

Key Points
Mice constitutively developing a JAK2V617F-induced PV exhibit a bleeding tendency combined with the accelerated formation of unstable clots.
Hemostatic defects are not concomitant with JAK2V617F expression, suggesting they are not directly caused by the mutation.
Abstract
Thrombosis is common in patients suffering from myeloproliferative neoplasm (MPN), whereas bleeding is less frequent. JAK2V617F, the main mutation involved in MPN, is considered as a risk factor for thrombosis, although the direct link between the mutation and hemostatic disorders is not strictly established. We investigated this question using conditional JAK2V617F knock-in mice with constitutive and inducible expression of JAK2V617F in hematopoietic cells, which develop a polycythemia vera (PV)-like disorder evolving into myelofibrosis. In vitro, thrombosis was markedly impaired with an 80% decrease in platelet-covered surface, when JAK2V617F blood was perfused at arterial shear over collagen. JAK2V617F platelets presented only a moderate glycoprotein (GP) VI deficiency not responsible for the defective platelet accumulation. In contrast, a decreased proportion of high-molecular-weight von Willebrand factor multimers could reduce platelet adhesion. Accordingly, the tail bleeding time was prolonged. In the FeCl3-induced thrombosis model, platelet aggregates formed rapidly but were highly unstable. Interestingly, vessels were considerably dilated. Thus, mice developing PV secondary to constitutive JAK2V617F expression exhibit a bleeding tendency combined with the accelerated formation of unstable clots, reminiscent of observations made in patients. Hemostatic defects were not concomitant with the induction of JAK2V617F expression, suggesting they were not directly caused by the mutation but were rather the consequence of perturbations in blood and vessel homeostasis.
Introduction
Myeloproliferative neoplasms (MPNs) are clonal diseases arising from hematopoietic stem cells harboring molecular defects that promote an excessive and cytokine-deregulated production of mature myeloid blood cells. According to the classification of the World Health Organization, classical BCR-ABL-negative MPN includes polycythemia vera (PV), essential thrombocythemia (ET), and primary myelofibrosis (PMF). The unique acquired somatic JAK2V617F mutation is present in >95% of PV and 50% of ET and PMF patients.1-4
The V617F mutation in the pseudokinase domain constitutively activates cytokine signaling via the formation of active complexes with type I receptors, such as the erythropoietin receptor or the thrombopoietin (TPO) receptor.1
Disease-related hemostatic complications impair life expectancy of patients with PV and ET. Arterial and venous thrombosis are the main cause of death and morbidity and are often the initial event leading to diagnosis.5 Their incidence ranges from 12% to 39% in PV and 11% to 25% in ET.6 Hemorrhages are reported with a low ∼3% incidence in ET and PV but more frequently (∼10%) in PMF patients.7
Mechanisms underlying the MPN thrombotic diathesis are still largely elusive, and biological studies have provided data that were often conflicting or of uncertain clinical or pathogenic relevance. However, thrombosis appears to be more frequent in patients with the JAK2V617F mutation. This feature has been critically reevaluated in 3 independent large meta-analyses that reported an increased risk of venous and arterial thrombosis (odds ratio 2.09 and 1.68, 1.96 and 2.5, respectively),8,9 of thrombosis at presentation (odds ratio 1.88),8 and an increased rate of thrombotic events (32% vs 20%)10 in patients with the JAK2V617F mutation compared with their wild-type (WT) counterparts. Furthermore, rare ET patients harboring a homozygous JAK2V617F mutation had a higher risk of thrombosis than WT and heterozygous subjects and presented an increased risk of thrombosis recurrence.11,12 Platelets are assumed to impact the risk of thrombosis, not only because they are increased in number but also because of their hyperreactivity, as suggested by the increased P-selectin exposure on their surface.13-15 Enhanced urinary excretion of thromboxane metabolites in untreated ET patients is also in favor of in vivo platelet activation.16 Therefore, an association between the JAK2V617F mutation and platelet activation is suggested, but the direct assessment of this mutation’s effect on platelet function in patients with MPNs is skewed by the variable amplification of the JAK2V617F mutant clone, additional molecular defects, and the frequently associated prophylactic treatment of thrombosis.
The recently described knock-in (KI) mouse MPN models, in which JAK2V617F expression is inducible and tissue specific, offer an attractive alternative for the precise analysis of the impact of the mutation on hemostatic functions.17-20 These mice faithfully model human PV evolving into myelofibrosis (MF), and reports of cardiac thrombosis and gangrenous bowel are consistent with clinical events observed in MPN patients.17,19,20 Our objective was to use these KI mice to assess the role of JAK2V617F in the deregulation of hemostasis, with a special interest in platelets.
Materials and methods
A detailed methods section can be found in the supplemental Methods available on the Blood Web site.
Animal models
All animal experiments were carried out at the animal facilities of the Institut Gustave Roussy and of Inserm U1148 and approved by the local review boards (protocol no. 2012-061), under the conditions established by the European Community (Directive 86/609/CCE).
JAK2V617F KI mice.
Conditional JAK2V617F KI mice were crossed with VavCre or SCLCreERt transgenic mice for fetal or tamoxifen-inducible adult expression of JAK2V617F in hematopoietic tissues, respectively. Bone marrow (BM) cells from VavCre/JAK2V617F KI or SCLCreERt /JAK2V617F KI mice were grafted into lethally irradiated WT recipients leading to constitutive or inducible heterozygous expression of the mutation in hematopoietic cells only. Recipients from both donor mice develop a PV-like disease evolving into MF.18,20 Studies were performed 9 weeks after transplantation of VavCre/JAK2V617F KI BM cells or 6, 13, 30, and 60 days after tamoxifen administration to SCLCreERt/JAK2V617F KI recipient mice. Control mice were WT mice grafted with WT BM cells or vehicle-treated SCLCreERt/JAK2V617F KI recipient mice.
TPOhigh model.
BM cells transduced with a retrovirus expressing the murine TPO gene were grafted into irradiated WT recipient mice resulting in an ET-like disease rapidly evolving into MF, as previously described.21 These mice were used 12 weeks posttransplantation.
Secondary polycythemia model.
WT mice were treated by intraperitoneal injection of human recombinant EPO (epoetine alfa 5000 UI/kg) every 2 days during 3 weeks.
Results
Hematologic parameters in JAK2V617F KI mice
As previously reported, a PV-like disorder was observed 2 months after transplantation in VavCre/JAK2V617F BM recipients20 but also in SCLCreERt/JAK2V617F BM recipient mice treated with tamoxifen (supplemental Table 1A-B). The size of the spleen was doubled in VavCre/JAK2V617F mice as compared with WT (supplemental Figure 1Ai).
Transplantation of TPO-transduced BM cells (TPOhigh model)21 resulted, 12 weeks after transplantation, in a reduction of the initial thrombocytosis to normal platelet levels (1029 ± 492 × 106/mL), variable leukocytosis (14 ± 12 × 106/mL), severe anemia (hematocrit 19 ± 6.6%), and a 1.7-fold enlargement of the spleen. EPO-treated mice exhibited isolated erythrocytosis (red blood cell [RBC] concentration: 1150 ± 66 × 109/mL, hematocrit 60 ± 5.5%). Only slight splenomegaly was observed in EPO-treated mice (supplemental Figure 1Aiii).
The tail bleeding time (TBT) from VavCre/JAK2V617F KI mice was assessed 2 months posttransplantation. It was characterized by frequent, short transient arrests followed by immediate intense and prolonged bleeding that required stopping by the experimenter after 15 minutes (Figure 1Ai). Accordingly, the volume of blood lost was significantly increased (Figure 1Aii). In vehicle-treated SCLCreERt/JAK2V617F KI mice or tamoxifen-treated WT mice (not shown), neither the TBT nor the blood loss significantly changed whatever the time after induction. In contrast, the TBT and the volume of blood lost progressively increased from 2 weeks after treatment by tamoxifen of SCLCreERt/JAK2V617F KI mice, and 2 months posttreatment, bleeding needed to be stopped as in VavCre/JAK2V617F KI mice (Figure 1Bi-ii). The frequency of transient bleeding interruptions increased from treatment to day 30 and decreased at day 60 when bleeding became continuous (Figure 1C).
Figure 1.

TBT. The TBT was measured 2 months post-BM grafting in WT and VavCre/JAK2V617F KI mice (A, n = 8) and at different times after administration of tamoxifen (TM, n = 8) or vehicle (VH, n = 22) in SCLCreERt/JAK2V617F KI mice (B). The arrow at 15 minutes indicates that bleeding was arrested by the experimenter. Each point represents an individual mouse. The volume of blood lost in 15 minutes was measured in VavCre/JAK2V617F KI (Aii) and in SCLCreERt/JAK2V617F KI mice (Bii). (C) The frequency of transient bleeding arrests observed in SCLCreERt/JAK2V617F KI mice at different times after administration of tamoxifen was quantified. *P < .05; **P < .004; ***P < .0001.
Thrombosis is impaired ex vivo in JAK2V617F mice
When whole blood was perfused at 1500 s−1 over collagen, WT platelets rapidly adhered to the surface and formed aggregates. In contrast, the adhesion of VavCre/JAK2V617F platelets was delayed and the formation of stable aggregates was markedly impaired (Figure 2A). After 5 minutes of flow, surface coverage was decreased by 80% for VavCre/JAK2V617F KI compared with WT platelets (Figure 2B). The capacity of platelets from SCLCreERt/JAK2V617F KI mice to adhere to collagen dramatically dropped 2 weeks after induction of the mutation and was comparable to that of platelets of VavCre/JAK2V617F KI (Figure 2C-D).
Figure 2.

Collagen-induced in vitro thrombosis. Whole blood was perfused for 5 minutes over an immobilized collagen surface at 1500 s−1. The adhesion of DiOC6-labeled platelets was recorded. (A) Views of platelet adhesion at different times (30 seconds, 1 minute, 2 minutes, and 5 minutes) of 1 out of 6 significant experiments from VavCre/JAK2V617F (KI) and WT mice are shown. Original magnification ×20 for all panels. Quantification of the covered surface obtained with VavCre/JAK2V617F platelets (KI, n = 6) and of SCLCreERt/JAK2V617F platelets (TM, n = 4 per group) and their matched controls is shown in panels B and C, respectively. **P < .007. (D) Mosaic views of SCLCreERt/JAK2V617F (TM) platelets and their controls (VH) are shown at day 6 (D6) and day 60 (D60).
Thrombus formation was further analyzed in vitro by thromboelastometry on whole blood from VavCre/JAK2V617F KI mice and their matched controls (supplemental Figure 2). The clotting time and clot formation time of JAK2V617F blood were both prolonged as compared with controls, whereas the maximum clot firmness was decreased, and maximum lysis increased.
JAK2V617F KI platelets show significantly reduced activation response
The platelet surface expression levels of the main glycoproteins (GPs), GPIbα, GPV, and integrins α2 and αIIbβ3, were similar to controls in VavCre/JAK2V617F, EPO-treated, TPOhigh, and SCLCreERt/JAK2V617F KI mice, whatever the time of analysis (supplemental Table 2).
In contrast, surface-expressed GPVI was systematically decreased in VavCre/JAK2V617F KI mice compared with control mice as indicated by a 40% mean reduction of the GPVI labeling, although variations between mice were observed (Figure 3A). Accordingly, on immunoblots, the intensity of the GPVI band was deceased to 45 ± 12% of the mean value obtained for WT platelets (Figure 3Biii). Furthermore, the FcRγ chain that is constitutively associated with GPVI was also decreased by 36 ± 10% in VavCre/JAK2V617F KI platelets (Figure 3Bi). The treatment of control platelets by N-ethylmaleimide (NEM) resulted in the appearance of a band corresponding to the intracellular GPVI remnant that reflects GPVI shedding. This band was not observed in the extracts of JAK2V617F platelets unless they were treated by NEM, ruling out the hypothesis that GPVI was cleaved in vivo by metalloproteases (Figure 3Bii). In SCLCreERt/JAK2V617F KI mice, the expression of GPVI remained normal until 2 months after tamoxifen administration and then dropped to the same level as observed on VavCre/JAK2V617F KI platelets (Figure 3C). Of note, GPVI was also decreased by 20% on platelets from TPOhigh mice but was normal on platelets from EPO-treated mice (Figure 3D).
Figure 3.

JAK2V617F platelets present a GPVI deficiency. (A) GPVI expression was quantified by flow cytometry (mean fluorescence intensity, MFI) on the platelets of WT (n = 11) and VavCre/JAK2V617F (n = 24) mice 2 months after BM graft. ***P < .0003. (Bi) Representative (of 3) immunoblots of platelet extracts (10 µg) from 4 different WT mice and 4 different VavCre/JAK2V617F (KI) mice are shown. The same membrane was stripped and reblotted with antibodies against GPVI (JAQ1), the FcRγ chain, and β-actin. (Bii) The shedding of GPVI was analyzed. One representative immunoblot is shown (n = 3). Platelets from WT mice and VavCre/JAK2 V617F (KI) mice were treated or not with NEM. Platelet extracts (10 µg) were immunoblotted using a polyclonal antibody to the intracellular domain of GPVI. GPVI-IC indicates the GPVI remnant cytoplasmic domain that remained associated with platelets after shedding of the extracellular domain. (Biii) Densitometric analysis of the GPVI band from 4 different mice per group. *P < .03. (C) Surface expression of GPVI was measured on SCLCreERt /JAK2 V617F (TM) at different times after administration of tamoxifen (n = 7 for each group). **P < .002. (D) Platelet surface expression of GPVI in TPOhigh (n = 9) and EPO-treated mice (n = 3) is shown.
In nonstimulated whole blood, the levels of P-selectin and activated integrin αIIbβ3 were low both on JAK2V167F and control platelets (Figure 4A). After stimulation by convulxin, rhodocytin, or thrombin receptor-activating peptide 4 (TRAP4), integrin αIIbβ3 became activated but to a lesser extent on VavCre/JAK2V617F platelets than in controls, and similar data were obtained for P-selectin exposure (data not shown). In SCLCreERt/JAK2V617F KI mice, alterations in P-selectin exposure in response to convulxin (Figure 4Bi) or rhodocytin (Figure 4Bii) were only observed 2 weeks after induction by tamoxifen. Similar data were obtained for the activation of integrin αIIbβ3, and dose-response curves showed that increasing convulxin doses did not restore a full response (Figure 4C).
Figure 4.

P-selectin exposure or JON/A binding on resting and activated platelets. Whole blood was incubated with buffer, convulxin (Cvx, 2.5 µg/mL), rhodocytin (rhodo, 50 nM), or TRAP4 (1000 µM) for 15 minutes at room temperature before measurement of P-selectin or JON/A (GPIIbIIa active form) exposure at the platelet surface. (A) Experiments performed in VavCre/JAK2V617F mice (KI) and their controls (WT). ***P < .0001. (B) Experiments were performed in SCLCreERt/JAK2V617F mice at different times after administration of tamoxifen (white squares) or vehicle (black circles), incubated with convulxin (i) or rhodocytin (ii). (C) Surface expression of the active form of integrin αIIbβ3 (JON/A) was quantified on SCLCreERt/JAK2V617F platelets (TM) activated in whole blood by 3 doses of convulxin (0.5, 1, and 2.5 µg/mL) at different times after administration of tamoxifen.
VWF proteolysis in JAK2V617F mice
Because neither the moderate GPVI deficiency nor the platelet hyporesponsiveness was sufficient to explain the defect in collagen-induced thrombosis in flow conditions, we analyzed plasma von Willebrand factor (VWF) in VavCre/JAK2V617F and SCLCreERt/JAK2V617F KI mice (Figure 5). The plasma concentration of VWF was not different in VavCre/JAK2V617F compared with WT mice, but electrophoretic analysis revealed a decreased intensity of the bands of high molecular weight, together with an increased intensity of medium- and low-sized multimers (Figure 5B). In SCLCreERt/JAK2V617F KI mice, the plasma concentration of VWF tended to increase with time after treatment by tamoxifen (Figure 5Aii). The proportion of low-molecular-weight multimers increased from day 13 posttreatment (Figure 5C). These data indicate an enhanced proteolysis of VWF in JAK2V617F-expressing mice.
Figure 5.
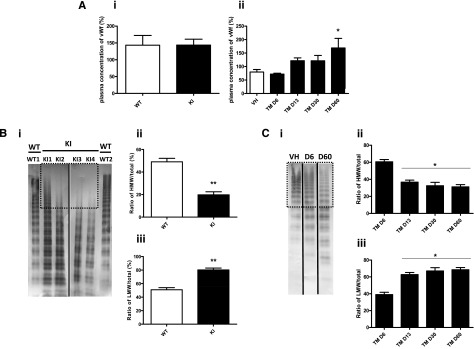
Von Willebrand analysis in JAK2V617F mice. (A) Plasma concentrations of VWF in VavCre/JAK2V617F (Ai, KI, n = 21), SCLCreERt/JAK2V617F (Aii, TM, n = 4 per group), and their controls. Mouse plasma samples were analyzed 2-3 months after BM graft. *P < .03. (Bi) Representative electrophoreses (n = 3) of the multimeric structure of VWF from 2 different WT mice and 4 different VavCre/JAK2V617F (KI). The dotted line square represents the decreased intensity of the high-molecular-weight bands (HMW). Quantification of HMW (Bii) and low-molecular-weight bands (LMW) (Biii) of VWF from 9 VavCre/JAK2V617F (KI) and WT. **P < .002. (Ci) Representative electrophoreses of the multimeric structure of VWF from SCLCreERt/JAK2V617F mice at D6 and D60 compared with controls (VH). Quantification of HMW (Cii) and LMW bands (Ciii) of VWF from 4 SCLCreERt/JAK2V617F per group at different times after treatment by tamoxifen.
Vascular abnormalities in JAK2V617F mice
When mesenteric vessels were exposed to study in vivo thrombosis, we observed that the diameter of the vessels was increased in VavCre/JAK2V617F KI mice as compared with controls (Figure 6A). In addition, a similar increased diameter of mesenteric vessels was observed in EPO-treated mice but not in TPOhigh mice, suggesting an adaptive process to the high hematocrit (Figure 6Ai). In SCLCreERt/JAK2V617F mice, the diameters of mesenteric vessels were normal 1 month after tamoxifen administration but were increased as in VavCre/JAK2V617F KI mice 2 months after tamoxifen administration (Figure 6B).
Figure 6.

Vasodilation in JAK2V617F mice. (Ai) Representative images of the macroscopic aspect of mesenteric vessels from VavCre/JAK2V617F (KI), EPO-treated, and TPOhigh mice. Bars represent 5 mm. (Aii) Diameters of mesenteric VavCre/JAK2V617F vessels (KI) and matched controls (CTR) (n = 12). ***P < .0001. (B) Representative images showing that the increase in diameter of mesenteric vessel of SCLCreERt/JAK2V617F mice appeared 60 days (D60) after treatment by tamoxifen. (C) Representative transverse sections (n = 3) of the aorta from 1 WT and 1 VavCre/JAK2V617F (hematoxylin and eosin staining). Bars represent 500 µm.
Increased vessel diameter was also observed in other vascular territories, such as the aorta and ear vessels. Histologic analysis of aortic sections from VavCre/JAK2V617F mice showed an increased luminal surface, and the vessel wall presented signs of distension: in the media, the numbers of smooth muscle cells and elastic laminae were unchanged compared with control mice, but they appeared distended (Figure 6C). We suspected that the increased vascular diameter represented a process of adaptation to the increased blood viscosity (approximately twofold) because of the high hematocrit. Indeed, VavCre/JAK2V617F KI mice had a normal heart rate (492 ± 15.28 in WT mice vs 509 ± 85.58 bpm in KI mice) and electrocardiogram. Furthermore, calculation of the shear indicated that, because of this dilatation, it was increased by only 35% in the aortas of VavCre/JAK2V617F KI mice compared with controls.
Thrombosis is accelerated in vivo, but thrombi are unstable in JAK2V617F mice
An FeCl3-induced thrombosis model was then used to evaluate the influence of JAK2V617F on localized thrombus formation. Vessels of similar diameter were selected in WT and KI mice. Platelet accumulation started very rapidly, only a few minutes after the injury in VavCre/JAK2V617F KI mice, and time to occlusion was shorter than in control mice (19.1 ± 5.0 minutes, n = 7 vs 39.2 ± 5.8 minutes, n = 6) (Figure 7A). In SCLCreERt/JAK2V617F KI mice, the rate of thrombus formation was identical to controls up to 1 month after tamoxifen administration; however, 1 month later, it was increased as in VavCre/JAK2V617F KI mice. Once the thrombus was formed, it was stable in control animals. However, in VavCre/JAK2V617F KI mice and SCLCreERt/JAK2V617F KI mice 2 months postinduction, we observed images of thrombolysis (Figure 7B) and also of embolization (Figure 7C).
Figure 7.
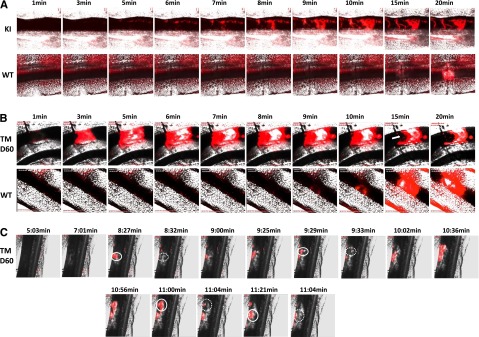
In vivo thrombosis. Representative images of n = 3 (for each mouse) of in vivo thrombosis experiments. FeCl3 injury of mesenteric vessels was performed in VavCre/JAK2V617F mice (A) or SCLCreERt/ JAK2V617F mice 2 months after treatment by tamoxifen (B and C). The accumulation of rhodamin 6G-stained platelets (in red) was recorded. The white arrow in panel B indicates signs of accelerated thrombus lysis, and the white circles in panel C indicate thrombus embolization. Note that the time to occlusion in JAK2V617F KI mice is reduced as compared with their controls. Original magnification ×5 for all panels.
It is noteworthy that platelet accumulation at the site of vascular injury occurred at a normal rate in TPOhigh mice. However, complete occlusion was hardly ever reached, whereas EPO- and vehicle-treated mice were indistinguishable with regard to occlusion time and stability of thrombi, strongly suggesting that erythrocytosis was not involved in the thrombus defects observed in JAK2V617F KI mice (supplemental Figure 3).
Discussion
Arterial and venous thrombosis is the main cause of death and morbidity in PV and ET, whereas hemorrhages are more often observed in PMF. However, because of the heterogeneity of patients and associated prophylactic treatment, these important health issues are difficult to investigate in human patients. JAK2V617F KI mice were generated, and several studies have shown that they faithfully model human PV, evolving into MF. Furthermore, lethality in these models is strongly suspected to be of hemostatic/thrombotic origin.17,19 Therefore, investigation of the role of JAK2V617F expression in hemostasis and thrombosis using this mouse model is justified. Both constitutive (VavCre) and inducible (SCLCreERt) JAK2V617F KI models were studied to determine the impact of JAK2V617F on these parameters with a special focus on platelet function. VavCre or SCLCreERt mice express the recombinase Cre in hematopoietic cells but also in some endothelial cells. Transplantations of BM cells taken from VavCre/JAK2V617F KI or SCLCreERt/JAK2V617F KI mice were performed in order to express the JAK2V617F mutation exclusively in hematopoietic cells. Furthermore, SCLCreERt/JAK2V617F KI, in contrast to VavCre/JAK2V617F KI, allows expression of the mutation in adult mice as in sporadic MPN with dissociation of the direct effect of JAK2 activation from the indirect effect of the resulting long-term disease. Finally, we evaluated the relative roles of thrombocytosis/MF or erythrocytosis in the thrombotic defects by using 2 non-JAK2V617F models: the TPOhigh retroviral (RV) model or EPO-treated mice, respectively.
We report an increased diameter of the vessels in JAK2V617F KI mice in different vascular territories including mesentery, ear, and aorta. Histology performed on transverse sections indicated that the increased diameter was attributable to distension of the vascular wall, which was not modified in terms of numbers of cells and elastic laminae, but both had a stretched appearance. Regarding the morphology of vessels in patients, most studies have been performed in the BM. Zetterberg et al observed vessels of larger perimeter in the BM of patients with MF.22 Also, interestingly, a reduction in flow-mediated vasodilatation was observed in PV patients consistent with constitutive dilatation.23 The observation of similarly dilated vessels in EPO-treated mice, but not in TPOhigh mice, is consistent with the proposal that vasodilatation represents a process of adaptation to the increased blood viscosity. Vasodilatation reduces vascular resistance and shear forces. Indeed, recording the electrocardiogram showed no change in heart rate or signs of overload in JAK2V617F KI mice. The observation that the vessels of EPO-treated mice are similarly dilated to those of JAK2V617F KI mice, whereas EPO-treated mice do not present hemostatic alterations, rules out the possibility that the changes in the vessel diameter are sufficient to impact hemostasis and thrombosis.
Hemostasis and thrombosis were analyzed in vitro and in vivo. Globally, VavCre/JAK2V617F KI mice were characterized by (1) a marked deficiency in thrombus formation in vitro in collagen-coated flow chambers, (2) a mild GPVI deficiency, (3) hyporesponsive platelets, (4) a decreased plasma content in large VWF multimers, (5) a prolonged bleeding time, and (6) the rapid formation of occlusive but unstable thrombi in FeCl3-injured mesenteric vessels. In SCLCreERt/JAK2V617F KI mice, impaired in vitro thrombosis, low platelet reactivity, VWF proteolysis, and prolonged bleeding time occurred 2 weeks after induction of the mutation by tamoxifen. A 2-month delay was necessary to observe the GPVI deficiency, vasodilation, and the rapid in vivo formation of unstable thrombi. These observations call for several comments: (1) Only a few JAK2V617F platelets attached to collagen, and they were unable to form thrombi when using whole blood at arterial shear rates; because the appearance of the moderate GPVI deficiency and of the vasodilatation is markedly delayed with respect to the defect in thrombus formation (day 60 vs day 13), their responsibility in the inability of platelets to form thrombi on collagen can be ruled out. (2) Controversial data have been reported regarding GP expression at the platelet surface in PV, ET,13,24-26 and PMF13 patients; however, we found only a mild GPVI deficiency in JAK2V617F KI mice. Such a defect has been previously reported in PV and ET patients27 and is unlikely to affect adhesion and activation in thrombus formation according to a previous report indicating that a 20% expression of GPVI is sufficient to permit platelet activation by collagen.27 Because the GPVI deficiency occurred late in the course of the disease when signs of MF were observed, we hypothesized that the GPVI deficiency could be related to fibrosis. The observation that platelets from TPOhigh mice also exhibited significant GPVI deficiency supports this hypothesis. Cleavage of the GPVI extracellular domain was undetectable in JAK2V617F platelets, arguing against GPVI shedding as a cause of platelet GPVI deficiency. GPVI appears late in megakaryocytic maturation, and its deficiency may result from impaired/dysmorphic megakaryopoiesis, as reported in MF. Alternatively, GPVI may be internalized and degraded, as described for antibody-induced GPVI deficiency, but requires platelet activation.28
The defective in vitro thrombosis and low responsiveness of JAK2V617F platelets occurred simultaneously in SCLCreERt/JAK2V617F KI mice. In PV and ET patients, platelets have been proposed to be less responsive in vitro as a consequence of their in vivo activation,29,30 with elevated markers of platelet activation reported in some, but not all, studies on PV,13,31 ET,16,32,33 and PMF13 patients. In contrast to these observations, no increased P-selectin expression was observed on platelets from VavCre/JAK2V617F and SCLCreERt/JAK2V617F KI mice. Accordingly, integrin αIIbβ3 was not activated on freshly drawn JAK2V617F platelets. The impaired responses of JAK2V617F platelets to several agonists are thus not the consequence of a desensitization process.
In VavCre/JAK2V617F, the content in hemostatically active VWF multimers was severely reduced. In SCLCreERt/JAK2V617F KI mice, the proportion of highly reactive multimers started to decrease 2 weeks after treatment by tamoxifen. The electrophoretic pattern of VWF indicated that it had been submitted to increased proteolysis in JAK2V617F mice. It is likely that VWF multimer deficiency is involved in the impaired adhesion of JAK2V617F platelets in flow conditions. This is reminiscent of the acquired von Willebrand syndrome (AVWS) described in MPN patients with extremely high platelet counts causing an increased clearance of platelet-bound large VWF multimers.34,35 To determine whether VWF proteolysis is caused by the disintegrin and metalloprotease with thrombospondin type I repeats-13 (ADAMTS-13), or by other proteases, in increased shear conditions requires additional studies.
The prolonged TBT of JAK2V617F KI mice, also observed in the JAK2V617F RV model mice,36,37 is consistent with in vitro defective thrombus formation. In MPN patients, bleeding has been reported to be the consequence of an AVWS.7,38-40 Indeed, thrombus formation in vitro was measured using flow adjusted to provide a shear rate of 1500 s−1 with WT blood such as found in medium-sized arteries, conditions in which VWF is assumed to trigger the first contact between platelets and collagen.41 VWF must be even more important for platelet adhesion in the case of JAK2V617F blood, as the approximately twofold increased blood viscosity produces higher shear rates.
In contrast to the defective thrombus formation in vitro, the formation of occlusive platelet aggregates was considerably accelerated in response to vascular injury by FeCl3 in JAK2V617F KI mice. However, once formed, thrombi were less stable in JAK2V617F KI than in WT mice. Frequent signs of embolization were observed as well as clot lysis, a process never observed previously.42 The observation of transient arrests of tail bleeding before the reoccurrence of persistent bleeding is consistent with clot instability in JAK2V617F KI mice.
The differences between in vitro (defective) and in vivo (accelerated) thrombus formation can have many causes, including, in particular, the initiation mechanisms. (1) In the FeCl3 model used here, the event triggering the formation of the thrombus is complex and still a subject of debate, particularly regarding the earliest stages.43,44 RBCs interacting with endothelial cells appear to have the ability to participate in these early stages by recruiting platelets. On one hand, we have not observed accelerated thrombosis in EPO-treated mice, suggesting that an increased RBC count is not sufficient to accelerate platelet adhesion. On the other hand, the recent observation that RBC adhesion to endothelial laminin is increased, because of the phosphorylation of the Lutheran/basal cell adhesion molecule (Lu/BCAM) by the JAK2V617F/Rap1/Akt pathway,45 suggests that RBCs could contribute to the rapid initiation of thrombosis by FeCl3 in JAK2V617F KI mice. (2) Although thrombin formation is blocked when thrombosis is analyzed in anticoagulated blood in vitro, it is known to be important in FeCl3-induced thrombosis.42 Unfortunately, thrombin generation could not be tested because not enough plasma could be collected from high-hematocrit JAK2V617F KI blood, but thromboelastometry on whole anticoagulated blood showed that, in vitro, tissue factor–induced coagulation was delayed. However, the possibility that the oxidative stress attributable to FeCl3 would injure leukocytes in vivo, leading to the formation of neutrophil extracellular traps and activation of coagulation, should be considered.46 Indeed, activation of leukocytes in MPN patients has been reported, and leukocytosis is a powerful independent risk factor for thrombosis.47-50
An interesting point is also that, once formed, clots are unstable in JAK2V617F KI mice. Decreased firmness and accelerated clot lysis was indeed observed in whole blood by thromboelastometry. Together with the increased proteolysis of VWF, these observations evoke the activation of proteases, possibly released by leukocytes, capable of destabilizing platelet aggregates and of activating fibrinolysis in JAK2V617F KI mice. Further studies are necessary to elucidate the role of leukocytes in clot formation and instability.
Importantly, the effect of JAK2V617F on hemostasis was delayed with respect to the expression of JAK2V617F induced by the administration of tamoxifen to SCLCreERt/JAK2V617F KI mice: a 2-week delay was required to observe the defective in vitro thrombosis, prolonged bleeding time, low platelet reactivity, and VWF proteolysis; and a 2-month delay was necessary to observe the GPVI deficiency, vasodilation, and rapid in vivo formation of unstable thrombi. Together, these observations rule out a direct effect of the JAK2 mutation on hemostasis but are in favor of its role in changes associated with the progression of MPN. The observations that (1) EPO-driven erythrocytosis resulted in vasodilatation and (2) thrombocytosis from TPOhigh RV mice was associated with a GPVI deficiency are also consistent with indirect JAK2V617F effects.
In contrast to our results, Hobbs et al using a different JAK2V617F KI mouse model showed increased platelet reactivity to some agonists (collagen and thrombin) and decreased tail bleeding volume.51 Differences may be attributable to the ET phenotype of this model instead of the PV/PMF phenotype of our model. On the other hand, Etheridge et al,34 using Tie2Cre/JAK2V617F TG mice (expressing human JAK2V617F in endothelial and hematopoietic cells) displaying an ET/PMF phenotype, showed attenuated arterial thrombosis after injury and an AVWS. The fact that RBCs were not increased in these mice argues in favor of a contribution of RBCs to the initiation of thrombus formation in FeCl3-induced thrombosis in our PV model. Furthermore, transplantation experiments showed that delayed occlusion after injury required JAK2V617F expression both in hematopoietic and endothelial cells arguing, in agreement with our data, against the responsibility of an isolated JAK2V617F platelet defect. Interestingly, decreased clot stability was observed in Tie2Cre/JAK2V617F as in VavCre and SCLCreERt/JAK2V617F mice (this study), all models displaying a PMF-like phenotype.34
These results (1) emphasize the role of MPN disease phenotype and not JAK2V617F in the hemostasis disorders observed in different mouse models of MPN, (2) are consistent with the variability of the disorders observed in MPN patients, and (3) offer the opportunity to develop biomarkers for the preclinical study of thrombosis and to determine the etiology of disturbed hemostasis in MPN.
Acknowledgments
The authors thank Dr Joachim Goethert (University of Duisburg, Essen, Germany) for providing the HSC-SCLCreERt mice; Dr Warren Alexander (Walter Elisa Hall Institute, Melbourne, Australia) for the VavCre mice; the staff of the animal facilities of the Institut Gustave Roussy, directed by Patrick Gonin; Paulette Legendre (Inserm U770 Le Kremlin Bicêtre, France) for VWF analysis, Dr Katsue Suzuki-Inoue (University of Yamanashi, Japan) for the kind gift of rhodocytin; and Mary Osborne-Pellegrin for help in editing the manuscript.
This work was supported by INSERM and Université Paris Diderot, the Cancéropôle Ile-de-France, and the Ligue Nationale Contre le Cancer (labeled team 2009), as well as the Ministère de la Recherche et de l’Enseignement Supérieur (L.L.) and the French Group on Thrombosis and Haemostasis (GEHT) (L.L.).
Footnotes
The online version of this article contains a data supplement.
There is an Inside Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: L.L. designed and performed experiments, analyzed data, and wrote the manuscript; C.L. generated KI mice; C.V.D., V.O., and B.H.T.N. performed experiments; E.G. provided crucial reagents and analyzed data; W.V. provided funding and scientific advice; and J.-L.V. and M.J.-P. conceived the study, designed experiments, analyzed data, and wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Martine Jandrot-Perrus, Inserm U1148, Hôpital Bichat, 46, rue Henri Huchard, 75877 Paris cedex 18, France; e-mail: martine.jandrot-perrus@inserm.fr; and Jean-Luc Villeval, Inserm U1009, Institut Gustave Roussy, PR1, 114 rue Edouard Vaillant, 94805 Villejuif, France; e-mail: jean-luc.villeval@gustaveroussy.fr.
References
- 1.James C, Ugo V, Le Couédic JP, et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature. 2005;434(7037):1144–1148. doi: 10.1038/nature03546. [DOI] [PubMed] [Google Scholar]
- 2.Baxter EJ, Scott LM, Campbell PJ, et al. Cancer Genome Project. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet. 2005;365(9464):1054–1061. doi: 10.1016/S0140-6736(05)71142-9. [DOI] [PubMed] [Google Scholar]
- 3.Kralovics R, Passamonti F, Buser AS, et al. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med. 2005;352(17):1779–1790. doi: 10.1056/NEJMoa051113. [DOI] [PubMed] [Google Scholar]
- 4.Levine RL, Wadleigh M, Cools J, et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell. 2005;7(4):387–397. doi: 10.1016/j.ccr.2005.03.023. [DOI] [PubMed] [Google Scholar]
- 5.Elliott MA, Tefferi A. Thrombosis and haemorrhage in polycythaemia vera and essential thrombocythaemia. Br J Haematol. 2005;128(3):275–290. doi: 10.1111/j.1365-2141.2004.05277.x. [DOI] [PubMed] [Google Scholar]
- 6.Tefferi A. Polycythemia vera and essential thrombocythemia: 2012 update on diagnosis, risk stratification, and management. Am J Hematol. 2012;87(3):284–293. doi: 10.1002/ajh.23135. [DOI] [PubMed] [Google Scholar]
- 7.Kessler CM. Propensity for hemorrhage and thrombosis in chronic myeloproliferative disorders. Semin Hematol. 2004;41(2 suppl 3):10–14. doi: 10.1053/j.seminhematol.2004.02.008. [DOI] [PubMed] [Google Scholar]
- 8.Ziakas PD. Effect of JAK2 V617F on thrombotic risk in patients with essential thrombocythemia: measuring the uncertain. Haematologica. 2008;93(9):1412–1414. doi: 10.3324/haematol.12970. [DOI] [PubMed] [Google Scholar]
- 9.Dahabreh IJ, Zoi K, Giannouli S, Zoi C, Loukopoulos D, Voulgarelis M. Is JAK2 V617F mutation more than a diagnostic index? A meta-analysis of clinical outcomes in essential thrombocythemia. Leuk Res. 2009;33(1):67–73. doi: 10.1016/j.leukres.2008.06.006. [DOI] [PubMed] [Google Scholar]
- 10.Lussana F, Caberlon S, Pagani C, Kamphuisen PW, Büller HR, Cattaneo M. Association of V617F Jak2 mutation with the risk of thrombosis among patients with essential thrombocythaemia or idiopathic myelofibrosis: a systematic review. Thromb Res. 2009;124(4):409–417. doi: 10.1016/j.thromres.2009.02.004. [DOI] [PubMed] [Google Scholar]
- 11.Vannucchi AM, Antonioli E, Guglielmelli P, et al. Clinical profile of homozygous JAK2 617V>F mutation in patients with polycythemia vera or essential thrombocythemia. Blood. 2007;110(3):840–846. doi: 10.1182/blood-2006-12-064287. [DOI] [PubMed] [Google Scholar]
- 12.De Stefano V, Za T, Rossi E, et al. GIMEMA CMD-Working Party. Recurrent thrombosis in patients with polycythemia vera and essential thrombocythemia: incidence, risk factors, and effect of treatments. Haematologica. 2008;93(3):372–380. doi: 10.3324/haematol.12053. [DOI] [PubMed] [Google Scholar]
- 13.Jensen MK, de Nully Brown P, Lund BV, Nielsen OJ, Hasselbalch HC. Increased platelet activation and abnormal membrane glycoprotein content and redistribution in myeloproliferative disorders. Br J Haematol. 2000;110(1):116–124. doi: 10.1046/j.1365-2141.2000.02030.x. [DOI] [PubMed] [Google Scholar]
- 14.Falanga A, Marchetti M, Vignoli A, Balducci D, Barbui T. Leukocyte-platelet interaction in patients with essential thrombocythemia and polycythemia vera. Exp Hematol. 2005;33(5):523–530. doi: 10.1016/j.exphem.2005.01.015. [DOI] [PubMed] [Google Scholar]
- 15.Arellano-Rodrigo E, Alvarez-Larrán A, Reverter JC, Villamor N, Colomer D, Cervantes F. Increased platelet and leukocyte activation as contributing mechanisms for thrombosis in essential thrombocythemia and correlation with the JAK2 mutational status. Haematologica. 2006;91(2):169–175. [PubMed] [Google Scholar]
- 16.Rocca B, Ciabattoni G, Tartaglione R, et al. Increased thromboxane biosynthesis in essential thrombocythemia. Thromb Haemost. 1995;74(5):1225–1230. [PubMed] [Google Scholar]
- 17.Akada H, Yan D, Zou H, Fiering S, Hutchison RE, Mohi MG. Conditional expression of heterozygous or homozygous Jak2V617F from its endogenous promoter induces a polycythemia vera-like disease. Blood. 2010;115(17):3589–3597. doi: 10.1182/blood-2009-04-215848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Marty C, Lacout C, Martin A, et al. Myeloproliferative neoplasm induced by constitutive expression of JAK2V617F in knock-in mice. Blood. 2010;116(5):783–787. doi: 10.1182/blood-2009-12-257063. [DOI] [PubMed] [Google Scholar]
- 19.Mullally A, Lane SW, Ball B, et al. Physiological Jak2V617F expression causes a lethal myeloproliferative neoplasm with differential effects on hematopoietic stem and progenitor cells. Cancer Cell. 2010;17(6):584–596. doi: 10.1016/j.ccr.2010.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hasan S, Lacout C, Marty C, et al. JAK2V617F expression in mice amplifies early hematopoietic cells and gives them a competitive advantage that is hampered by IFNα. Blood. 2013;122(8):1464–1477. doi: 10.1182/blood-2013-04-498956. [DOI] [PubMed] [Google Scholar]
- 21.Villeval JL, Cohen-Solal K, Tulliez M, et al. High thrombopoietin production by hematopoietic cells induces a fatal myeloproliferative syndrome in mice. Blood. 1997;90(11):4369–4383. [PubMed] [Google Scholar]
- 22.Zetterberg E, Vannucchi AM, Migliaccio AR, et al. Pericyte coverage of abnormal blood vessels in myelofibrotic bone marrows. Haematologica. 2007;92(5):597–604. doi: 10.3324/haematol.11013. [DOI] [PubMed] [Google Scholar]
- 23.Neunteufl T, Heher S, Stefenelli T, Pabinger I, Gisslinger H. Endothelial dysfunction in patients with polycythaemia vera. Br J Haematol. 2001;115(2):354–359. doi: 10.1046/j.1365-2141.2001.03092.x. [DOI] [PubMed] [Google Scholar]
- 24.Thibert V, Bellucci S, Cristofari M, Gluckman E, Legrand C. Increased platelet CD36 constitutes a common marker in myeloproliferative disorders. Br J Haematol. 1995;91(3):618–624. doi: 10.1111/j.1365-2141.1995.tb05357.x. [DOI] [PubMed] [Google Scholar]
- 25.Clezardin P, McGregor JL, Dechavanne M, Clemetson KJ. Platelet membrane glycoprotein abnormalities in patients with myeloproliferative disorders and secondary thrombocytosis. Br J Haematol. 1985;60(2):331–344. doi: 10.1111/j.1365-2141.1985.tb07419.x. [DOI] [PubMed] [Google Scholar]
- 26.Bolin RB, Okumura T, Jamieson GA. Changes in distribution of platelet membrane glycoproteins in patients with myeloproliferative disorders. Am J Hematol. 1977;3(1):63–71. doi: 10.1002/ajh.2830030108. [DOI] [PubMed] [Google Scholar]
- 27.Best D, Senis YA, Jarvis GE, et al. GPVI levels in platelets: relationship to platelet function at high shear. Blood. 2003;102(8):2811–2818. doi: 10.1182/blood-2003-01-0231. [DOI] [PubMed] [Google Scholar]
- 28.Rabie T, Varga-Szabo D, Bender M, et al. Diverging signaling events control the pathway of GPVI down-regulation in vivo. Blood. 2007;110(2):529–535. doi: 10.1182/blood-2006-11-058107. [DOI] [PubMed] [Google Scholar]
- 29.Michiels JJ, Berneman Z, Schroyens W, Finazzi G, Budde U, van Vliet HH. The paradox of platelet activation and impaired function: platelet-von Willebrand factor interactions, and the etiology of thrombotic and hemorrhagic manifestations in essential thrombocythemia and polycythemia vera. Semin Thromb Hemost. 2006;32(6):589–604. doi: 10.1055/s-2006-949664. [DOI] [PubMed] [Google Scholar]
- 30.Castaldi PA, Berndt MC, Booth W, Gregory C, Bull H, Greaves M. Evidence for a platelet membrane defect in the myeloproliferative syndromes. Thromb Res. 1982;27(5):601–609. doi: 10.1016/0049-3848(82)90307-3. [DOI] [PubMed] [Google Scholar]
- 31.Santilli F, Romano M, Recchiuti A, et al. Circulating endothelial progenitor cells and residual in vivo thromboxane biosynthesis in low-dose aspirin-treated polycythemia vera patients. Blood. 2008;112(4):1085–1090. doi: 10.1182/blood-2007-11-123091. [DOI] [PubMed] [Google Scholar]
- 32.Arellano-Rodrigo E, Alvarez-Larrán A, Reverter JC, et al. Platelet turnover, coagulation factors, and soluble markers of platelet and endothelial activation in essential thrombocythemia: relationship with thrombosis occurrence and JAK2 V617F allele burden. Am J Hematol. 2009;84(2):102–108. doi: 10.1002/ajh.21338. [DOI] [PubMed] [Google Scholar]
- 33.Griesshammer M, Beneke H, Nussbaumer B, Grünewald M, Bangerter M, Bergmann L. Increased platelet surface expression of P-selectin and thrombospondin as markers of platelet activation in essential thrombocythaemia. Thromb Res. 1999;96(3):191–196. doi: 10.1016/s0049-3848(99)00095-x. [DOI] [PubMed] [Google Scholar]
- 34.Tatewaki W, Takahashi H, Shibata A. Multimeric composition of plasma von Willebrand factor in chronic myeloproliferative disorders. Clin Lab Haematol. 1988;10(4):417–425. doi: 10.1111/j.1365-2257.1988.tb01190.x. [DOI] [PubMed] [Google Scholar]
- 35.Budde U, Scharf RE, Franke P, Hartmann-Budde K, Dent J, Ruggeri ZM. Elevated platelet count as a cause of abnormal von Willebrand factor multimer distribution in plasma. Blood. 1993;82(6):1749–1757. [PubMed] [Google Scholar]
- 36.Etheridge SL, Roh ME, Cosgrove ME, et al. JAK2V617F-positive endothelial cells contribute to clotting abnormalities in myeloproliferative neoplasms. Proc Natl Acad Sci USA. 2014;111(6):2295–2300. doi: 10.1073/pnas.1312148111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zaleskas VM, Krause DS, Lazarides K, et al. Molecular pathogenesis and therapy of polycythemia induced in mice by JAK2 V617F. PLoS ONE. 2006;1(1):e18. doi: 10.1371/journal.pone.0000018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Landolfi R, Rocca B, Patrono C. Bleeding and thrombosis in myeloproliferative disorders: mechanisms and treatment. Crit Rev Oncol Hematol. 1995;20(3):203–222. doi: 10.1016/1040-8428(94)00164-O. [DOI] [PubMed] [Google Scholar]
- 39.Castaman G, Lattuada A, Ruggeri M, Tosetto A, Mannucci PM, Rodeghiero F. Platelet von Willebrand factor abnormalities in myeloproliferative syndromes. Am J Hematol. 1995;49(4):289–293. doi: 10.1002/ajh.2830490406. [DOI] [PubMed] [Google Scholar]
- 40.Budde U, Schaefer G, Mueller N, et al. Acquired von Willebrand’s disease in the myeloproliferative syndrome. Blood. 1984;64(5):981–985. [PubMed] [Google Scholar]
- 41.Konstantinides S, Ware J, Marchese P, Almus-Jacobs F, Loskutoff DJ, Ruggeri ZM. Distinct antithrombotic consequences of platelet glycoprotein Ibalpha and VI deficiency in a mouse model of arterial thrombosis. J Thromb Haemost. 2006;4(9):2014–2021. doi: 10.1111/j.1538-7836.2006.02086.x. [DOI] [PubMed] [Google Scholar]
- 42.Boulaftali Y, Lamrani L, Rouzaud MC, et al. The mouse dorsal skinfold chamber as a model for the study of thrombolysis by intravital microscopy. Thromb Haemost. 2012;107(5):962–971. doi: 10.1160/TH11-10-0705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Eckly A, Hechler B, Freund M, et al. Mechanisms underlying FeCl3-induced arterial thrombosis. J Thromb Haemost. 2011;9(4):779–789. doi: 10.1111/j.1538-7836.2011.04218.x. [DOI] [PubMed] [Google Scholar]
- 44.Barr JD, Chauhan AK, Schaeffer GV, Hansen JK, Motto DG. Red blood cells mediate the onset of thrombosis in the ferric chloride murine model. Blood. 2013;121(18):3733–3741. doi: 10.1182/blood-2012-11-468983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.De Grandis M, Cambot M, Wautier MP, et al. JAK2V617F activates Lu/BCAM-mediated red cell adhesion in polycythemia vera through an EpoR-independent Rap1/Akt pathway. Blood. 2013;121(4):658–665. doi: 10.1182/blood-2012-07-440487. [DOI] [PubMed] [Google Scholar]
- 46.von Brühl ML, Stark K, Steinhart A, et al. Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo. J Exp Med. 2012;209(4):819–835. doi: 10.1084/jem.20112322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Carobbio A, Finazzi G, Antonioli E, et al. Thrombocytosis and leukocytosis interaction in vascular complications of essential thrombocythemia. Blood. 2008;112(8):3135–3137. doi: 10.1182/blood-2008-04-153783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vannucchi AM, Antonioli E, Guglielmelli P, et al. MPD Research Consortium. Prospective identification of high-risk polycythemia vera patients based on JAK2(V617F) allele burden. Leukemia. 2007;21(9):1952–1959. doi: 10.1038/sj.leu.2404854. [DOI] [PubMed] [Google Scholar]
- 49.Passamonti F. Prognostic factors and models in polycythemia vera, essential thrombocythemia, and primary myelofibrosis. Clin Lymphoma Myeloma Leuk. 2011;11(suppl 1):S25–S27. doi: 10.1016/j.clml.2011.01.002. [DOI] [PubMed] [Google Scholar]
- 50.Falanga A, Marchetti M. Thrombotic disease in the myeloproliferative neoplasms. Hematology Am Soc Hematol Educ Program. 2012;2012(1):571–581. doi: 10.1182/asheducation-2012.1.571. [DOI] [PubMed] [Google Scholar]
- 51.Hobbs CM, Manning H, Bennett C, et al. JAK2V617F leads to intrinsic changes in platelet formation and reactivity in a knock-in mouse model of essential thrombocythemia. Blood. 2013;122(23):3787–3797. doi: 10.1182/blood-2013-06-501452. [DOI] [PMC free article] [PubMed] [Google Scholar]