatt JE, Merriman TR, Førre ØT, Helgetveit K, Kvien TK, Lie BA. Association analysis of the interleukin 17A gene in Caucasian rheumatoid arthritis patients from Norway and New Zealand. Rheumatology (Oxford) 2009;48:367–370. doi: 10.1093/rheumatology/ken512. [DOI] [PubMed] [Google Scholar]
- 136.Furuya T, Hakoda M, Ichikawa N, Higami K, Nanke Y, Yago T, Kamatani N, Kotake S. Associations between HLA-DRB1, RANK, RANKL, OPG, and IL-17 genotypes and disease severity phenotypes in Japanese patients with early rheumatoid arthritis. Clin Rheumatol. 2007;26:2137–2141. doi: 10.1007/s10067-007-0745-4. [DOI] [PubMed] [Google Scholar]
- 137.Emonts M, Hazes MJ, Houwing-Duistermaat JJ, van der Gaast-de Jongh CE, de Vogel L, Han HK, Wouters JM, Laman JD, Dolhain RJ. Polymorphisms in genes controlling inflammation and tissue repair in rheumatoid arthritis: a case control study. BMC Med Genet. 2011;12:36. doi: 10.1186/1471-2350-12-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Smolen JS, van der Heijde D, Machold KP, Aletaha D, Landewé R. Proposal for a new nomenclature of disease-modifying antirheumatic drugs. Ann Rheum Dis. 2014;73:3–5. doi: 10.1136/annrheumdis-2013-204317. [DOI] [PubMed] [Google Scholar]
- 139.Shanahan JC, Moreland LW, Carter RH. Upcoming biologic agents for the treatment of rheumatic diseases. Curr Opin Rheumatol. 2003;15:226–236. doi: 10.1097/00002281-200305000-00009. [DOI] [PubMed] [Google Scholar]
- 140.Smolen JS, Landewé R, Breedveld FC, Buch M, Burmester G, Dougados M, Emery P, Gaujoux-Viala C, Gossec L, Nam J, et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2013 update. Ann Rheum Dis. 2014;73:492–509. doi: 10.1136/annrheumdis-2013-204573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Saag KG, Teng GG, Patkar NM, Anuntiyo J, Finney C, Curtis JR, Paulus HE, Mudano A, Pisu M, Elkins-Melton M, et al. American College of Rheumatology 2008 recommendations for the use of nonbiologic and biologic disease-modifying antirheumatic drugs in rheumatoid arthritis. Arthritis Rheum. 2008;59:762–784. doi: 10.1002/art.23721. [DOI] [PubMed] [Google Scholar]
- 142.Singh JA, Furst DE, Bharat A, Curtis JR, Kavanaugh AF, Kremer JM, Moreland LW, O’Dell J, Winthrop KL, Beukelman T, et al. 2012 update of the 2008 American College of Rheumatology recommendations for the use of disease-modifying antirheumatic drugs and biologic agents in the treatment of rheumatoid arthritis. Arthritis Care Res (Hoboken) 2012;64:625–639. doi: 10.1002/acr.21641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Felson DT, Anderson JJ, Boers M, Bombardier C, Furst D, Goldsmith C, Katz LM, Lightfoot R, Paulus H, Strand V. American College of Rheumatology. Preliminary definition of improvement in rheumatoid arthritis. Arthritis Rheum. 1995;38:727–735. doi: 10.1002/art.1780380602. [DOI] [PubMed] [Google Scholar]
- 144.Fries JF, Spitz P, Kraines RG, Holman HR. Measurement of patient outcome in arthritis. Arthritis Rheum. 1980;23:137–145. doi: 10.1002/art.1780230202. [DOI] [PubMed] [Google Scholar]
- 145.van der Heijde DM, van ‘t Hof M, van Riel PL, van de Putte LB. Development of a disease activity score based on judgment in clinical practice by rheumatologists. J Rheumatol. 1993;20:579–581. [PubMed] [Google Scholar]
- 146.Deighton C, O’Mahony R, Tosh J, Turner C, Rudolf M. Management of rheumatoid arthritis: summary of NICE guidance. BMJ. 2009;338:b702. doi: 10.1136/bmj.b702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Gonzalez-Juanatey C, Testa A, Garcia-Castelo A, Garcia-Porrua C, Llorca J, Gonzalez-Gay MA. Active but transient improvement of endothelial function in rheumatoid arthritis patients undergoing long-term treatment with anti-tumor necrosis factor alpha antibody. Arthritis Rheum. 2004;51:447–450. doi: 10.1002/art.20407. [DOI] [PubMed] [Google Scholar]
- 148.Black RA, Rauch CT, Kozlosky CJ, Peschon JJ, Slack JL, Wolfson MF, Castner BJ, Stocking KL, Reddy P, Srinivasan S, et al. A metalloproteinase disintegrin that releases tumour-necrosis factor-alpha from cells. Nature. 1997;385:729–733. doi: 10.1038/385729a0. [DOI] [PubMed] [Google Scholar]
- 149.Peschon JJ, Torrance DS, Stocking KL, Glaccum MB, Otten C, Willis CR, Charrier K, Morrissey PJ, Ware CB, Mohler KM. TNF receptor-deficient mice reveal divergent roles for p55 and p75 in several models of inflammation. J Immunol. 1998;160:943–952. [PubMed] [Google Scholar]
- 150.Mikuls TR, Moreland LW. TNF blockade in the treatment of rheumatoid arthritis: infliximab versus etanercept. Expert Opin Pharmacother. 2001;2:75–84. doi: 10.1517/14656566.2.1.75. [DOI] [PubMed] [Google Scholar]
- 151.Rau R. Adalimumab (a fully human anti-tumour necrosis factor alpha monoclonal antibody) in the treatment of active rheumatoid arthritis: the initial results of five trials. Ann Rheum Dis. 2002;61 Suppl 2:ii70–ii73. doi: 10.1136/ard.61.suppl_2.ii70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Newton RC, Solomon KA, Covington MB, Decicco CP, Haley PJ, Friedman SM, Vaddi K. Biology of TACE inhibition. Ann Rheum Dis. 2001;60 Suppl 3:iii25–iii32. doi: 10.1136/ard.60.90003.iii25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Eastgate JA, Symons JA, Wood NC, Grinlinton FM, di Giovine FS, Duff GW. Correlation of plasma interleukin 1 levels with disease activity in rheumatoid arthritis. Lancet. 1988;2:706–709. doi: 10.1016/s0140-6736(88)90185-7. [DOI] [PubMed] [Google Scholar]
- 154.Dinarello CA. The interleukin-1 family: 10 years of discovery. FASEB J. 1994;8:1314–1325. [PubMed] [Google Scholar]
- 155.Bresnihan B, Alvaro-Gracia JM, Cobby M, Doherty M, Domljan Z, Emery P, Nuki G, Pavelka K, Rau R, Rozman B, et al. Treatment of rheumatoid arthritis with recombinant human interleukin-1 receptor antagonist. Arthritis Rheum. 1998;41:2196–2204. doi: 10.1002/1529-0131(199812)41:12<2196::AID-ART15>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 156.Cohen S, Hurd E, Cush J, Schiff M, Weinblatt ME, Moreland LW, Kremer J, Bear MB, Rich WJ, McCabe D. Treatment of rheumatoid arthritis with anakinra, a recombinant human interleukin-1 receptor antagonist, in combination with methotrexate: results of a twenty-four-week, multicenter, randomized, double-blind, placebo-controlled trial. Arthritis Rheum. 2002;46:614–624. doi: 10.1002/art.10141. [DOI] [PubMed] [Google Scholar]
- 157.Church LD, McDermott MF. Canakinumab, a fully-human mAb against IL-1beta for the potential treatment of inflammatory disorders. Curr Opin Mol Ther. 2009;11:81–89. [PubMed] [Google Scholar]
- 158.Svenson M, Nedergaard S, Heegaard PM, Whisenand TD, Arend WP, Bendtzen K. Differential binding of human interleukin-1 (IL-1) receptor antagonist to natural and recombinant soluble and cellular IL-1 type I receptors. Eur J Immunol. 1995;25:2842–2850. doi: 10.1002/eji.1830251020. [DOI] [PubMed] [Google Scholar]
- 159.Schoels MM, van der Heijde D, Breedveld FC, Burmester GR, Dougados M, Emery P, Ferraccioli G, Gabay C, Gibofsky A, Gomez-Reino JJ, et al. Blocking the effects of interleukin-6 in rheumatoid arthritis and other inflammatory rheumatic diseases: systematic literature review and meta-analysis informing a consensus statement. Ann Rheum Dis. 2013;72:583–589. doi: 10.1136/annrheumdis-2012-202470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Mease P, Strand V, Shalamberidze L, Dimic A, Raskina T, Xu LA, Liu Y, Smith J. A phase II, double-blind, randomised, placebo-controlled study of BMS945429 (ALD518) in patients with rheumatoid arthritis with an inadequate response to methotrexate. Ann Rheum Dis. 2012;71:1183–1189. doi: 10.1136/annrheumdis-2011-200704. [DOI] [PubMed] [Google Scholar]
- 161.Weinblatt M, Combe B, Covucci A, Aranda R, Becker JC, Keystone E. Safety of the selective costimulation modulator abatacept in rheumatoid arthritis patients receiving background biologic and nonbiologic disease-modifying antirheumatic drugs: A one-year randomized, placebo-controlled study. Arthritis Rheum. 2006;54:2807–2816. doi: 10.1002/art.22070. [DOI] [PubMed] [Google Scholar]
- 162.Groom J, Kalled SL, Cutler AH, Olson C, Woodcock SA, Schneider P, Tschopp J, Cachero TG, Batten M, Wheway J, et al. Association of BAFF/BLyS overexpression and altered B cell differentiation with Sjögren’s syndrome. J Clin Invest. 2002;109:59–68. doi: 10.1172/JCI14121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Kyttaris VC. Kinase inhibitors: a new class of antirheumatic drugs. Drug Des Devel Ther. 2012;6:245–250. doi: 10.2147/DDDT.S25426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.van der Heijde D, Tanaka Y, Fleischmann R, Keystone E, Kremer J, Zerbini C, Cardiel MH, Cohen S, Nash P, Song YW, et al. Tofacitinib (CP-690,550) in patients with rheumatoid arthritis receiving methotrexate: twelve-month data from a twenty-four-month phase III randomized radiographic study. Arthritis Rheum. 2013;65:559–570. doi: 10.1002/art.37816. [DOI] [PubMed] [Google Scholar]
- 165.Plater-Zyberk C, Joosten LA, Helsen MM, Sattonnet-Roche P, Siegfried C, Alouani S, van De Loo FA, Graber P, Aloni S, Cirillo R, et al. Therapeutic effect of neutralizing endogenous IL-18 activity in the collagen-induced model of arthritis. J Clin Invest. 2001;108:1825–1832. doi: 10.1172/JCI12097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Leung BP, Xu D, Culshaw S, McInnes IB, Liew FY. A novel therapy of murine collagen-induced arthritis with soluble T1/ST2. J Immunol. 2004;173:145–150. doi: 10.4049/jimmunol.173.1.145. [DOI] [PubMed] [Google Scholar]
- 167.Carriere V, Roussel L, Ortega N, Lacorre DA, Americh L, Aguilar L, Bouche G, Girard JP. IL-33, the IL-1-like cytokine ligand for ST2 receptor, is a chromatin-associated nuclear factor in vivo. Proc Natl Acad Sci USA. 2007;104:282–287. doi: 10.1073/pnas.0606854104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Verri WA, Guerrero AT, Fukada SY, Valerio DA, Cunha TM, Xu D, Ferreira SH, Liew FY, Cunha FQ. IL-33 mediates antigen-induced cutaneous and articular hypernociception in mice. Proc Natl Acad Sci USA. 2008;105:2723–2728. doi: 10.1073/pnas.0712116105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Firestein GS, Xu WD, Townsend K, Broide D, Alvaro-Gracia J, Glasebrook A, Zvaifler NJ. Cytokines in chronic inflammatory arthritis. I. Failure to detect T cell lymphokines (interleukin 2 and interleukin 3) and presence of macrophage colony-stimulating factor (CSF-1) and a novel mast cell growth factor in rheumatoid synovitis. J Exp Med. 1988;168:1573–1586. doi: 10.1084/jem.168.5.1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Schrohenloher RE, Koopman WJ, Woodworth TG, Moreland LW. Suppression of in vitro IgM rheumatoid factor production by diphtheria toxin interleukin 2 recombinant fusion protein (DAB 486IL-2) in patients with refractory rheumatoid arthritis. J Rheumatol. 1996;23:1845–1848. [PubMed] [Google Scholar]
- 171.Brok HP, Tekoppele JM, Hakimi J, Kerwin JA, Nijenhuis EM, De Groot CW, Bontrop RE, ‘t Hart BA. Prophylactic and therapeutic effects of a humanized monoclonal antibody against the IL-2 receptor (DACLIZUMAB) on collagen-induced arthritis (CIA) in rhesus monkeys. Clin Exp Immunol. 2001;124:134–141. doi: 10.1046/j.1365-2249.2001.01487.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Gonzalez-Alvaro I, Ortiz AM, Garcia-Vicuña R, Balsa A, Pascual-Salcedo D, Laffon A. Increased serum levels of interleukin-15 in rheumatoid arthritis with long- term disease. Clin Exp Rheumatol. 2003;21:639–642. [PubMed] [Google Scholar]
- 173.Baslund B, Tvede N, Danneskiold-Samsoe B, Larsson P, Panayi G, Petersen J, Petersen LJ, Beurskens FJ, Schuurman J, van de Winkel JG, et al. Targeting interleukin-15 in patients with rheumatoid arthritis: a proof-of-concept study. Arthritis Rheum. 2005;52:2686–2692. doi: 10.1002/art.21249. [DOI] [PubMed] [Google Scholar]
- 174.Waldmann TA. Targeting the interleukin-15/interleukin-15 receptor system in inflammatory autoimmune diseases. Arthritis Res Ther. 2004;6:174–177. doi: 10.1186/ar1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Niu X, He D, Zhang X, Yue T, Li N, Zhang JZ, Dong C, Chen G. IL-21 regulates Th17 cells in rheumatoid arthritis. Hum Immunol. 2010;71:334–341. doi: 10.1016/j.humimm.2010.01.010. [DOI] [PubMed] [Google Scholar]
- 176.Young DA, Hegen M, Ma HL, Whitters MJ, Albert LM, Lowe L, Senices M, Wu PW, Sibley B, Leathurby Y, et al. Blockade of the interleukin-21/interleukin-21 receptor pathway ameliorates disease in animal models of rheumatoid arthritis. Arthritis Rheum. 2007;56:1152–1163. doi: 10.1002/art.22452. [DOI] [PubMed] [Google Scholar]
- 177.Jüngel A, Distler JH, Kurowska-Stolarska M, Seemayer CA, Seibl R, Forster A, Michel BA, Gay RE, Emmrich F, Gay S, et al. Expression of interleukin-21 receptor, but not interleukin-21, in synovial fibroblasts and synovial macrophages of patients with rheumatoid arthritis. Arthritis Rheum. 2004;50:1468–1476. doi: 10.1002/art.20218. [DOI] [PubMed] [Google Scholar]
- 178.Monteleone G, Pallone F, Macdonald TT. Interleukin-21 as a new therapeutic target for immune-mediated diseases. Trends Pharmacol Sci. 2009;30:441–447. doi: 10.1016/j.tips.2009.05.006. [DOI] [PubMed] [Google Scholar]
- 179.Wendling D. Interleukin 23: a key cytokine in chronic inflammatory disease. Joint Bone Spine. 2008;75:517–519. doi: 10.1016/j.jbspin.2008.03.004. [DOI] [PubMed] [Google Scholar]
- 180.Kageyama Y, Kobayashi H, Kato N. Infliximab treatment reduces the serum levels of interleukin-23 in patients with rheumatoid arthritis. Mod Rheumatol. 2009;19:657–662. doi: 10.1007/s10165-009-0217-6. [DOI] [PubMed] [Google Scholar]
- 181.Ju JH, Cho ML, Moon YM, Oh HJ, Park JS, Jhun JY, Min SY, Cho YG, Park KS, Yoon CH, et al. IL-23 induces receptor activator of NF-kappaB ligand expression on CD4+ T cells and promotes osteoclastogenesis in an autoimmune arthritis model. J Immunol. 2008;181:1507–1518. doi: 10.4049/jimmunol.181.2.1507. [DOI] [PubMed] [Google Scholar]
- 182.Tang C, Chen S, Qian H, Huang W. Interleukin-23: as a drug target for autoimmune inflammatory diseases. Immunology. 2012;135:112–124. doi: 10.1111/j.1365-2567.2011.03522.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Krausz S, Boumans MJ, Gerlag DM, Lufkin J, van Kuijk AW, Bakker A, de Boer M, Lodde BM, Reedquist KA, Jacobson EW, et al. Brief report: a phase IIa, randomized, double-blind, placebo-controlled trial of apilimod mesylate, an interleukin-12/interleukin-23 inhibitor, in patients with rheumatoid arthritis. Arthritis Rheum. 2012;64:1750–1755. doi: 10.1002/art.34339. [DOI] [PubMed] [Google Scholar]
- 184.Hwang SY, Kim HY. Expression of IL-17 homologs and their receptors in the synovial cells of rheumatoid arthritis patients. Mol Cells. 2005;19:180–184. [PubMed] [Google Scholar]
- 185.Chabaud M, Durand JM, Buchs N, Fossiez F, Page G, Frappart L, Miossec P. Human interleukin-17: A T cell-derived proinflammatory cytokine produced by the rheumatoid synovium. Arthritis Rheum. 1999;42:963–970. doi: 10.1002/1529-0131(199905)42:5<963::AID-ANR15>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 186.Kirkham BW, Lassere MN, Edmonds JP, Juhasz KM, Bird PA, Lee CS, Shnier R, Portek IJ. Synovial membrane cytokine expression is predictive of joint damage progression in rheumatoid arthritis: a two-year prospective study (the DAMAGE study cohort) Arthritis Rheum. 2006;54:1122–1131. doi: 10.1002/art.21749. [DOI] [PubMed] [Google Scholar]
- 187.Genovese MC, Van den Bosch F, Roberson SA, Bojin S, Biagini IM, Ryan P, Sloan-Lancaster J. LY2439821, a humanized anti-interleukin-17 monoclonal antibody, in the treatment of patients with rheumatoid arthritis: A phase I randomized, double-blind, placebo-controlled, proof-of-concept study. Arthritis Rheum. 2010;62:929–939. doi: 10.1002/art.27334. [DOI] [PubMed] [Google Scholar]
- 188.Hueber W, Patel DD, Dryja T, Wright AM, Koroleva I, Bruin G, Antoni C, Draelos Z, Gold MH, Durez P, et al. Effects of AIN457, a fully human antibody to interleukin-17A, on psoriasis, rheumatoid arthritis, and uveitis. Sci Transl Med. 2010;2:52ra72. doi: 10.1126/scitranslmed.3001107. [DOI] [PubMed] [Google Scholar]
- 189.van Roon JA, van Roy JL, Gmelig-Meyling FH, Lafeber FP, Bijlsma JW. Prevention and reversal of cartilage degradation in rheumatoid arthritis by interleukin-10 and interleukin-4. Arthritis Rheum. 1996;39:829–835. doi: 10.1002/art.1780390516. [DOI] [PubMed] [Google Scholar]
- 190.Chomarat P, Vannier E, Dechanet J, Rissoan MC, Banchereau J, Dinarello CA, Miossec P. Balance of IL-1 receptor antagonist/IL-1 beta in rheumatoid synovium and its regulation by IL-4 and IL-10. J Immunol. 1995;154:1432–1439. [PubMed] [Google Scholar]
- 191.Kamiya S, Okumura M, Chiba Y, Fukawa T, Nakamura C, Nimura N, Mizuguchi J, Wada S, Yoshimoto T. IL-27 suppresses RANKL expression in CD4+ T cells in part through STAT3. Immunol Lett. 2011;138:47–53. doi: 10.1016/j.imlet.2011.02.022. [DOI] [PubMed] [Google Scholar]
- 192.Chaturvedi V, Collison LW, Guy CS, Workman CJ, Vignali DA. Cutting edge: Human regulatory T cells require IL-35 to mediate suppression and infectious tolerance. J Immunol. 2011;186:6661–6666. doi: 10.4049/jimmunol.1100315. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 193.Kochetkova I, Golden S, Holderness K, Callis G, Pascual DW. IL-35 stimulation of CD39+ regulatory T cells confers protection against collagen II-induced arthritis via the production of IL-10. J Immunol. 2010;184:7144–7153. doi: 10.4049/jimmunol.0902739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Han Z, Boyle DL, Chang L, Bennett B, Karin M, Yang L, Manning AM, Firestein GS. c-Jun N-terminal kinase is required for metalloproteinase expression and joint destruction in inflammatory arthritis. J Clin Invest. 2001;108:73–81. doi: 10.1172/JCI12466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Feldmann M, Andreakos E, Smith C, Bondeson J, Yoshimura S, Kiriakidis S, Monaco C, Gasparini C, Sacre S, Lundberg A, et al. Is NF-kappaB a useful therapeutic target in rheumatoid arthritis? Ann Rheum Dis. 2002;61 Suppl 2:ii13–ii18. doi: 10.1136/ard.61.suppl_2.ii13. [DOI] [PMC free article] [PubMed] [Google Scholar]