

Key Points
In people with cystic fibrosis, defective CFTR function alters neutrophil cytosolic ion homeostasis leading to impaired degranulation.
By normalizing CFTR function, ivacaftor treatment corrects neutrophil degranulation resulting in normalized killing of bacteria.
Abstract
Studies have endeavored to reconcile whether dysfunction of neutrophils in people with cystic fibrosis (CF) is a result of the genetic defect or is secondary due to infection and inflammation. In this study, we illustrate that disrupted function of the CF transmembrane conductance regulator (CFTR), such as that which occurs in patients with ∆F508 and/or G551D mutations, correlates with impaired degranulation of antimicrobial proteins. We demonstrate that CF blood neutrophils release less secondary and tertiary granule components compared with control cells and that activation of the low-molecular-mass GTP-binding protein Rab27a, involved in the regulation of granule trafficking, is defective. The mechanism leading to impaired degranulation involves altered ion homeostasis caused by defective CFTR function with increased cytosolic levels of chloride and sodium, yet decreased magnesium measured in CF neutrophils. Decreased magnesium concentration in vivo and in vitro resulted in significantly decreased levels of GTP-bound Rab27a. Treatment of G551D patients with the ion channel potentiator ivacaftor resulted in normalized neutrophil cytosolic ion levels and activation of Rab27a, thereby leading to increased degranulation and bacterial killing. Our results confirm that intrinsic alterations of circulating neutrophils from patients with CF are corrected by ivacaftor, thus illustrating additional clinical benefits for CFTR modulator therapy.
Introduction
Cystic fibrosis (CF) is a fatal genetic disease caused by mutations in the CF transmembrane conductance regulator gene (CFTR) encoding a protein kinase (PK)A-regulated chloride (Cl−) channel. More than 1000 mutations in the CFTR gene have been identified that result in defective regulation of Cl− transport. Reported mutations are categorized according to the effect on CFTR protein and function. Class II mutations are caused by a defect in CFTR processing and include the ∆F508 mutation, of which 90% of people with CF (PWCF) carry 1 copy.1 Class III defects include the G551D mutation, which encodes a protein localized to the membrane containing a primary defect in channel gating. Ivacaftor (VX-770) has been developed as a CFTR potentiator for class III mutations and has been shown to improve Cl− secretion and decrease sodium (Na+) absorption in vitro.2 Recent clinical trials with ivacaftor in PWCF with ≥1 G551D-CFTR mutation have shown improvements in lung function compared with placebo,3 decreased pulmonary exacerbations, weight gain, and decrease of sweat Cl− concentration to the normal range.4
The exact etiology of lung disease associated with CF is unclear, yet 1 definitive characteristic is sustained: neutrophil recruitment and neutrophil-dominated inflammation.5,6 Our understanding of the pathophysiology of CF is undergoing a reassessment, and focus has turned to defective immune cell function and, in particular, dysregulated neutrophil activity. Although CFTR protein expression on neutrophil membranes, secretary vesicles, and phagocytic vacuoles has been described,7,8 multiple studies have endeavored to reconcile whether neutrophil dysfunction in CF is a result of the genetic defect or provoked by chronic bacterial infiltration and inflammation.9 For example, defective microbial killing has been reported to be both dependent on10 and independent of the inflammatory environment.11 Additional studies have revealed a disparity in neutrophil microbial uptake,10 increased degranulation of primary granule contents,12,13 altered chemotaxis,14 increased recruitment,15 defective PKC activity,16 and increased oxidant formation.17,18 Observations of neutrophil-dominated airway inflammation in children with CF,19,20 delayed cell apoptosis in heterozygote individuals,21,22 and impaired CFTR-dependent vacuolar Cl− transport and microbial killing7,23-25 support an innate defect rather than inflammatory cell reprogramming in the control of neutrophil functionality in CF; yet further studies are required to corroborate this theory.
The aim of this study was to shed further light on this pivotal area and to investigate dysregulated neutrophil degranulation capacity in CF. We found that disrupted function of CFTR in vivo, or pharmacological CFTR inhibition in vitro, results in changes in cytosolic ion concentrations leading to abnormal degranulation responses and impaired bacterial killing, an innate defect rectified by CFTR potentiator therapy.
Materials and methods
Chemicals and reagents
All chemicals and reagents were of the highest purity available and were purchased from Sigma-Aldrich (St. Louis, MO) unless otherwise indicated.
Study design
The study was approved by the Beaumont Hospital Ethics Committee, conducted in accordance with the Declaration of Helsinki, and informed written consent was obtained from all participants. For the proteomics study, clinically stable and exacerbating PWCF (ΔF508 homozygous genotype) and non-CF bronchiectasis (inflammatory control) and healthy control (HC) individuals (n = 6 for each cohort) were recruited as previously described.26 Clinically stable CF patients (n = 33; mean age, 24.2 ± 1.0 years; forced expiratory volume in 1 second (FEV1), 59.2 ± 5.0% predicted) with the genotype ∆F508/∆F508 were recruited for the study of degranulation and Rab27a activation. To assess the effect of ivacaftor on neutrophil degranulation and protein activation, clinically stable CF patients with the genotype ∆F508/G551D receiving 150 mg ivacaftor twice daily (n = 6; mean age, 21.7 ± 1.5 years; FEV1, 63.0 ± 19.7% predicted) and control individuals not receiving the drug (n = 4; mean age, 19.7 ± 0.3 years; FEV1, 50.3 ± 14.7% predicted) were recruited. HC volunteers were age and gender matched, had no respiratory symptoms, and were not receiving medication.
Neutrophil isolation
Neutrophils were isolated as previously described.27 The purity of isolated neutrophils was validated by flow cytometric analysis using a monoclonal antibody against CD16b and was >96%.28,29 Neutrophil viability was assessed by Trypan blue exclusion assay, and analysis of apoptotic and dead cells was evaluated by using an Annexin V-FITC Apoptosis Kit (BioVision, Milpitas, CA). Results confirmed viability of neutrophils >98%. Neutrophil cytosolic Ca2+, pH, and GTP levels30; degranulation; apoptosis; migration and bacterial killing assays31; subcellular fractionation32; and western blotting are detailed in the supplemental Methods available on the Blood Web site.
Proteomic analysis
Proteomic analysis and two-dimensional difference gel electrophoresis (2D-DIGE) were performed as previously described.26 Labeled samples were pooled and separated by 2D-DIGE. Gel images were scanned using a Typhoon 9400 variable mode imager (GE Healthcare, Little Chalfont, Buckinghamshire, UK), and Decyder software version 6.2 (GE Healthcare) was used for quantitative analysis. Proteins were identified by liquid chromatography (LC)-tandem mass spectroscopy (MS/MS) using the Ultimate 3000 nanoLC system (Dionex) and were interfaced to an LTQ Orbitrap XL (Thermo Fisher Scientific, Sunnyvale, CA), and data were acquired by Xcalibur 2.0.7 software (Thermo Fisher Scientific). A database search was performed using the online database SWISSPROT.
Measurements of GTP-bound Rab27a
Neutrophils (2 × 107 cells/mL) were stimulated with tumor necrosis factor (TNF)-α (10 ng/mL), formyl-methionyl-leucyl phenylalanine (fMLF; 1 μM), or phorbol-12-myristate-13-acetate (PMA; 1 μg/mL) for 0, 5, 10, or 20 minutes, and quantification of Rab27a-bound GTP was performed as previously described.33 GTP bound to immunoprecipitated Rab27a was converted to ATP using nucleoside diphosphate kinase, and ATP concentration was quantified employing the PhosphoWorks Luminometric ATP Assay Kit *Bright Glow* (AAT Bioquest, Sunnyvale, CA), according to the manufacturer’s instructions. GTP levels were normalized to the amount of immunoprecipitated Rab27a as determined by western blot analysis.
Measurement of cytosolic ion concentrations
For electron probe X-ray microanalysis (EPXMA), resting neutrophils were snap frozen in liquid propane cooled to −185°C using a Rapid Immersion Plunge Freezer (EMS). Frozen pellets were ultra-cryosectioned using a EM UC6 ultratome (Leica), and sections were transferred onto nickel grids and freeze-dried using the K775 freeze dryer (Emitech). Sections were examined by transmission electron microscopy (TEM) using a Tecnai T12 (FEI) at 120 kV. X-ray spectra of cytosols were generated using a Sapphire Si(Li) Detector for TEM, and Kα peaks were analyzed by Genesis software (both EDAX). The obtained ion concentrations were normalized to phosphor concentrations as previously described to account for varying thickness of the specimen34 and related to previously shown ion concentrations.35,36
In additional experiments, neutrophils from HC subjects were resuspended in N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid (HEPES) buffer (10 mM HEPES, 150 mM NaCl, 5 mM KCl, 1.2 mM MgCl2, 1.3 mM CaCl2, and 5 mM glucose, pH 7.4) and loaded with the Cl− indicator N-(ethoxycarbonylmethyl)-6-methoxyquinolinium bromide (MQAE) (1 mM), the Na+ indicator 1,3-benzenedicarboxylic acid, 4,4′-[1,4,10-trioxa-7,13-diazacyclopentadecane-7,13-diylbis(5-methoxy-6,12-benzofurandiyl)]bis-, tetrakis[(acetyloxy)methyl] ester (SBFI-AM) (10 μM with 0.025% w/v Pluronic F), or the Mg2+ indicator Mag-Fluo-4-AM (5 μM) at 37°C for 30 minutes (all dyes were purchased from Life Technologies, Thermo Fisher Scientific). Cells were washed and resuspended in fresh buffer before being treated with either CFTR(inh)-172 (10 μM), GlyH-101 (25 μM),25 or the vehicle control dimethylsulfoxide (DMSO) (0.1% v/v). CFTR(inh)-172 is a specific inhibitor of CFTR and functions via binding Arg347 of the sixth transmembrane helix of the CFTR protein.37,38 Fluorescence was measured at the following excitation/emission wavelengths: Cl− at 355 nm/460 nm, Na+ at 340 nm/510 nm and 380 nm/510 nm, and Mg2+ at 490 nm/535 nm. To measure absolute Cl− concentrations of neutrophil cytosols, cells were loaded with MQAE as described and resuspended in HEPES buffer containing varying concentrations of Cl− (0, 20, 40, 60, 80 100, 120, and 140 mM NaCl, sodium gluconate was added to obtain 140 mM extracellular Na+ concentration) and the ionophores nigericin (7 μM) and tributyltin chloride (10 μM) as previously described.39 For experiments involving altered Mg2+ cytosolic levels, neutrophils were resuspended in HEPES buffer containing varying concentrations of Mg2+ (0.5, 0.6, 0.7, and 0.8 mM MgCl2) and were treated with the ionophore A23187 (20 μM) for 10 minutes at room temperature prior to treatment with TNF-α.40 Serum Mg2+ levels of individuals recruited to this study were analyzed by standard clinical laboratory testing using a 5400 Analyzer (Beckman Coulter).
Statistical analysis
Results are expressed as mean ± standard error of the mean (SEM) of n separate biological replicates as stated in the figure legends. Statistical analysis was performed with GraphPad Prism (version 4.03 for Windows). For statistical comparison of small datasets (n < 6), the Student t test was performed to determine P values.41 For larger datasets, the D'Agostino and Pearson omnibus normality test was carried out to determine whether data were normally distributed. When normally distributed, groups were compared by the Student t test; otherwise, the nonparametric Mann-Whitney U test was used. For comparison of ≥3 groups, 1-way analysis of variance (ANOVA) was performed. P values were considered statistically significant at P < .05. Differential expression of proteins identified by proteomic analysis was defined as a >1.5-fold change in expression with P < .05 or 1.2-fold with P < .001.
Results
Altered membrane protein expression in CF neutrophils
Expression of CFTR protein was confirmed in HC neutrophils and was severely impaired in neutrophils isolated from PWCF carrying the ∆F508 or G551D mutation (supplemental Figure 1A-C). To evaluate additional CF neutrophil membrane alterations, quantitative changes of neutrophil membrane proteins were analyzed using 2D-DIGE. Neutrophils were isolated from PWCF homozygous for the ∆F508 mutation during an exacerbation and when stable and from HCs and non-CF bronchiectasis patients when stable (inflammatory control) (n = 6 for each cohort). Five proteins of particular interest to this study were revealed to be differentially expressed on comparative analysis (Figure 1A). All were identified as neutrophil granule proteins with 4 down-regulated and 1 up-regulated (Table 1). Alterations to the degranulation pattern of neutrophil primary granules in CF have been previously reported12,13 and are further supported by this study, which showed increased levels of myeloperoxidase (MPO) on the circulating CF neutrophil membrane. In contrast, significant down-regulation of lactoferrin, serotransferrin, and dermicidin of secondary granules and matrix metalloprotease 9 (MMP-9) of tertiary granules was observed (Table 1). The average of 6 different samples is illustrated in Figure 1B and demonstrates persistent down-regulation of lactoferrin and MMP-9 during CF exacerbation and when stable compared with HC and non-CF bronchiectasis patient control membranes (P < .05 by ANOVA). Although lactoferrin and MMP-9 are equally expressed in whole cell lysates (supplemental Figure 2A-B), western blotting of CF and HC membrane fractions for lactoferrin and MMP-9 (Figure 1C) and quantification of immuno-bands by densitometry (Figure 1D-E) confirmed the proteomic results. Collectively, these results indicate changes in the levels of membrane associated proteins of circulating neutrophils from individuals with clinically stable CF, with evidence of decreased levels of proteins from secondary and tertiary granules forming the basis of this study.
Figure 1.
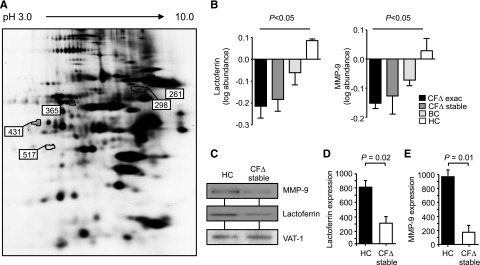
Intrinsic and inflammatory changes to proteins present on CF neutrophil membranes. (A) Comparative analysis of proteins extracted from membranes of ΔF508 homozygous PWCF during an exacerbation (CF∆ exac), the same individuals after exacerbation (CF∆ stable), HC, or non-CF bronchiectasis patient control cells (BC) was performed by 2D-DIGE. Differentially expressed proteins (numbered) illustrated within the representative 2D gel were analyzed by LC-MS/MS. (B) The log protein abundance illustrates persistent down-regulation of lactoferrin and MMP-9 on CFΔ exac and CFΔ stable, compared with HC and BC, samples (P < .05, 1-way ANOVA, n = 6 subjects per group). (C) Neutrophil membrane fractions from HC and CF∆ stable samples were subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis and western blot analysis for MMP-9 and lactoferrin. By 2D-DIGE, VAT-1 was found equally expressed between the different membrane types and was therefore used as a loading control. Band intensity for (D) lactoferrin and (E) MMP-9 was quantified by densitometry and normalized to VAT-1 (P = .02 and P = .01, respectively, Student t test, n = 5 subjects per group). All measurements are means ± SEM from biological replicates.
Table 1.
Granular proteins with altered membrane expression in CF neutrophil membranes.
| Protein function | Spot number | Accession number | Identified protein | Mass (kDa) | Coverage (%) |
|---|---|---|---|---|---|
| Primary granules | 431 | gi129825 | MPO ↑ | 53.81 | 13.47 |
| Secondary granules | 298 | gi8570015 | Lactoferrin ↓ | 78.13 | 52.11 |
| 365 | gi136191 | Serotransferrin ↓ | 77.0 | 19.77 | |
| 517 | gi120141302 | Dermicidin ↓ | 11.27 | 20.00 | |
| Tertiary granules | 261 | gi269849668 | MMP-9 ↓ | 74.40 | 35.00 |
Up- or down-regulation of protein expression is indicated by the arrows (↑ and ↓, respectively).
Impaired degranulation of secondary and tertiary granules by CF neutrophils
Following the finding of decreased levels of granule proteins associated with the plasma membrane of CF neutrophils, the kinetics of degranulation were compared between CF and control cells. Within this set of experiments, TNF-α was the chosen stimulus, as elevated levels are found associated with CF,42 and it has been shown to cause degranulation of secondary and tertiary but not primary granules.43 Control experiments confirmed that there was equal surface expression of TNF receptor 1 (TNFR1) and TNFR2 on control and CF neutrophil membranes (supplemental Figure 2C-D) and that the concentration of TNF-α used did not induce cell apoptosis (supplemental Figure 3).
On stimulation, TNF-α-induced release of granule proteins was quantified in the extracellular supernatant by immunoblotting and enzyme-linked immunosorbent assay. Levels of released hCAP-18 (Figure 2A,D; P = .007 and P = .002) and lactoferrin (Figure 2B,E; P = .03) from secondary and MMP-9 (Figure 2C; P = .002) from tertiary granules were significantly reduced by CF neutrophils after 10-minute stimulation. This defect was also observed after stimulation with fMLF or fMLF in combination with TNF-α, indicating that the observed degranulation impairment was not stimuli specific (supplemental Figure 4).
Figure 2.
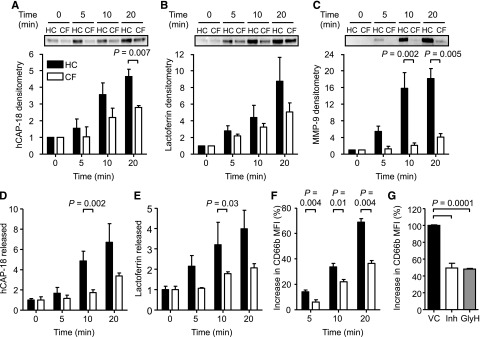
Impaired degranulation of secondary and tertiary granules from CF neutrophils and CFTR inhibited cells. HC and CF neutrophils were stimulated with TNF-α (10 ng/2 × 107 cells/mL), and the extracellular supernatants were obtained after 0, 5, 10, and 20 minutes. (A-C) (Top) Representative western blots of supernatants probed for (A) hCAP-18, (B) lactoferrin, and (C) MMP-9. Immunoband intensity was quantified by densitometry, expressed as a multiple of the time zero value of unstimulated cells and illustrated in the corresponding bar graphs (P = .007 at 20-minute time point for hCAP-18; P = .002 at 10-minute time point and P = .005 at 20-minute time point for MMP-9, Student t test, n = 3 subjects per group). Levels of (D) hCAP-18 and (E) lactoferrin in extracellular supernatants were also assessed by enzyme-linked immunosorbent assay (P = .002 and P = .03 at 10 minutes, respectively, Student t test, n=3 subjects per group). Values presented are relative to time 0 of unstimulated cells. (F) HC and CF neutrophils were fixed in 4% (w/v) paraformaldehyde following 0, 5, 10, and 20 minutes of TNF-α stimulation (10 ng/mL) and analyzed by flow cytometry using a fluorescein isothiocyanate-labeled CD66b antibody. Data are represented as the percent increase in mean fluorescence intensity (MFI) from unstimulated control cells (P = .004, P = .01, and P = .004 at 5, 10, and 20 minutes, respectively, Student t test, n=8 subjects per group). (G) HC neutrophils were treated with vehicle control (0.1% v/v DMSO; VC) or the CFTR inhibitors CFTRinh-172 (10 μM; Inh) or GlyH-101 (25 μM; GlyH) and then stimulated with TNF-α for 10 minutes. The level of degranulation was compared by flow cytometry using a fluorescein isothiocyanate-labeled CD66b antibody. Data are represented as increase of MFI relative to vehicle-treated control cells (P = .0001, Student t test, n = 4 subjects per group). All measurements are means ± SEM from biological replicates.
As an alternative approach, we investigated the membrane expression of CD66b.44 As illustrated in Figure 2F, up-regulation of CD66b to the plasma membrane was greatly decreased in CF neutrophils compared with HC cells (n = 8; P = .004, P = .01, and P = .004 after 5, 10, and 20 minutes, respectively). Moreover, experiments investigated primary granule exocytosis using MPO as a marker. The level of MPO release by CF neutrophils in response to fMLF and TNF-α (P = .05) or the nonphysiological stimulus PMA (P = .03) was significantly higher compared with control cells (supplemental Figure 5).
We next investigated whether CFTR function was related to impaired degranulation by exposing control neutrophils to the CFTR inhibitors CFTR(inh)-17237,38 or GlyH-101.25 After a 10-minute stimulation, membrane levels of CD66b in cells treated with either CFTR inhibitor were significantly diminished compared with vehicle control-treated samples (P = .0001; Figure 2G). In contrast, CFTR(inh)-172 was found to have no effect on neutrophil primary granule release (P = .84; supplemental Figure 6), neutrophil apoptosis (supplemental Figure 3), or migration (P = .9; supplemental Figure 7).
Collectively, these results illustrate that CFTR Cl− channel function is important for efficient degranulation, and impaired release of secondary and tertiary granules is an intrinsic defect of CF neutrophils.
Decreased levels of GTP-bound Rab27a as a cause of aberrant degranulation by CF neutrophils
The small GTPase Rab27a has been shown to regulate degranulation of secondary and tertiary granules.45 Rab27a interacts with its effector protein Munc13-4,46 and the SNARE protein complex reportedly required for release of secondary and tertiary granules is composed of VAMP2 on the granule membrane and SNAP23 and syntaxin 4 (STX4) on the plasma membrane.47-49 To rule out differential protein expression as a possible cause for impaired degranulation, expression levels of Rab27a, Munc13-4, and the SNAREs VAMP2, SNAP23, and STX4 were determined by immuno-blotting (Figure 3A). Results revealed equal expression of all proteins in whole cell lysates and the respective compartments (secondary and tertiary granules and plasma membranes) of control and CF cells (Figure 3A). When translocation of Rab27a, the central regulatory protein involved in secondary and tertiary granule trafficking, was explored after TNF-α stimulation, results revealed severely impaired membrane translocation and docking of Rab27a in CF cells (Figure 3B). In HC cells, Rab27a membrane binding was increased by approximately twofold after a 5-minute TNF-α stimulation, and in contrast, significantly lower levels were detected over the entire time course up to 20 minutes in CF neutrophils (n = 3; P = .05; Figure 3B). Moreover, significantly reduced levels of Rab27a membrane docking in response to fMLF were observed (P = .01), indicating that this impairment was not stimuli specific (supplemental Figure 8). In contrast, however, stimulation with PMA resulted in equal Rab27a membrane translocation in control and CF cells (P = .34).
Figure 3.

Impaired translocation and reduced GTP activation of Rab27a in CF neutrophils. (A) HC and ΔF508 homozygous CF (CFΔ) neutrophil whole cell lysates, secondary and tertiary granules, or isolated plasma membranes were analyzed by immunoblotting for protein expression of Rab27a, VAMP2, Munc13-4, SNAP23, and STX4. Glyceraldehyde-3-phosphate dehydrogenase and Na+/K+-ATPase were used as a loading control for whole cell lysates and plasma membranes, respectively. Representative blot images are shown (n = 3 subjects per group). (B) HC or ΔF508 homozygous CF (CFΔ) neutrophils were stimulated with TNF-α (10 ng/mL), and membrane proteins were assayed for Rab27a by immunoblotting. Quantification of immunoblots by densitometry revealed decreased levels of Rab27a on membranes of CF cells (P = .05, paired Student t test, n = 3 subjects per group). (C) To assess the activation levels of Rab27a, cell lysates from TNF-α-stimulated cells were immunoprecipitated with polyclonal rabbit anti-Rab27a antibodies. GTP was quantified using a coupled enzymatic assay and normalized to precipitated Rab27a levels and the 0 time point value (P = .004, P = .03, and P = .003 at 0.5, 1, and 4 mintutes, respectively, Student t test, n = 5 subjects per group). Each measurement in B and C are mean ± SEM from biological replicates.
Translocation to the membrane fraction requires Rab27a to be in an active GTP-bound form.50 Levels of GTP-bound Rab27a in HC neutrophils reached a maximum 1 minute after TNF-α stimulation, and then after, decreased to unstimulated basal levels at 4 minutes. In contrast, at the earlier time points of 30 seconds and 1 minute, significantly lower levels of GTP-bound Rab27a were recorded in CF cells compared with control cells, with maximum levels attained after a 4-minute stimulation (P = .004 at 30 seconds and P = .03 at 1 minute). Subsequent to 4-minute TNF-α activation, GTP-Rab27a levels in both cell types fell below those found in respective unstimulated cells. Collectively, these results demonstrate that Rab27a regulation and activation is altered in CF neutrophils, which could contribute significantly to impaired degranulation of secondary and tertiary granules.45
Disturbed ion homeostasis in CF neutrophils
The mechanism leading to decreased degranulation of secondary and tertiary granules, and altered levels of GTP-bound Rab27a by CF neutrophils were next explored. No link between Ca2+ flux and pH as a cause for this defect was observed (supplemental Figure 9A-B); thus, ensuing experiments investigated whether a lack of CFTR function in CF cells impacted on cellular ion homeostasis. EPXMA of resting cells was used to compare intracellular ion concentrations between CF and HC neutrophils, and the obtained relative values were correlated to known cytosolic ion concentrations.35,36 Although the concentrations of Cl− and Na+ were significantly increased in CF cells from 100 to 120 mM and from 12 to 15 mM (n = 3, P = .006), respectively, the level of Mg2+ in CF cells was decreased from 0.8 to 0.7 mM (n = 3, P = .01; Figure 4A,C,E). Interestingly, HC cells treated with CFTR(inh)-172 illustrated similar changes in cytosolic ion concentrations compared with neutrophils isolated from PWCF with increased Cl− and Na+ levels of 122 and 17.5 mM, respectively, and a decreased Mg2+ concentration of 0.7 mM (n = 3).
Figure 4.

Altered ion homeostasis due to impaired CFTR function in CF neutrophils. Intracellular levels of (A) Cl−, (C) Na+, and (E) Mg2+ in neutrophils from HCs, patients with CF of the ΔF508/ΔF508 (CFΔ) or G551D/ΔF508 genotype following ivacaftor treatment (CFG+iva), and HC cells treated with CFTRinh-172 (10 μM; Inh) were analyzed using electron probe X-ray microanalysis. Relative values of CFΔ or Inh normalized to HC or CFG+iva, respectively, are shown on the left y-axis and are related to recorded ion concentrations in cytosols of HC cells on the right y-axis.35,36 (A) Cytosolic Cl− was increased in CFΔ or inhibitor-treated cells (P = .006 between HC and CFΔ and P = .005 between CFG+iva and Inh, Student t test, n = 3 subjects per group). Na+ was also increased in these samples (P = .006 between HC and CFΔ and P = .0001 between CFG+iva and Inh, Student t test, n = 3 subjects per group). Mg2+ levels were decreased in CFΔ and Inh treated cells (P = .04 between HC and CFΔ and P = .04 between CFG+iva and Inh, Student t test, n=3 subjects per group). (B,D,F) Relative changes in cytosolic (B) Cl−, (D) Na+, and (F) Mg2+ levels in CFTRinh-172 (10 μM) treated HC neutrophils compared with vehicle control (0.05% v/v DMSO) treated cells were measured for 16 minutes using MQAE, SBFI-AM, and Mag-Fluo-4-AM, respectively (P = .0001, paired Student t test, n = 3 subjects per group). All measurement are mean ± SEM from biological replicates.
To confirm that the observed alterations in ion levels are due to a lack of CFTR function rather than inflammation, HC neutrophils were loaded with the Cl−, Na+, or Mg2+ sensitive fluorescent dyes MQAE, SBFI-AM, or Mag-Fluo-4-AM, respectively, and treated with CFTR(inh)-172. CFTR inhibition resulted in a significant increase in Cl− and Na+ levels but decreased Mg2+ levels over the time course studied (n = 3, P = .0001; Figure 4B,D,F), confirming the results obtained by EPXMA. Moreover, the increase in cytosolic Cl− due to CFTR inhibition was further confirmed using GlyH-101 (n = 3, P = .0001; supplemental Figure 10).
Ensuing experiments investigated whether altered ion concentrations could negatively affect Rab27a activation, with focus on reduced Mg2+ levels as the GTP-Rab27a complex has previously been reported to be stabilized by Mg2+ through ionic interactions.51 As CFTR(inh)-172 treatment altered ion levels to those observed in CF cells, we determined whether CFTR inhibition affected levels of GTP-bound Rab27a. TNF-α stimulated cells treated with CFTR(inh)-172 exhibited a significant 63 ± 3% reduction in the maximum level of GTP-bound Rab27a compared with vehicle control treatment (n = 3, P = .01; Figure 5A). The observed reduction in response to CFTR inhibition was comparable to the 52 ± 8% decrease in GTP-Rab27a observed in CF neutrophils (n = 3, P = .2; Figure 5A). Moreover, incremental reduction of cytosolic Mg2+ levels from 0.8 to 0.5 mM from the use of the ionophore A23187 in healthy neutrophils resulted in a dose-dependent decrease in the levels of GTP-bound Rab27a (n = 3, P = .0001 by ANOVA; Figure 5B). A significant reduction of GTP-bound Rab27a levels by 25 ± 6% was observed in cells with 0.7 mM Mg2+, the concentration detected in CF cells (n = 3, P = .008). Intracellular GTP as a cause of perturbed GTP-Rab27a levels in CF neutrophils was excluded as no significant difference between HC and CF neutrophils was detected (n = 3, P = .90; Figure 5C). To rule out hypomagnesaemia52 as a potential cause for lower cytosolic Mg2+ levels in circulating CF neutrophils, serum Mg2+ levels of PWCF recruited to this study were analyzed. No statistical difference in serum Mg2+ levels was detected between HC (0.83 ± 0.02 mM) and CF samples (0.82 ± 0.01 mM) (n = 28, P = .6), and the mean values of both groups were higher than the 0.75 mM reference for hypomagnesaemia (Figure 5D). Collectively, these results indicate that lack of CFTR Cl− channel function significantly perturbed the Mg2+ ion homeostasis of neutrophils, in turn decreasing GTP-bound Rab27a levels.
Figure 5.

Reduced Mg2+ in CF cells impairs GTP activation of Rab27a. (A) Maximum increase in GTP-bound Rab27a following TNF-α treatment was compared between HC and ΔF508/ΔF508 CF (CFΔ) neutrophils and between vehicle control treated (0.05% v/v DMSO; VC) or CFTRinh-172 (10 μM; Inh) treated HC cells (CFΔ and Inh expressed relative to HC and VC, respectively). Reduction in maximum Rab27a activity in inhibitor-treated HC cells is similar to CFΔ cells (P = .03 between HC and CFΔ, P = .01 between VC and Inh, and P = .2 between CFΔ and Inh, Student t test, n = 3 subjects per group). (B) The cytosolic Mg2+ concentration in HC cells was incrementally decreased from 0.8 to 0.5 mM using the ionophore A23187 (20 μM), and maximum increase in GTP-bound Rab27a following TNF-α treatment was analyzed and expressed relative to 0.8 mM (P = .0001 between 0.5 and 0.6 mM, P = .005 between 0.6 and 0.7 mM, and P = .008 between 0.7 and 0.8 mM, Student t test, P = .0001 by 1-way ANOVA, n = 3 subjects per group). (C) Cytosolic GTP content of HC neutrophils or cells from patients with CF of the ΔF508/ΔF508 (CFΔ) or G551D/ΔF508 (CFG) genotype was equal as measured by high-performance liquid chromotography (P = .9, 1-way ANOVA, n = 3 subjects per group). (D) Serum Mg2+ levels of HC and PWCF were not statistically different (P = .6, Student t test, n = 28 subjects per group) and were within the normal range (hashed lines). All measurements are means ± SEM from biological replicates.
Ivacaftor treatment corrects activation of Rab27a and degranulation
Using EPXMA, we evaluated the effect of ivacaftor on cytosolic ion levels of CF resting neutrophils. Results revealed that the concentrations of cytosolic Cl− (100 ± 5 mM), Na+ (12.0 ± 0.5 mM), and Mg2+ (0.80 ± 0.04 mM) ions in neutrophils of PWCF heterozygous for the G551D mutation (G551D/∆F508) who were receiving ivacaftor was similar to HC cells (Figure 4A,C,E, respectively). Moreover, GTP-bound Rab27a levels in PWCF homozygous for the ∆F508 mutation (∆F508/∆F508) or heterozygous for the G551D mutation (G551D/∆F508) remained relatively unchanged following TNF-α simulation for 2 minutes (Figure 6A) compared with HC cells, which reached a maximal twofold increase after 1 minute. Moreover, statistical analysis revealed that CF neutrophils donated by PWCF on ivacaftor therapy illustrated Rab27a activation levels that were similar to control cells (Figure 6A) and that increased compared with homozygous ∆F508 patients or untreated G551D/∆F508 heterozygote patients after TNF-α stimulation for 30 seconds (Figure 6B).
Figure 6.

Treatment with the ion channel potentiator ivacaftor corrects impaired Rab27a activation and neutrophil secondary and tertiary granule exocytosis resulting in normalized bacterial killing. (A) Rab27a was immunoprecipitated from neutrophils of HC donors, from CF neutrophils of the ΔF508/ΔF508 (CFΔ) or G551D/ΔF508 (CFG) genotypes, or from G551D/ΔF508 neutrophils donated by PWCF treated with ivacaftor (CFG+iva). Levels of immunoprecipitated GTP-bound Rab27a were enzymatically detected and normalized to precipitated Rab27a levels and the 0 time point value (n = 4 subjects per group). (B) GTP-Rab27a levels at the 30-second time point of TNF-α stimulation were significantly lower in neutrophils from untreated PWCF (CFΔ, CFG) compared with levels in HCs and ivacaftor-treated individuals (CFG+iva) (P = .03 between HC and CFΔ, P = .04 between HC and CFG, P = .4 between HC and CFG+iva, Student t test, n = 4 subjects per group). (C) Neutrophils isolated from healthy volunteers, from PWCF with the ΔF508/ΔF508 or G551D/ΔF508 genotypes, or from G551D/ΔF508 individuals after ivacaftor treatment were stimulated with TNF-α (10 ng/mL) for 0, 5, 10, or 20 minutes. Degranulation was analyzed by measuring up-regulation of CD66b to the cell surface by flow cytometry and was expressed as the percentage increase in MFI. CD66b levels on CFΔ and CFG cells were significantly decreased compared with HCs (P = .01 between HC and CFΔ and P = .02 between HC and CFG, Student t test, n = 4 subjects per group), whereas ivacaftor treatment corrected CD66b levels (P = .03 between CFG and CFG+iva and P = .5 between HC and CFG+iva, Student t test, n = 4 subjects per group). (D) Survival of P aeruginosa after a 4-minute incubation with degranulated proteins obtained from unstimulated control neutrophils of healthy volunteers (ctrl), TNF-α (10 ng/mL, 10 minutes) stimulated control cells (HCs), CF cells of the ΔF508/ΔF508 or G551D/ΔF508 genotypes, or during ivacaftor treatment. The bacterial survival is expressed as percentage survival of the initial bacterial count at time 0. Bacterial survival was significantly decreased following treatment with TNF-α in HC cells (P = .0001 between ctrl and HC, Student t test, n = 3), but was significantly increased in CF samples compared with the HCs or during ivacaftor treatment (P = .0001 between HC and CFΔ and between HC and CFG, P = .2 between HC and CFG+iva, Student t test, n = 3 subjects per group). All measurements are means ± SEM from biological replicates.
Subsequent experiments were designed to assess the effect of corrected Rab27a activation on granule release by CF cells. Degranulation was determined by analyzing CD66b up-regulation following 5-, 10-, and 20-minute TNF-α stimulation. In HC cells, surface CD66b expression increased by 94 ± 15% after 20 minutes, and in contrast, neutrophils from ∆F508 homozygous and G551D/∆F508 heterozygous PWCF illustrated significantly decreased up-regulation (33 ± 11% and 42 ± 8%, respectively; P = .01 and P = .02, respectively; Figure 6C). In addition, a significant increase (96 ± 17%) in the level of CD66b was detected in G551D/∆F508 heterozygote patients after ivacaftor treatment compared with untreated individuals (P = .03; Figure 6C).
Impaired intravacuolar bacterial killing by neutrophils from PWCF has been reported7 and confirmed in this study (supplemental Figure 11). However, after release, neutrophil proteases also contribute to the extracellular killing of microorganisms,53 and in support of this theory, the potent antibacterial protein hCAP-18 can be processed to its active form in the extracellular environment.54 For this reason, ensuing experiments investigated the clinical significance and association between defective extracellular degranulation by CF neutrophils and microbial killing of Pseudomonas aeruginosa, the predominant pathogen in CF.55 For this experiment, neutrophils from HCs and ∆F508 homozygous or G551D/∆F508 heterozygous PWCF with or without ivacaftor treatment were stimulated with TNF-α for 10 minutes or left unstimulated. Subsequently, bacteria were exposed to degranulated supernatants for 4 minutes, and Pseudomonas survival was determined (Figure 6D). The percentage of P aeruginosa survival when incubated with supernatants from unstimulated HC neutrophils was 94%; in turn, exposure of bacteria to supernatants from TNF-α-stimulated HC neutrophils reduced bacterial survival to 11% (P = .0001). In contrast to the HCs, bacteria treated with supernatants from ∆F508 homozygous and G551D/∆F508 heterozygous patients indicated significantly increased bacterial survival (23% and 20%, respectively, P = .0001). This impairment of bacterial killing by CF neutrophils was also confirmed in cells stimulated with fMLF for 10 minutes (P = .0001; supplemental Figure 12). Of major importance, degranulated proteins in supernatants from patients during ivacaftor treatment showed enhanced killing ability and successfully reduced bacterial survival to 10%, similar to the 11% survival observed for HCs (Figure 6D; P = .2). In summary, these results indicate that the reduction in degranulation of secondary and tertiary granules by CF neutrophils results in ineffective bacterial killing; a defect corrected by ivacaftor treatment.
Discussion
It has been reported that CFTR is involved directly in myeloid cell function,56 and CFTR mRNA transcripts have been reported in neutrophils at an expression level similar to those found in monocytes and alveolar macrophages.7,57 Studies performed on neutrophils of heterozygous parents of children with CF have aided in deciphering the molecular mechanisms underlying dysregulation of neutrophil function due to the CFTR mutation in the absence of infection. Indeed, neutrophils of heterozygotes subjects have been shown to have increased survival21 and disturbed oxidant generation.17 In the present study, we report that CFTR function is required for ion homeostasis within the circulating neutrophil. In PWCF, ivacaftor treatment functions to correct ion concentrations and levels of GTP-bound Rab27a, thereby facilitating the degranulation process of CF blood neutrophils.
In support of neutrophil-associated CFTR protein, immuno-analysis of the receptor identified the mature and fully glycosylated form of the protein in purified neutrophils,7 and recruitment of CFTR to the phagosome on phagocytosis of serum-opsonized latex beads has been reported.8 Within our own study, the expression of CFTR protein by neutrophils was confirmed with decreased levels detected in CF neutrophils from PWCF. The lower detection of CFTR in CF cells has been described in nasal polyps and the intestine of ∆F508 homozygous CF patients.58 Moreover, the expression of CFTR in healthy neutrophils, and lack or inactivity thereof in CF cells, corroborates an intrinsic defect leading to neutrophil dysfunction.
It has previously been shown that CF neutrophils release greater levels of primary granules compared with HC cells,12,13 and data from the present study further support these findings with increased levels of MPO detected on membranes of CF cells. It is recognized that granule proteins remain bound to the cell surface of neutrophils when granules fuse with the plasma membrane during exocytosis. Indeed, both lactoferrin28 and MMP-959 have been detected on plasma membranes of human neutrophils, and it has been shown that MPO binds the membranes via a CD11b/CD18 integrin-dependent mechanism.60 In contrast to increased primary granule release, however, a significant reduction in membrane CD66b and extracellular released lactoferrin and MMP-9 from CF neutrophils compared with HC cells were observed. In addition, pharmacological inhibition with CFTR(inh)-172 is an accepted model of CF,61 and our CFTR inhibitory studies are in agreement with the data from PWCF whereby degranulation was clearly down-regulated compared with matching controls. These results indicate that a functional CFTR protein is required for optimal degranulation of secondary and tertiary granules. Impaired degranulation of lactoferrin may have important implications in CF due to its effect on bacterial biofilm formation.62 Moreover, hCAP-18 is a major antibacterial protein against Pseudomonas,31 and MMP-9 is essential for clearance of Escherichia coli and Streptococcus pneumoniae infections.63,64
Neutrophil degranulation of secondary and tertiary granules is regulated by Rab27a,45 and in the present study, measurements of GTP-bound Rab27a illustrated significantly decreased and delayed activity in CF neutrophils. The importance of Rab27a has been demonstrated in a number of cell types as it is involved in the release of dense granules by platelets, melanosomes by melanocytes, and lysosomes from cytotoxic T lymphocytes.65 Consequently, ashen mice, which express a mutant Rab27a, have increased bleeding times due to defective degranulation of dense core granules by platelets and greatly reduced activity of cytotoxic T cells.66 Similarly, a Rab27a mutation in humans can cause Griscelli syndrome type 2, resulting in partial albinism and immunodeficiency.67 The active GTP-bound Rab27a complex is stabilized by Mg2+, which is coordinated by the β- and γ-phosphate oxygen atoms of GTP and serine and threonin residues in the nucleotide-binding domain.51 Indeed, in the present study, a direct concentration-dependent effect of Mg2+ on levels of GTP-bound Rab27a was observed. Interestingly, the observed degranulation defect is in contrast to the previously illustrated increased release of MPO12 and neutrophil elastase13 from primary granules, indicating distinct mechanisms for regulating release of individual granule types. The small GTPase rac2 has been shown necessary for the release of primary granules,68 but of interest, a subpopulation of Rab27a-positive primary granules was also found,69 with different Rab27a effectors (Slp1 and Munc13-4) reported necessary for primary and tertiary granules release, respectively.70 Observations that Rab27b knockout mice also demonstrate impaired primary granule release and that up-regulation of Rab27b expression fails to compensate for the degranulation impairment observed in Rab27a-deficient cells71 highlight the different roles that Rab proteins play in the regulation of neutrophil degranulation72 and the need for further studies in this field.
Results illustrated significant increases in cytosolic Cl− and Na+, and decreased Mg2+, levels in CF cells, indicating altered ion homeostasis. The CFTR channel has been shown to interact with and regulate epithelial sodium channel (ENaC),73 the outwardly rectifying Cl− channel,74 and renal outer medullary potassium channel,75 thereby contributing to cellular ion homeostasis. However, the exact mechanism of CFTR-mediated regulation of these channels is still not fully understood, and various hypotheses including direct interaction of channels, indirect interaction through adaptor proteins, and regulation by Cl− sensing are still being investigated.76 In the present study, inhibition of CFTR function in HC neutrophils mirrored the altered ion levels observed in CF cells. These results indicate that altered ion homeostasis is due to defective CFTR function rather than a secondary effect caused by bacterial infection and chronic inflammation. This finding is supported by a previous study illustrating increased cytosolic Cl− levels following CFTR inhibitor treatment of human bronchial epithelial cells.77 It has further been shown that CFTR function is important for inhibition of ENaC activity and absence or dysfunction of CFTR results in overactive ENaC,73 in part explaining increased Na+ levels in CF and CFTR inhibitor-treated cells.78 However, to the best of our knowledge, the effect of CFTR dysfunction on Mg2+ levels has not been studied, and the underlying mechanism remains unclear. One explanation may involve cytosolic Na+ and Mg2+ levels, as increased cytosolic Na+ has been reported to result in increased efflux of Mg2+ via Na+/Mg2+ antiport in erythrocytes from PWCF.79
The ivacaftor compound has been shown to increase CFTR-mediated Cl− flux of bronchial epithelial cells in vitro.80 In our study, ivacaftor treatment resulted in normalized ion homeostasis and corrected Rab27a activation as indicated by the increased level of bound GTP. In line with the ability of ivacaftor to correct Rab27a activation and neutrophil degranulation, extracellular pseudomonal killing by CF neutrophils obtained from PWCF during treatment was significantly increased.
In conclusion, neutrophils from PWCF with either the ∆F508 and/or G551D mutations illustrate altered cytosolic ion concentrations and Rab27a activation, imparting impaired degranulation. This intrinsic defect was corrected by treatment with the ion channel potentiator ivacaftor, the effects of which significantly improved microbial killing.
Acknowledgments
The authors thank Sidney Whiteheart (Department of Molecular and Cellular Biochemistry, College of Medicine, University of Kentucky, Lexington, KY) for kindly providing the Munc13-4 antibody; D. Gudmundur Bergsson (Royal College of Surgeons in Ireland) for technical help on initial degranulation assays; Professor Alice Warley (Centre for Ultrastructural Imaging, King’s College London, London, UK) for assistance with electron probe X-ray microanalysis studies; Fergal Somerville, Gary Walsh, and Lynzie Ritchie for their tremendous support for cystic fibrosis research; and all the patients with cystic fibrosis who participated in this study.
The authors acknowledge our funding sources, including the Health Research Board Ireland (grant PHD/2007/11) (to K.P.), Science Foundation Ireland (grant 11/RFP/BMT/3094) (to E.P.R. and M.M.W.), 3U Biomedical Research (DCU-NUI Maynooth-Royal College of Surgeons in Ireland) (to P.M., M.C., N.G.M., and E.P.R.), the US Cystic Fibrosis Foundation (to N.G.M.), Vertex Pharmaceuticals (to N.G.M.), Beaumont Hospital Foundation (Dublin, Ireland), and the Programme for Research in Third Level Institutes administered by the Higher Education Authority (to N.G.M.).
Footnotes
The online version of this article contains a data supplement.
There is an Inside Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: E.P.R., N.G.M. and K.P., conceived and planned the study design, designed experiments, performed quality assurance, interpreted the data, and wrote the manuscript; K.P., E.H., M.H., M.M.W., D.A.B., O.J.M., and B.J. carried out experiments; J.K., P.M., and M.C. assisted with proteomic and LC-MS/MS analysis and data interpretation; K.P., E.H., and J.K. performed statistical analysis; and K.M. and C.M. contributed to patient accrual and performed clinical data collection.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Emer P. Reeves, Respiratory Research Division, Department of Medicine, Royal College of Surgeons in Ireland, Education and Research Centre, Beaumont Hospital, Dublin 9, Ireland; e-mail: emerreeves@rcsi.ie.
References
- 1.Zielenski J, Tsui LC. Cystic fibrosis: genotypic and phenotypic variations. Annu Rev Genet. 1995;29:777–807. doi: 10.1146/annurev.ge.29.120195.004021. [DOI] [PubMed] [Google Scholar]
- 2.Ley K, Laudanna C, Cybulsky MI, Nourshargh S. Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat Rev Immunol. 2007;7(9):678–689. doi: 10.1038/nri2156. [DOI] [PubMed] [Google Scholar]
- 3.Accurso FJ, Rowe SM, Clancy JP, et al. Effect of VX-770 in persons with cystic fibrosis and the G551D-CFTR mutation. N Engl J Med. 2010;363(21):1991–2003. doi: 10.1056/NEJMoa0909825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ramsey BW, Davies J, McElvaney NG, et al. VX08-770-102 Study Group. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N Engl J Med. 2011;365(18):1663–1672. doi: 10.1056/NEJMoa1105185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Birrer P, McElvaney NG, Rüdeberg A, et al. Protease-antiprotease imbalance in the lungs of children with cystic fibrosis. Am J Respir Crit Care Med. 1994;150(1):207–213. doi: 10.1164/ajrccm.150.1.7912987. [DOI] [PubMed] [Google Scholar]
- 6.McElvaney NG, Nakamura H, Birrer P, et al. Modulation of airway inflammation in cystic fibrosis. In vivo suppression of interleukin-8 levels on the respiratory epithelial surface by aerosolization of recombinant secretory leukoprotease inhibitor. J Clin Invest. 1992;90(4):1296–1301. doi: 10.1172/JCI115994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Painter RG, Valentine VG, Lanson NA, Jr, et al. CFTR expression in human neutrophils and the phagolysosomal chlorination defect in cystic fibrosis. Biochemistry. 2006;45(34):10260–10269. doi: 10.1021/bi060490t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhou Y, Song K, Painter RG, et al. Cystic fibrosis transmembrane conductance regulator recruitment to phagosomes in neutrophils. J Innate Immun. 2013;5(3):219–230. doi: 10.1159/000346568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hayes E, Pohl K, McElvaney NG, Reeves EP. The cystic fibrosis neutrophil: a specialized yet potentially defective cell. Arch Immunol Ther Exp (Warsz) 2011;59(2):97–112. doi: 10.1007/s00005-011-0113-6. [DOI] [PubMed] [Google Scholar]
- 10.Alexis NE, Muhlebach MS, Peden DB, Noah TL. Attenuation of host defense function of lung phagocytes in young cystic fibrosis patients. J Cyst Fibros. 2006;5(1):17–25. doi: 10.1016/j.jcf.2005.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Moraes TJ, Plumb J, Martin R, et al. Abnormalities in the pulmonary innate immune system in cystic fibrosis. Am J Respir Cell Mol Biol. 2006;34(3):364–374. doi: 10.1165/rcmb.2005-0146OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Koller DY, Urbanek R, Götz M. Increased degranulation of eosinophil and neutrophil granulocytes in cystic fibrosis. Am J Respir Crit Care Med. 1995;152(2):629–633. doi: 10.1164/ajrccm.152.2.7633718. [DOI] [PubMed] [Google Scholar]
- 13.Taggart C, Coakley RJ, Greally P, Canny G, O’Neill SJ, McElvaney NG. Increased elastase release by CF neutrophils is mediated by tumor necrosis factor-alpha and interleukin-8. Am J Physiol Lung Cell Mol Physiol. 2000;278(1):L33–L41. doi: 10.1152/ajplung.2000.278.1.L33. [DOI] [PubMed] [Google Scholar]
- 14.Brennan S, Cooper D, Sly PD. Directed neutrophil migration to IL-8 is increased in cystic fibrosis: a study of the effect of erythromycin. Thorax. 2001;56(1):62–64. doi: 10.1136/thorax.56.1.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Davis PB, Drumm M, Konstan MW. Cystic fibrosis. Am J Respir Crit Care Med. 1996;154(5):1229–1256. doi: 10.1164/ajrccm.154.5.8912731. [DOI] [PubMed] [Google Scholar]
- 16.Corvol H, Fitting C, Chadelat K, et al. Distinct cytokine production by lung and blood neutrophils from children with cystic fibrosis. Am J Physiol Lung Cell Mol Physiol. 2003;284(6):L997–L1003. doi: 10.1152/ajplung.00156.2002. [DOI] [PubMed] [Google Scholar]
- 17.Witko-Sarsat V, Allen RC, Paulais M, et al. Disturbed myeloperoxidase-dependent activity of neutrophils in cystic fibrosis homozygotes and heterozygotes, and its correction by amiloride. J Immunol. 1996;157(6):2728–2735. [PubMed] [Google Scholar]
- 18.Witko-Sarsat V, Delacourt C, Rabier D, Bardet J, Nguyen AT, Descamps-Latscha B. Neutrophil-derived long-lived oxidants in cystic fibrosis sputum. Am J Respir Crit Care Med. 1995;152(6 Pt 1):1910–1916. doi: 10.1164/ajrccm.152.6.8520754. [DOI] [PubMed] [Google Scholar]
- 19.Armstrong DS, Hook SM, Jamsen KM, et al. Lower airway inflammation in infants with cystic fibrosis detected by newborn screening. Pediatr Pulmonol. 2005;40(6):500–510. doi: 10.1002/ppul.20294. [DOI] [PubMed] [Google Scholar]
- 20.Rosenfeld M, Gibson RL, McNamara S, et al. Early pulmonary infection, inflammation, and clinical outcomes in infants with cystic fibrosis. Pediatr Pulmonol. 2001;32(5):356–366. doi: 10.1002/ppul.1144. [DOI] [PubMed] [Google Scholar]
- 21.Moriceau S, Lenoir G, Witko-Sarsat V. In cystic fibrosis homozygotes and heterozygotes, neutrophil apoptosis is delayed and modulated by diamide or roscovitine: evidence for an innate neutrophil disturbance. J Innate Immun. 2010;2(3):260–266. doi: 10.1159/000295791. [DOI] [PubMed] [Google Scholar]
- 22.Moriceau S, Kantari C, Mocek J, et al. Coronin-1 is associated with neutrophil survival and is cleaved during apoptosis: potential implication in neutrophils from cystic fibrosis patients. J Immunol. 2009;182(11):7254–7263. doi: 10.4049/jimmunol.0803312. [DOI] [PubMed] [Google Scholar]
- 23.Bonvillain RW, Painter RG, Adams DE, Viswanathan A, Lanson NA, Jr, Wang G. RNA interference against CFTR affects HL60-derived neutrophil microbicidal function. Free Radic Biol Med. 2010;49(12):1872–1880. doi: 10.1016/j.freeradbiomed.2010.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Painter RG, Bonvillain RW, Valentine VG, et al. The role of chloride anion and CFTR in killing of Pseudomonas aeruginosa by normal and CF neutrophils. J Leukoc Biol. 2008;83(6):1345–1353. doi: 10.1189/jlb.0907658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Painter RG, Marrero L, Lombard GA, Valentine VG, Nauseef WM, Wang G. CFTR-mediated halide transport in phagosomes of human neutrophils. J Leukoc Biol. 2010;87(5):933–942. doi: 10.1189/jlb.1009655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Reeves EP, Williamson M, O'Neill SJ, Greally P, McElvaney NG. Nebulised hypertonic saline decreases interleukin-8 in sputum of patients with cystic fibrosis. Am J Respir Crit Care Med. 2011;183(11):1517–1523. doi: 10.1164/rccm.201101-0072OC. [DOI] [PubMed] [Google Scholar]
- 27.Reeves EP, Lu H, Jacobs HL, et al. Killing activity of neutrophils is mediated through activation of proteases by K+ flux. Nature. 2002;416(6878):291–297. doi: 10.1038/416291a. [DOI] [PubMed] [Google Scholar]
- 28.Bergin DA, Reeves EP, Hurley K, et al. The circulating proteinase inhibitor alpha-1 antitrypsin regulates neutrophil degranulation and autoimmunity. Sci Transl Med. 2014;6(217):217ra211. [DOI] [PubMed]
- 29.Saeki K, Saeki K, Nakahara M, et al. A feeder-free and efficient production of functional neutrophils from human embryonic stem cells. Stem Cells. 2009;27(1):59–67. doi: 10.1634/stemcells.2007-0980. [DOI] [PubMed] [Google Scholar]
- 30.Meshkini A, Yazdanparast R, Nouri K. Intracellular GTP level determines cell’s fate toward differentiation and apoptosis. Toxicol Appl Pharmacol. 2011;253(3):188–196. doi: 10.1016/j.taap.2011.02.021. [DOI] [PubMed] [Google Scholar]
- 31.Bergsson G, Reeves EP, McNally P, et al. LL-37 complexation with glycosaminoglycans in cystic fibrosis lungs inhibits antimicrobial activity, which can be restored by hypertonic saline. J Immunol. 2009;183(1):543–551. doi: 10.4049/jimmunol.0803959. [DOI] [PubMed] [Google Scholar]
- 32.Bergin DA, Reeves EP, Meleady P, et al. α-1 Antitrypsin regulates human neutrophil chemotaxis induced by soluble immune complexes and IL-8. J Clin Invest. 2010;120(12):4236–4250. doi: 10.1172/JCI41196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Scheele JS, Rhee JM, Boss GR. Determination of absolute amounts of GDP and GTP bound to Ras in mammalian cells: comparison of parental and Ras-overproducing NIH 3T3 fibroblasts. Proc Natl Acad Sci USA. 1995;92(4):1097–1100. doi: 10.1073/pnas.92.4.1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McLaughlin CW, Peart D, Purves RD, Carré DA, Macknight AD, Civan MM. Effects of HCO3- on cell composition of rabbit ciliary epithelium: a new model for aqueous humor secretion. Invest Ophthalmol Vis Sci. 1998;39(9):1631–1641. [PubMed] [Google Scholar]
- 35.Simchowitz L, De Weer P. Chloride movements in human neutrophils. Diffusion, exchange, and active transport. J Gen Physiol. 1986;88(2):167–194. doi: 10.1085/jgp.88.2.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lodish HBA, Zipursky SL, Matsudaira P, Baltimore D. Molecular Cell Biology, 4th Ed. W.H. Freeman, New York; 2000. [Google Scholar]
- 37.Caci E, Caputo A, Hinzpeter A, et al. Evidence for direct CFTR inhibition by CFTR(inh)-172 based on Arg347 mutagenesis. Biochem J. 2008;413(1):135–142. doi: 10.1042/BJ20080029. [DOI] [PubMed] [Google Scholar]
- 38.Ma T, Thiagarajah JR, Yang H, et al. Thiazolidinone CFTR inhibitor identified by high-throughput screening blocks cholera toxin-induced intestinal fluid secretion. J Clin Invest. 2002;110(11):1651–1658. doi: 10.1172/JCI16112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pilas B, Durack G. A flow cytometric method for measurement of intracellular chloride concentration in lymphocytes using the halide-specific probe 6-methoxy-N-(3-sulfopropyl) quinolinium (SPQ). Cytometry. 1997;28(4):316–322. [PubMed] [Google Scholar]
- 40.Tashiro M, Konishi M. Basal intracellular free Mg2+ concentration in smooth muscle cells of guinea pig tenia cecum: intracellular calibration of the fluorescent indicator furaptra. Biophys J. 1997;73(6):3358–3370. doi: 10.1016/S0006-3495(97)78360-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Janusonis S. Comparing two small samples with an unstable, treatment-independent baseline. J Neurosci Methods. 2009;179(2):173–178. doi: 10.1016/j.jneumeth.2009.01.017. [DOI] [PubMed] [Google Scholar]
- 42.Kronborg G, Hansen MB, Svenson M, Fomsgaard A, Høiby N, Bendtzen K. Cytokines in sputum and serum from patients with cystic fibrosis and chronic Pseudomonas aeruginosa infection as markers of destructive inflammation in the lungs. Pediatr Pulmonol. 1993;15(5):292–297. doi: 10.1002/ppul.1950150506. [DOI] [PubMed] [Google Scholar]
- 43.Richter J, Ng-Sikorski J, Olsson I, Andersson T. Tumor necrosis factor-induced degranulation in adherent human neutrophils is dependent on CD11b/CD18-integrin-triggered oscillations of cytosolic free Ca2+. Proc Natl Acad Sci USA. 1990;87(23):9472–9476. doi: 10.1073/pnas.87.23.9472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Niessen HW, Verhoeven AJ. Differential up-regulation of specific and azurophilic granule membrane markers in electropermeabilized neutrophils. Cell Signal. 1992;4(5):501–509. doi: 10.1016/0898-6568(92)90019-5. [DOI] [PubMed] [Google Scholar]
- 45.Herrero-Turrión MJ, Calafat J, Janssen H, Fukuda M, Mollinedo F. Rab27a regulates exocytosis of tertiary and specific granules in human neutrophils. J Immunol. 2008;181(6):3793–3803. doi: 10.4049/jimmunol.181.6.3793. [DOI] [PubMed] [Google Scholar]
- 46.Neeft M, Wieffer M, de Jong AS, et al. Munc13-4 is an effector of rab27a and controls secretion of lysosomes in hematopoietic cells. Mol Biol Cell. 2005;16(2):731–741. doi: 10.1091/mbc.E04-10-0923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Borregaard N, Sørensen OE, Theilgaard-Mönch K. Neutrophil granules: a library of innate immunity proteins. Trends Immunol. 2007;28(8):340–345. doi: 10.1016/j.it.2007.06.002. [DOI] [PubMed] [Google Scholar]
- 48.Mollinedo F, Martín-Martín B, Calafat J, Nabokina SM, Lazo PA. Role of vesicle-associated membrane protein-2, through Q-soluble N-ethylmaleimide-sensitive factor attachment protein receptor/R-soluble N-ethylmaleimide-sensitive factor attachment protein receptor interaction, in the exocytosis of specific and tertiary granules of human neutrophils. J Immunol. 2003;170(2):1034–1042. doi: 10.4049/jimmunol.170.2.1034. [DOI] [PubMed] [Google Scholar]
- 49.Stow JL, Manderson AP, Murray RZ. SNAREing immunity: the role of SNAREs in the immune system. Nat Rev Immunol. 2006;6(12):919–929. doi: 10.1038/nri1980. [DOI] [PubMed] [Google Scholar]
- 50.Seabra MC, Wasmeier C. Controlling the location and activation of Rab GTPases. Curr Opin Cell Biol. 2004;16(4):451–457. doi: 10.1016/j.ceb.2004.06.014. [DOI] [PubMed] [Google Scholar]
- 51.Sprang SR, Coleman DE. Invasion of the nucleotide snatchers: structural insights into the mechanism of G protein GEFs. Cell. 1998;95(2):155–158. doi: 10.1016/s0092-8674(00)81746-8. [DOI] [PubMed] [Google Scholar]
- 52.Gupta A, Eastham KM, Wrightson N, Spencer DA. Hypomagnesaemia in cystic fibrosis patients referred for lung transplant assessment. J Cyst Fibros. 2007;6(5):360–362. doi: 10.1016/j.jcf.2007.01.004. [DOI] [PubMed] [Google Scholar]
- 53.Pham CT. Neutrophil serine proteases: specific regulators of inflammation. Nat Rev Immunol. 2006;6(7):541–550. doi: 10.1038/nri1841. [DOI] [PubMed] [Google Scholar]
- 54.Sørensen OE, Follin P, Johnsen AH, et al. Human cathelicidin, hCAP-18, is processed to the antimicrobial peptide LL-37 by extracellular cleavage with proteinase 3. Blood. 2001;97(12):3951–3959. doi: 10.1182/blood.v97.12.3951. [DOI] [PubMed] [Google Scholar]
- 55.Burns JL, Emerson J, Stapp JR, et al. Microbiology of sputum from patients at cystic fibrosis centers in the United States. Clin Infect Dis. 1998;27(1):158–163. doi: 10.1086/514631. [DOI] [PubMed] [Google Scholar]
- 56.Bonfield TL, Hodges CA, Cotton CU, Drumm ML. Absence of the cystic fibrosis transmembrane regulator (Cftr) from myeloid-derived cells slows resolution of inflammation and infection. J Leukoc Biol. 2012;92(5):1111–1122. doi: 10.1189/jlb.0412188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yoshimura K, Nakamura H, Trapnell BC, et al. Expression of the cystic fibrosis transmembrane conductance regulator gene in cells of non-epithelial origin. Nucleic Acids Res. 1991;19(19):5417–5423. doi: 10.1093/nar/19.19.5417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kälin N, Claass A, Sommer M, Puchelle E, Tümmler B. DeltaF508 CFTR protein expression in tissues from patients with cystic fibrosis. J Clin Invest. 1999;103(10):1379–1389. doi: 10.1172/JCI5731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Owen CA, Hu Z, Barrick B, Shapiro SD. Inducible expression of tissue inhibitor of metalloproteinases-resistant matrix metalloproteinase-9 on the cell surface of neutrophils. Am J Respir Cell Mol Biol. 2003;29(3 Pt 1):283–294. doi: 10.1165/rcmb.2003-0034OC. [DOI] [PubMed] [Google Scholar]
- 60.Lau D, Mollnau H, Eiserich JP, et al. Myeloperoxidase mediates neutrophil activation by association with CD11b/CD18 integrins. Proc Natl Acad Sci USA. 2005;102(2):431–436. doi: 10.1073/pnas.0405193102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Perez A, Issler AC, Cotton CU, Kelley TJ, Verkman AS, Davis PB. CFTR inhibition mimics the cystic fibrosis inflammatory profile. Am J Physiol Lung Cell Mol Physiol. 2007;292(2):L383–L395. doi: 10.1152/ajplung.00403.2005. [DOI] [PubMed] [Google Scholar]
- 62.Kamiya H, Ehara T, Matsumoto T. Inhibitory effects of lactoferrin on biofilm formation in clinical isolates of Pseudomonas aeruginosa. J Infect Chemother. 2012;18(1):47–52. doi: 10.1007/s10156-011-0287-1. [DOI] [PubMed] [Google Scholar]
- 63.Böttcher T, Spreer A, Azeh I, Nau R, Gerber J. Matrix metalloproteinase-9 deficiency impairs host defense mechanisms against Streptococcus pneumoniae in a mouse model of bacterial meningitis. Neurosci Lett. 2003;338(3):201–204. doi: 10.1016/s0304-3940(02)01406-4. [DOI] [PubMed] [Google Scholar]
- 64.Renckens R, Roelofs JJ, Florquin S, et al. Matrix metalloproteinase-9 deficiency impairs host defense against abdominal sepsis. J Immunol. 2006;176(6):3735–3741. doi: 10.4049/jimmunol.176.6.3735. [DOI] [PubMed] [Google Scholar]
- 65.Kuroda TS, Fukuda M, Ariga H, Mikoshiba K. The Slp homology domain of synaptotagmin-like proteins 1-4 and Slac2 functions as a novel Rab27A binding domain. J Biol Chem. 2002;277(11):9212–9218. doi: 10.1074/jbc.M112414200. [DOI] [PubMed] [Google Scholar]
- 66.Novak EK, Gautam R, Reddington M, et al. The regulation of platelet-dense granules by Rab27a in the ashen mouse, a model of Hermansky-Pudlak and Griscelli syndromes, is granule-specific and dependent on genetic background. Blood. 2002;100(1):128–135. doi: 10.1182/blood.v100.1.128. [DOI] [PubMed] [Google Scholar]
- 67.Ménasché G, Pastural E, Feldmann J, et al. Mutations in RAB27A cause Griscelli syndrome associated with haemophagocytic syndrome. Nat Genet. 2000;25(2):173–176. doi: 10.1038/76024. [DOI] [PubMed] [Google Scholar]
- 68.Abdel-Latif D, Steward M, Macdonald DL, Francis GA, Dinauer MC, Lacy P. Rac2 is critical for neutrophil primary granule exocytosis. Blood. 2004;104(3):832–839. doi: 10.1182/blood-2003-07-2624. [DOI] [PubMed] [Google Scholar]
- 69.Munafó DB, Johnson JL, Ellis BA, Rutschmann S, Beutler B, Catz SD. Rab27a is a key component of the secretory machinery of azurophilic granules in granulocytes. Biochem J. 2007;402(2):229–239. doi: 10.1042/BJ20060950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Brzezinska AA, Johnson JL, Munafo DB, et al. The Rab27a effectors JFC1/Slp1 and Munc13-4 regulate exocytosis of neutrophil granules. Traffic. 2008;9(12):2151–2164. doi: 10.1111/j.1600-0854.2008.00838.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Johnson JL, Brzezinska AA, Tolmachova T, et al. Rab27a and Rab27b regulate neutrophil azurophilic granule exocytosis and NADPH oxidase activity by independent mechanisms. Traffic. 2010;11(4):533–547. doi: 10.1111/j.1600-0854.2009.01029.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Catz SD. Regulation of vesicular trafficking and leukocyte function by Rab27 GTPases and their effectors. J Leukoc Biol. 2013;94(4):613–622. doi: 10.1189/jlb.1112600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lazrak A, Jurkuvenaite A, Chen L, et al. Enhancement of alveolar epithelial sodium channel activity with decreased cystic fibrosis transmembrane conductance regulator expression in mouse lung. Am J Physiol Lung Cell Mol Physiol. 2011;301(4):L557–L567. doi: 10.1152/ajplung.00094.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ando-Akatsuka Y, Abdullaev IF, Lee EL, Okada Y, Sabirov RZ. Down-regulation of volume-sensitive Cl- channels by CFTR is mediated by the second nucleotide-binding domain. Pflugers Arch. 2002;445(2):177–186. doi: 10.1007/s00424-002-0920-z. [DOI] [PubMed] [Google Scholar]
- 75.Yoo D, Flagg TP, Olsen O, Raghuram V, Foskett JK, Welling PA. Assembly and trafficking of a multiprotein ROMK (Kir 1.1) channel complex by PDZ interactions. J Biol Chem. 2004;279(8):6863–6873. doi: 10.1074/jbc.M311599200. [DOI] [PubMed] [Google Scholar]
- 76.Berdiev BK, Qadri YJ, Benos DJ. Assessment of the CFTR and ENaC association. Mol Biosyst. 2009;5(2):123–127. doi: 10.1039/b810471a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Oglesby IK, Chotirmall SH, McElvaney NG, Greene CM. Regulation of cystic fibrosis transmembrane conductance regulator by microRNA-145, -223, and -494 is altered in ΔF508 cystic fibrosis airway epithelium. J Immunol. 2013;190(7):3354–3362. doi: 10.4049/jimmunol.1202960. [DOI] [PubMed] [Google Scholar]
- 78.Mall M, Bleich M, Greger R, Schreiber R, Kunzelmann K. The amiloride-inhibitable Na+ conductance is reduced by the cystic fibrosis transmembrane conductance regulator in normal but not in cystic fibrosis airways. J Clin Invest. 1998;102(1):15–21. doi: 10.1172/JCI2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Vormann J, Magdorf K, Günther T, Wahn U. Increased Na+/Mg2+ antiport in erythrocytes of patients with cystic fibrosis. Eur J Clin Chem Clin Biochem. 1994;32(11):833–836. doi: 10.1515/cclm.1994.32.11.833. [DOI] [PubMed] [Google Scholar]
- 80.Van Goor F, Hadida S, Grootenhuis PD, et al. Rescue of CF airway epithelial cell function in vitro by a CFTR potentiator, VX-770. Proc Natl Acad Sci USA. 2009;106(44):18825–18830. doi: 10.1073/pnas.0904709106. [DOI] [PMC free article] [PubMed] [Google Scholar]