

Abstract
Studies have demonstrated that reactive oxygen species (ROS) are closely related to inflammatory disorders. Nicotinamide adenine dinucleotide phosphate oxidase (NOX), originally found in phagocytes, is the main source of ROS in nonphagocytic cells. Besides directly producing the detrimental highly reactive ROS to act on biomolecules (lipids, proteins, and nucleic acids), NOX can also activate multiple signal transduction pathways, which regulate cell growth, proliferation, differentiation and apoptosis by producing ROS. Recently, research on pancreatic NOX is no longer limited to inflammatory cells, but extends to the aspect of pancreatic acinar cells and pancreatic stellate cells, which are considered to be potentially associated with pancreatitis. In this review, we summarize the literature on NOX protein structure, activation, function and its role in the pathogenesis of pancreatitis.
Keywords: Nicotinamide adenine dinucleotide phosphate oxidase, Reactive oxygen species, Pancreatitis, Pancreatic acinar cells, Pancreatic stellate cells
Core tip: Besides directly producing the detrimental highly reactive reactive oxygen species (ROS) to act on biomolecules, nicotinamide adenine dinucleotide phosphate (NADPH) oxidase can also activate multiple signal transduction pathways, which regulate cell growth, proliferation, differentiation and apoptosis by producing ROS. Recently, research on pancreatic NADPH oxidase is no longer limited to inflammatory cells, but extends to the aspect of pancreatic acinar cells and pancreatic stellate cells, which are considered to be potentially associated with pancreatitis.
INTRODUCTION
Studies have demonstrated that reactive oxygen species (ROS) are involved in the pathogenesis of pancreatitis[1]. Nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (NOX), a transmembrane flavoprotein enzyme, uses NADPH as an electron donor to catalyze the univalent reduction of oxygen, resulting in the production of superoxide free radical, which might be a source of oxidants in injured pancreas[1]. NOX is mainly distributed in the phagocytic cell membrane with cytochrome C and flavin adenine dinucleotide groups, which can produce ROS, scavenging pathogenic microorganisms such as bacteria[2]. ROS, being generated by NOX, also participate in intracellular signaling processes in the pancreas. Recently, research on NOX is no longer limited to inflammatory cells, but extends to the aspect of pancreatic acinar cells and pancreatic stellate cells (PSCs) in pancreatitis patients[2]. The function of NOX, which is involved in the pathogenesis of inflammation in pancreatic acinar cells and PSCs, has become the hotspot of research. Non-phagocytic NOX derived ROS function as a messenger molecule to participate in the modulation of cell differentiation, proliferation and apoptosis in the pancreas. In this review, we summarize the literature on NOX protein structure, activation, function and its role in the pathogenesis of pancreatitis.
STRUCTURE, LOCATION AND FUNCTION OF NOX IN THE PANCREAS
NOX is a multicomponent enzyme consisting of five different subunits, including the subunits p22phox and gp91phox (also known as NOX2) located in the membrane, together with the cytosolic subunits p40phox, p47phox and p67phox. The participation of Rac would elicit full oxidase activity[3-5]. Relative to gp91phox (the catalytic subunit of NOX), p22phox, p47phox, p40phox and p67phox are regulatory subunits. Gp91phox in different types of cells has other six homologues, termed NOX1, NOX3, NOX4, NOX5, DUOX1 and DUOX2, which constitute the NOX family proteins[6-8]. NOX is an enzyme which was initially discovered in phagocytes[4,5]. NOX in neutrophils is composed of constitutive subunits (p22phox and gp91phox) positioned in membrane and regulatory subunits (p47phox and p67phox, and possibly p40phox) stationed in the cytosol[9]. In recent years, NOX has been discovered in several nonphagocytic cells such as fibroblasts[10], vascular smooth muscle cells[11] and hepatic stellate cells[12]. More recently, it has been found that NOX was present in pancreatic β cells[13,14], pancreatic acinar cells[15-18] and PSCs[19,20]. The main intrinsic components of NOX comprising the NOX2 isoform are present in human pancreatic islets[14]. Cytosolic subunits p47phox and p67phox as well as membrane-bound subunits p22phox and NOX1 are constitutively expressed in pancreatic acinar AR42J cells[16,21,22]. The key subunits of NOX including p22phox, p47phox, NOX activator 1 (a homologue of p67phox), NOX1, NOX4, and NOX2 (gp91phox) are expressed in PSCs[19,20]. The activation of non-phagocytic NOX is similar to that in neutrophils[23]. Upon activation of NOX, p47 translocates to the membrane and then recruits p67 to interact with the p22 subunit, thus facilitating NADPH-dependent formation of superoxide (O2-), which increases the production of secondary ROS such as hydrogen peroxide (H2O2)[21]. Non-phagocytic NOX derived ROS function as a messenger molecule to participate in the modulation of cell differentiation, proliferation and apoptosis[6-8]. NOX protein family can be activated quickly under pathophysiological conditions, leading to high production of ROS, which contributes to oxidative stress and a wide range of diseases.
ACTIVATION AND INHIBITION FACTORS OF NOX IN THE PATHOGENESIS OF PANCREATITIS
Cholecystokinin analogues
Cerulein, an analogue of cholecystokinin (CCK), can stimulate the pancreatic exocrine secretion by binding CCK receptors, causing the autolysis of pancreatic acinar cell[24]. There are two kinds of CCK receptor subtypes, CCK1 and CCK2 receptors. CCK1 receptors regulate pancreatic digestive enzymes, satiety and feeding behavior, while CCK2 receptors enhance the level of gastric acid, as well as gastrin which has anti-apoptotic effects on pancreatic cells[25]. Experimental pancreatitis induced with high dosages of cerulean, similar to human edematous pancreatitis, is characterized by cytoplasmic vacuolization, formation of edema and acinar cell death as well as elevation in serum levels of digestive enzymes caused by unconventional secretion of digestive enzymes[26]. ROS are involved in the activation of oxidant-sensitive nuclear transcription factor (NF-κB), expression of cytokine, apoptosis and further occurrence of pancreatitis[27]. P47phox, p67phox, NOX1 and p22phox in pancreatic AR42J cells could produce ROS after cerulein stimulation[21]. Intrapancreatic trypsin is not only activated by high-dose cerulein, but also regulated by neutrophils via NADPH oxidase[28]. The mechanism for the activation of NF-κB and expression of cytokines in pancreatic acinar cells stimulated by cerulein may be summarized as the following steps. Cerulein binds to the CCK receptor, a G-protein-coupled receptor, to activate phospholipase C (PLC) and inositol 1,4,5-trisphosphate (IP3), triggering transient Ca2+ release from the endoplasmic reticulum in pancreatic acinar cells. NOX activated by Ca2+ produces ROS to activate IκB kinase and then to phosphorylate IκB. Phosphorylated IκB can be ubiquitinated and degraded in a proteasome dependent manner to eliminate the inhibition of NF-κB, a p65/p50 heterodimer in the cytosol. NF-κB then translocates to the nucleus to mediate the expression of cytokines which are involved in the pathogenesis of pancreatitis (Figures 1 and 2)[27].
Figure 1.
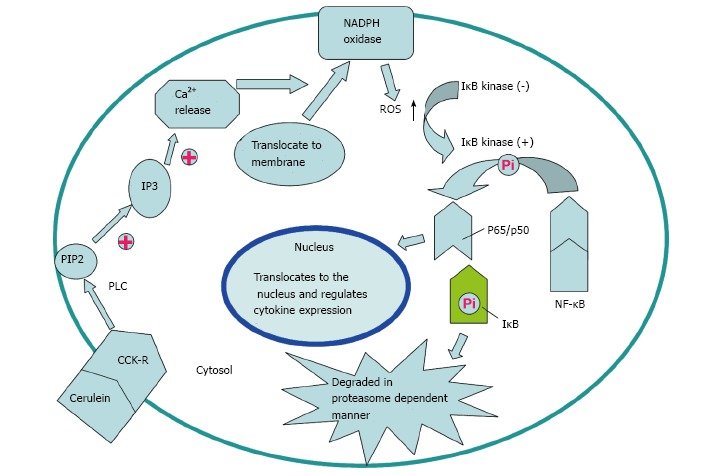
Potential mechanism of nicotinamide adenine dinucleotide phosphate oxidase activation via cholecystokinin receptor. Cerulein and cholecystokinin (CCK) receptor binding triggers transient Ca2+ release from the endoplasmic reticulum to activate nicotinamide adenine dinucleotide phosphate (NADPH) oxidase, which is mediated by PLC and IP3. Reactive oxygen species (ROS) generated by NADPH oxidase activate IκB kinase to phosphorylate IκB in the cytosol. Phosphorylated IκB is ubiquitinated and degraded in a proteasome-dependent manner. NF-κB translocates to the nucleus and regulates expression of cytokines to participate in the pathogenesis of pancreatitis.
Figure 2.
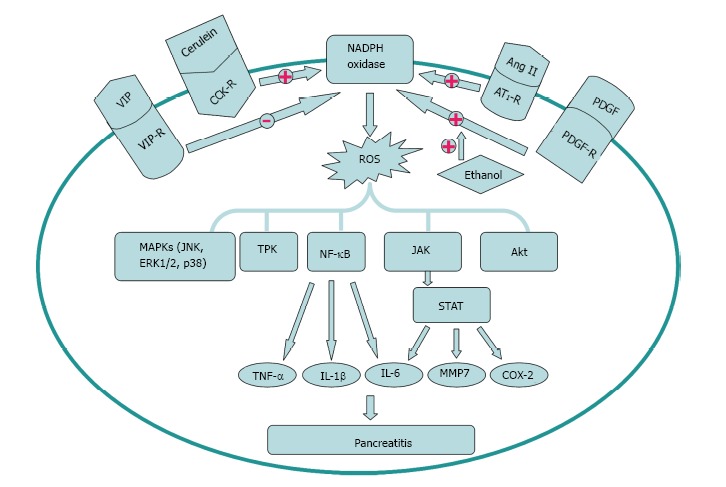
Activation and inhibition factors of nicotinamide adenine dinucleotide phosphate oxidase signal transduction in the pathogenesis of pancreatitis. Cerulein, Ang II and platelet derived growth factor (PDGF) can enhance, while VIP can decrease the activation of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase. Ethanol can augment the activation of the cell’s NADPH oxidase system stimulated by PDGF. The downstream signal molecules including MAPKs, TPK, NF-κB, JAK/STAT and Akt participate in the pathogenesis of pancreatitis. TNF: Tumor necrosis factor; IL: Interleukin; TPK: Tyrosine protein kinase; MAPK: Mitogen activated protein kinase.
Renin-angiotensin system
The Renin-angiotensin system (RAS) is generally considered to regulate blood pressure and body fluid homeostasis[29]. The pancreatic RAS activation that is related to the production of ROS might contribute to oxidative stress and tissue injury[30,31]. Angiotensin II, an active mediator of RAS, is transformed from angiotensin I by the angiotensin-converting enzyme (ACE)[32]. The effect of angiotensin II is regulated by its receptors, including angiotensin II type 1 receptor (AT1R) and angiotensin II type 2 receptor (AT2R)[32]. Many reports indicate that interaction of angiotensin II with AT1R promotes superoxide anion production through NOX system[30,31,33,34]. Inhibition of the AT1R, but not AT2R, may play a significant role in decreasing the severity of acute pancreatitis. Mechanism of NOX activation by AT1R and AT2R might contribute to different effects of AT1R and AT2R inhibitors on pancreatic injury induced by cerulein. Activation of pancreatic NOX was associated with oxidative stress which can be indicated by the level of protein oxidation in rats stimulated with cerulein[30,35]. However, further investigations about the potential application of RAS inhibitors including AT1R in treating acute pancreatitis are needed in the future (Figure 2).
Ethanol and platelet derived growth factor
Alcohol abuse has long been recognized as the most common factor leading to chronic pancreatitis[36]. Activated stellate cells are viewed as vital regulators of chronic alcoholic pancreatitis or fibrosis. Hu et al[20] investigated the mechanisms of action of alcohol on PSCs to determine the correlation of NOX system and alcohol with the proliferation of PSCs. The results demonstrated that NOX activity was predominantly located in the cell membrane fraction (95%) compared to the cytosolic fraction (5%) of the stellate cells. platelet derived growth factor (PDGF) could increase NOX activity in a dose- and time-dependent manner. PSC proliferation caused by alcohol is mediated by the activation of PDGF induced NADPH oxidase system. However, ethanol did not show a significant effect on stellate cell DNA synthesis, which provides a new perspective for the mechanism of fibrosis stimulated with alcohol (Figure 2)[20].
Vasoactive intestinal peptide
Previous reports found that vasoactive intestinal peptide (VIP) could decrease the production of cytokines to alleviate experimental acute pancreatitis[37]. VIP could decrease the level of ROS significantly and increase cell viability in acini cells in a dose dependent manner. NOX1 and NOX2 markedly increased following treatment with H2O2 in pancreatic acini. Besides, H2O2 can stimulate the activation of NOX. The production of ROS was affected by VIP via NADPH oxidase and the cAMP/PKA pathway because decreased NOX activity by administration of VIP could be abolished by PKA inhibitor H89. Oxidative stress and tissue injury in acini can be decreased by VIP through NOX inhibition (Figure 2)[38].
NOX SIGNAL TRANSDUCTION IN THE PATHOGENESIS OF PANCREATITIS
NOX protein family can be activated quickly under pathophysiological conditions, leading to high production of ROS, which contributes to oxidative stress and a wide range of diseases. Furthermore, ROS can act as an intracellular second messenger or chemoattractant to enhance the level of cytokines, resulting in the aggravation of pancreatitis[38]. Studies indicate that pro-inflammation cytokines such as IL-1β, IL-6 and TNF-α mediate the local or systemic manifestations of acute pancreatitis. IL-1β and TNF-α released from activated pancreatic macrophages respond to local tissue damage. Locally, these cytokines may aggravate the severity of acute pancreatitis. Systemically, IL-6 can increase the capillary permeability and accelerate the leukocyte adherence, leading to multiple organ failure (Figure 2)[27].
NF-κB and Janus kinase/signal transducers and activators of transcription
NF-κB, a member of the Rel family of transcription factors, can regulate the activation of cellular stress-related genes or early response genes such as growth factors, cytokines, adhesion molecules, and acute-phase proteins[39,40]. The Janus kinase/signal transducers and activators of transcription (JAK/STAT) signaling pathway was relevant to the immune response mediated by numerous cytokines and non-immune response mediated by hormones and growth factors. The JAK/STAT pathway activated by the family of cytokine receptors regulate a variety of biological processes, such as immune response, cell survival, differentiation, proliferation and oncogenesis[41]. Recently, reports indicated that cerulein could activate the JAK2/STAT3 pathway through NOX in pancreatic acinar cells[27].
NOX may be the source of ROS in pancreatic acinar cells during pancreatitis. ROS can induce expression of cytokines, apoptosis, NF-κB and JAK/STAT pathway activation, thus regulating the inflammation and apoptosis in pancreatic acinar cells. Consequently, NOX, NF-κB and JAK2/STAT3 may be involved in the pathogenesis of acute pancreatitis[27]. Inflammation and apoptosis in pancreatic acinar cells during pancreatitis may be alleviated by inhibition of NOX, NF-κB and JAK/STAT through suppression of inflammatory cytokines, apoptosis and caspase-3 activity. Ju et al[23] found that NOX inhibition suppresses STAT3-DNA binding, JAK2/STAT3 activation and TGF-β1 level in AR42J cells stimulated by cerulein. Therefore, ROS may activate NF-κB to induce cytokine production in pancreatic acinar cells through activation of NOX during pancreatitis[21]. NOX, NF-κB and JAK/STAT may be potential targets for treatment of acute pancreatitis.
Mitogen activated protein kinase and tyrosine protein kinase
Recently, studies found that mitogen activated protein kinase (MAPK) and tyrosine protein kinase (TPK) might be involved in NOX signal transduction pathway. ROS induced by the family of NOX can cause protein phosphorylation and cell apoptosis directly or indirectly.
In the direct way, ROS mediate the activation of the MAPK pathway and TPK pathway to promote protein phosphorylation in pancreatic acinar cells. ROS activate the signal transduction pathway which consists of different MAPK family members probably owing to the activation of the upstream ERK1/2 kinase pathway. ROS stimulate TPK signaling pathway through increasing the TPK activity, thereby promoting protein tyrosine phosphorylation and affecting signal transduction to regulate cell proliferation, differentiation, metabolism and apoptosis. Inhibition of NOX or ROS significantly reduced the p38MAPK signaling cascade[42]. Activation of the MAPK signaling pathway including SAPK/JNK, ERK1/2 and p38 by ROS induce cell apoptosis. The activation of the MAPK pathway is mainly dependent on the inhibition of tyrosine phosphatase by ROS[43].
In the indirect way, ROS reduce phosphatase activity, decrease protein dephosphorylation, and thus indirectly increase protein phosphorylation. ROS injure DNA, lipid and protein, thus indirectly inducing apoptosis. In some cases, NOX family can also inhibit cell apoptosis through ROS, which activate the pathway of NF-κB and Akt/ASK1, thereby reducing cell apoptosis[44].
NOX ACTIVATION IN DIFFERENT PANCREATIC CELLS INVOLVED IN THE PATHOGENESIS OF PANCREATITIS
Phagocytes
In support of the involvement of oxygen free radicals in acute pancreatitis, studies have addressed the possibility that the severity of pancreatitis can be reduced by inhibiting the activity of oxygen-derived free radicals[45]. ROS could have different origins, and the role of the NOX system in neutrophils but not pancreatic acinar tissue is originally considered essential. The phagocytic NOX is a multicomponent enzyme complex that is composed of membranous and cytosolic proteins in the resting cell. During activation, approximately 10% of cytosolic proteins including p47phox and p67phox are phosphorylated and translocate to the cell membrane to form active catalytic complexes with p22phox and gp91phox, resulting in the generation of ROS[4]. Intrapancreatic trypsin activation and acinar cell trypsin-activation peptide (TAP) labeling induced by high dose cerulein were significantly decreased in neutrophil depleted rats. NOX deficient mice displayed attenuation of the cerulean-induced trypsin activation, while myeloperoxidase (MPO) deficient mice did not. Neutrophils have been considered to be implicated in pathologic activation of digestive enzymes by infiltrating the pancreas in acute pancreatitis, which is mediated by products of NOX[28].
Evidence suggests that inflammatory cell infiltration is an early and vital event in acute pancreatitis, which will lead to local and systemic complications[46]. Many of the pathological failures of acute pancreatitis may be a consequence of the overstimulation of leukocytes[47]. The argument put forward was that once pancreatitis has been initiated, chemoattractants for polymorphonuclear leukocytes, macrophages and platelets are released, possibly via the action of oxygen derived free radicals. The chemoattractants induce leukocytes and macrophages to adhere to the endothelium of the postcapillary venule and to migrate into the interstitial spaces. Stimulus-secretion coupling causes synthesis of a range of enzymes including elastase, cathepsins, phospholipase A2, phospholipase C, platelet-activating factor (PAF) and MPO. When the quantity of material to be digested is excessive, phagocytosis may become so vigorous that the contents of leukocyte and macrophage granules are spilled outside the cell where they increase the severity of inflammation. As a result, large amounts of oxygen-derived free radicals are produced and may exceed the capacity of superoxide dismutase (SOD) and catalase to inactivate them[48].
Pancreatic acinar cells
ROS and apoptosis can be observed in pancreatic acinar cells in cerulein induced pancreatitis[49,50]. NADPH has been considered to be the major source of ROS in pancreatitis[18,21,22]. Oxidative stress induced inflammation and apoptosis have been implicated in pancreatitis[51,52]. Cerulein induced the expression of apoptosis-inducing factor (AIF). AIF is located in the mitochondrial membrane of pancreatic acinar cells. During apoptosis, AIF translocates from mitochondria to the cytoplasm and then enters into the nucleus, resulting in nuclear DNA aggregation and breakage to induce apoptosis of pancreatic acinar cells[53,54]. Antisense oligonucleotides (AS ODN) transfection or Ca2+ chelator treatment decreased the expression of AIF induced by cerulein in AR42J cells. These results suggested that intracellular Ca2+ increase and NOX activation might be the upstream events of AIF expression, which result in cerulein induced apoptosis of AR42J cells[18,55].
The activation of NOX was inhibited and the production of ROS was decreased when cerulein-stimulated pancreatic acinar cells were treated with Ca2+ chelator, which indicates that Ca2+ activate NOX and ROS. Transfection with AS ODN for NOX subunits p22phox and p47phox can inhibit the ROS generation, illustrating that NOX mediates the production of ROS. The apoptotic indices including apoptotic genes bax and p53, DNA fragmentation, caspase 3 activity, TUNEL staining and cell viability were inhibited by treatment with Ca2+ chelator or AS ODN transfection, indicating that NOX regulates ROS-induced apoptosis in a Ca2+ dependent manner in pancreatic acinar cells[22]. Diphenyleneiodonium (DPI), an inhibitor of NOX, reduces the AIF expression and caspase-3 activation, and thus inhibits apoptosis of AR42J cells[16]. During the stimulation with cerulein, the increase of NOX accelerates the formation of ROS in cells and mitochondria, thus further inducing the apoptosis of acinar cells[56,57]. ROS generated by pancreatic acinar cells stimulated with bile acids or cerulein can induce apoptosis and, at the same time, induce pancreatitis[58-60].
Research indicates that JAK2/STAT3 activation and increases of MAPKs and TGF-β1 induced by administration of cerulein were inhibited by AS ODN transfection in AR42J cells, which shows that NOX can activate JAK2/STAT3, MAPKs and TGF-β1[23]. NOX may regulate the production of cytokines by activating NF-κB in AR42J cells stimulated with cerulein. Rebamipide, an antiulcer agent, can scavenge ROS and decrease the level of superoxide[61,62]. Transfection with AS ODN for NOX subunits or administration of DPI or rebamipide inhibited cerulein induced NF-κB activation and IL-6 expression[21]. Cerulein also could produce large amounts of ROS to activate NF-κB and thus stimulate the expression of cytokines in freshly isolated pancreatic acinar cells without inflammation[63].
Numerous studies have shown that increases of ROS and peroxidation products are accompanied with endogenous antioxidant depletion in the early stage of pancreatitis. Many preclinical antioxidant treatments, including genetic manipulation, significantly reduce pancreatic injury and inflammation[1,64-66]. However, randomized clinical trials of antioxidants have produced conflicting results[67], and treatment of pancreatitis with antioxidants has even been discontinued because of adverse events[68]. Moreover, several studies indicated that NOX was only present in neutrophils but not in pancreatic acinar cells[28,69].
PSCs
PSCs are the major fibrogenic cells in chronic injury of the pancreas, which encircle the acinus[70,71]. PSCs account for approximately 4% of the total pancreatic cells[72]. PSCs are quiescent in normal pancreas and can be identified by the character of vitamin A containing lipid droplets in the cytoplasm. When chronic pancreatitis happens, PSCs are activated and transformed into myofibroblast-like cells. As a result, intracellular lipid droplets disappear and α-smooth muscle actin (α-SMA) and extracellular components such as fibronectin and collagen arise[19,73]. Besides, PSCs may be involved in the pathogenesis of acute pancreatitis[72]. Therefore, suppression of PSC activation is a potential target to treat pancreatic inflammation and fibrosis.
Studies showed that p22phox, p47phox, NOX1, gp91phox (NOX2), and NOX4 were expressed in rat quiescent and culture-activated PSCs as well as human activated PSCs, while p67phox and NOX3 were not detected. NOX activator 1 was present in human PSCs, while NOX organizer 1 was not detected. NOX can activate PSCs, which can be verified by DPI inhibition experiments. Studies showed that DPI could inhibit the activation of PSCs, that is, to inhibit proliferation, chemokine production, α-SMA and collagen expression. Platelet-derived growth factor BB (PDGF-BB) promoted proliferation of rat PSCs, which was inhibited by DPI in a dose-dependent manner, showing that NOX underlies the PDGF induced PSC proliferation. DPI decreased the chemokine production, which indicates that NOX also regulates the production of chemokines. DPI decreased the levels of α-SMA and collagen, once again, proving that NOX activate PSCs. DPI also inhibited interleukin 1β (IL-1β) and PDGF induced activation of MAPKs in PSCs, and this evidence indicates that NOX mediates the activation of MAPKs induced by IL-1β and PDGF in PSCs[19].
FUTURE RESEARCH ON THE PATHOGENESIS OF PANCREATITIS IN NOX
Accumulated evidence suggested that ROS induced by NOX play a significant role in pancreatitis. The activation of ROS mediates the activation of many cytokines[56,57]. ROS can induce cell apoptosis through direct and indirect pathways[43,44]. ROS induced by bile acids and cerulein can promote apoptosis of pancreatic acinar cells[18,69]. NOX is usually induced by cerulein, inflammatory factors, cytokines and growth factors as well as other stimuli in pancreatic acinar cells and PSCs. NOX can generate ROS, which in turn increase cytokines levels downstream to initiate the next activation cycle. The positive feedback of activation process might be one of the causes of pancreatitis. Although many scholars have made a great deal of research about the pathogenic mechanisms of NOX in the inflammation of pancreatic acinar cells and stellate cells, the relative importance of different pathogenic mechanisms of NOX in the pathogenesis of pancreatitis, the relationship between various pathogenic mechanisms of NOX, the specific pathways involved in each mechanism of NOX in pancreatitis, and the feasibility of NOX targeted therapy applied to pancreatitis are all needed to be studied in the future.
Footnotes
Supported by The National Natural Science Foundation of China, No. 81173393; the Natural Science Foundation of Tianjin City, No. 12YFJZJC00800; the Scientific Research Foundation (No. WHM201222, FYM201114) and the Innovation Team Program (No. WHTD201310) from Logistics University of the Chinese People’s Armed Police Forces
P- Reviewer: Coelho AMM S- Editor: Wen LL L- Editor: Wang TQ E- Editor: Lu YJ
References
- 1.Leung PS, Chan YC. Role of oxidative stress in pancreatic inflammation. Antioxid Redox Signal. 2009;11:135–165. doi: 10.1089/ars.2008.2109. [DOI] [PubMed] [Google Scholar]
- 2.Brown DI, Griendling KK. Nox proteins in signal transduction. Free Radic Biol Med. 2009;47:1239–1253. doi: 10.1016/j.freeradbiomed.2009.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lambeth JD. NOX enzymes and the biology of reactive oxygen. Nat Rev Immunol. 2004;4:181–189. doi: 10.1038/nri1312. [DOI] [PubMed] [Google Scholar]
- 4.Babior BM. NADPH oxidase: an update. Blood. 1999;93:1464–1476. [PubMed] [Google Scholar]
- 5.Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev. 2007;87:245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- 6.Babior BM. The respiratory burst oxidase. Curr Opin Hematol. 1995;2:55–60. doi: 10.1097/00062752-199502010-00008. [DOI] [PubMed] [Google Scholar]
- 7.Lambeth JD, Cheng G, Arnold RS, Edens WA. Novel homologs of gp91phox. Trends Biochem Sci. 2000;25:459–461. doi: 10.1016/s0968-0004(00)01658-3. [DOI] [PubMed] [Google Scholar]
- 8.Cheng G, Cao Z, Xu X, van Meir EG, Lambeth JD. Homologs of gp91phox: cloning and tissue expression of Nox3, Nox4, and Nox5. Gene. 2001;269:131–140. doi: 10.1016/s0378-1119(01)00449-8. [DOI] [PubMed] [Google Scholar]
- 9.Pongnimitprasert N, El-Benna J, Foglietti MJ, Gougerot-Pocidalo MA, Bernard M, Braut-Boucher F. Potential role of the “NADPH oxidases” (NOX/DUOX) family in cystic fibrosis. Ann Biol Clin (Paris) 2008;66:621–629. doi: 10.1684/abc.2008.0285. [DOI] [PubMed] [Google Scholar]
- 10.Meier B, Cross AR, Hancock JT, Kaup FJ, Jones OT. Identification of a superoxide-generating NADPH oxidase system in human fibroblasts. Biochem J. 1991;275(Pt 1):241–245. doi: 10.1042/bj2750241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Griendling KK, Sorescu D, Ushio-Fukai M. NAD(P)H oxidase: role in cardiovascular biology and disease. Circ Res. 2000;86:494–501. doi: 10.1161/01.res.86.5.494. [DOI] [PubMed] [Google Scholar]
- 12.Bataller R, Schwabe RF, Choi YH, Yang L, Paik YH, Lindquist J, Qian T, Schoonhoven R, Hagedorn CH, Lemasters JJ, et al. NADPH oxidase signal transduces angiotensin II in hepatic stellate cells and is critical in hepatic fibrosis. J Clin Invest. 2003;112:1383–1394. doi: 10.1172/JCI18212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mohammed AM, Syeda K, Hadden T, Kowluru A. Upregulation of phagocyte-like NADPH oxidase by cytokines in pancreatic beta-cells: attenuation of oxidative and nitrosative stress by 2-bromopalmitate. Biochem Pharmacol. 2013;85:109–114. doi: 10.1016/j.bcp.2012.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rebelato E, Mares-Guia TR, Graciano MF, Labriola L, Britto LR, Garay-Malpartida HM, Curi R, Sogayar MC, Carpinelli AR. Expression of NADPH oxidase in human pancreatic islets. Life Sci. 2012;91:244–249. doi: 10.1016/j.lfs.2012.07.004. [DOI] [PubMed] [Google Scholar]
- 15.Criddle DN, Gillies S, Baumgartner-Wilson HK, Jaffar M, Chinje EC, Passmore S, Chvanov M, Barrow S, Gerasimenko OV, Tepikin AV, et al. Menadione-induced reactive oxygen species generation via redox cycling promotes apoptosis of murine pancreatic acinar cells. J Biol Chem. 2006;281:40485–40492. doi: 10.1074/jbc.M607704200. [DOI] [PubMed] [Google Scholar]
- 16.Yu JH, Kim KH, Kim DG, Kim H. Diphenyleneiodonium suppresses apoptosis in cerulein-stimulated pancreatic acinar cells. Int J Biochem Cell Biol. 2007;39:2063–2075. doi: 10.1016/j.biocel.2007.05.021. [DOI] [PubMed] [Google Scholar]
- 17.Kim H. Inhibitory mechanism of lycopene on cytokine expression in experimental pancreatitis. Ann N Y Acad Sci. 2011;1229:99–102. doi: 10.1111/j.1749-6632.2011.06107.x. [DOI] [PubMed] [Google Scholar]
- 18.Yu JH, Kim KH, Kim H. Role of NADPH oxidase and calcium in cerulein-induced apoptosis: involvement of apoptosis-inducing factor. Ann N Y Acad Sci. 2006;1090:292–297. doi: 10.1196/annals.1378.031. [DOI] [PubMed] [Google Scholar]
- 19.Masamune A, Watanabe T, Kikuta K, Satoh K, Shimosegawa T. NADPH oxidase plays a crucial role in the activation of pancreatic stellate cells. Am J Physiol Gastrointest Liver Physiol. 2008;294:G99–G108. doi: 10.1152/ajpgi.00272.2007. [DOI] [PubMed] [Google Scholar]
- 20.Hu R, Wang YL, Edderkaoui M, Lugea A, Apte MV, Pandol SJ. Ethanol augments PDGF-induced NADPH oxidase activity and proliferation in rat pancreatic stellate cells. Pancreatology. 2007;7:332–340. doi: 10.1159/000105499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yu JH, Lim JW, Kim H, Kim KH. NADPH oxidase mediates interleukin-6 expression in cerulein-stimulated pancreatic acinar cells. Int J Biochem Cell Biol. 2005;37:1458–1469. doi: 10.1016/j.biocel.2005.02.004. [DOI] [PubMed] [Google Scholar]
- 22.Yu JH, Lim JW, Kim KH, Morio T, Kim H. NADPH oxidase and apoptosis in cerulein-stimulated pancreatic acinar AR42J cells. Free Radic Biol Med. 2005;39:590–602. doi: 10.1016/j.freeradbiomed.2005.04.019. [DOI] [PubMed] [Google Scholar]
- 23.Ju KD, Lim JW, Kim KH, Kim H. Potential role of NADPH oxidase-mediated activation of Jak2/Stat3 and mitogen-activated protein kinases and expression of TGF-β1 in the pathophysiology of acute pancreatitis. Inflamm Res. 2011;60:791–800. doi: 10.1007/s00011-011-0335-4. [DOI] [PubMed] [Google Scholar]
- 24.Yu JH, Seo JY, Kim KH, Kim H. Differentially expressed proteins in cerulein-stimulated pancreatic acinar cells: implication for acute pancreatitis. Int J Biochem Cell Biol. 2008;40:503–516. doi: 10.1016/j.biocel.2007.09.001. [DOI] [PubMed] [Google Scholar]
- 25.Noble F, Wank SA, Crawley JN, Bradwejn J, Seroogy KB, Hamon M, Roques BP. International Union of Pharmacology. XXI. Structure, distribution, and functions of cholecystokinin receptors. Pharmacol Rev. 1999;51:745–781. [PubMed] [Google Scholar]
- 26.Lerch MM, Adler G. Experimental animal models of acute pancreatitis. Int J Pancreatol. 1994;15:159–170. [PubMed] [Google Scholar]
- 27.Kim H. Cerulein pancreatitis: oxidative stress, inflammation, and apoptosis. Gut Liver. 2008;2:74–80. doi: 10.5009/gnl.2008.2.2.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gukovskaya AS, Vaquero E, Zaninovic V, Gorelick FS, Lusis AJ, Brennan ML, Holland S, Pandol SJ. Neutrophils and NADPH oxidase mediate intrapancreatic trypsin activation in murine experimental acute pancreatitis. Gastroenterology. 2002;122:974–984. doi: 10.1053/gast.2002.32409. [DOI] [PubMed] [Google Scholar]
- 29.Oparil S, Haber E. The renin-angiotensin system (first of two parts) N Engl J Med. 1974;291:389–401. doi: 10.1056/NEJM197408222910805. [DOI] [PubMed] [Google Scholar]
- 30.Sakurai T, Kudo M, Fukuta N, Nakatani T, Kimura M, Park AM, Munakata H. Involvement of angiotensin II and reactive oxygen species in pancreatic fibrosis. Pancreatology. 2011;11 Suppl 2:7–13. doi: 10.1159/000323478. [DOI] [PubMed] [Google Scholar]
- 31.Chan YC, Leung PS. Angiotensin II type 1 receptor-dependent nuclear factor-kappaB activation-mediated proinflammatory actions in a rat model of obstructive acute pancreatitis. J Pharmacol Exp Ther. 2007;323:10–18. doi: 10.1124/jpet.107.124891. [DOI] [PubMed] [Google Scholar]
- 32.Timmermans PB, Wong PC, Chiu AT, Herblin WF, Benfield P, Carini DJ, Lee RJ, Wexler RR, Saye JA, Smith RD. Angiotensin II receptors and angiotensin II receptor antagonists. Pharmacol Rev. 1993;45:205–251. [PubMed] [Google Scholar]
- 33.Dijkhorst-Oei LT, Stroes ES, Koomans HA, Rabelink TJ. Acute simultaneous stimulation of nitric oxide and oxygen radicals by angiotensin II in humans in vivo. J Cardiovasc Pharmacol. 1999;33:420–424. doi: 10.1097/00005344-199903000-00012. [DOI] [PubMed] [Google Scholar]
- 34.Landmesser U, Cai H, Dikalov S, McCann L, Hwang J, Jo H, Holland SM, Harrison DG. Role of p47(phox) in vascular oxidative stress and hypertension caused by angiotensin II. Hypertension. 2002;40:511–515. doi: 10.1161/01.hyp.0000032100.23772.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tsang SW, Ip SP, Leung PS. Prophylactic and therapeutic treatments with AT 1 and AT 2 receptor antagonists and their effects on changes in the severity of pancreatitis. Int J Biochem Cell Biol. 2004;36:330–339. doi: 10.1016/s1357-2725(03)00257-7. [DOI] [PubMed] [Google Scholar]
- 36.Lowenfels AB, Maisonneuve P, Cavallini G, Ammann RW, Lankisch PG, Andersen JR, Dimagno EP, Andrén-Sandberg A, Domellöf L. Pancreatitis and the risk of pancreatic cancer. International Pancreatitis Study Group. N Engl J Med. 1993;328:1433–1437. doi: 10.1056/NEJM199305203282001. [DOI] [PubMed] [Google Scholar]
- 37.Kojima M, Ito T, Oono T, Hisano T, Igarashi H, Arita Y, Kawabe K, Coy DH, Jensen RT, Nawata H. VIP attenuation of the severity of experimental pancreatitis is due to VPAC1 receptor-mediated inhibition of cytokine production. Pancreas. 2005;30:62–70. [PubMed] [Google Scholar]
- 38.Fujimori N, Oono T, Igarashi H, Ito T, Nakamura T, Uchida M, Coy DH, Jensen RT, Takayanagi R. Vasoactive intestinal peptide reduces oxidative stress in pancreatic acinar cells through the inhibition of NADPH oxidase. Peptides. 2011;32:2067–2076. doi: 10.1016/j.peptides.2011.08.027. [DOI] [PubMed] [Google Scholar]
- 39.Wulczyn FG, Krappmann D, Scheidereit C. The NF-kappa B/Rel and I kappa B gene families: mediators of immune response and inflammation. J Mol Med (Berl) 1996;74:749–769. doi: 10.1007/s001090050078. [DOI] [PubMed] [Google Scholar]
- 40.Barnes PJ, Karin M. Nuclear factor-kappaB: a pivotal transcription factor in chronic inflammatory diseases. N Engl J Med. 1997;336:1066–1071. doi: 10.1056/NEJM199704103361506. [DOI] [PubMed] [Google Scholar]
- 41.Rawlings JS, Rosler KM, Harrison DA. The JAK/STAT signaling pathway. J Cell Sci. 2004;117:1281–1283. doi: 10.1242/jcs.00963. [DOI] [PubMed] [Google Scholar]
- 42.Corbi G, Conti V, Russomanno G, Longobardi G, Furgi G, Filippelli A, Ferrara N. Adrenergic signaling and oxidative stress: a role for sirtuins? Front Physiol. 2013;4:324. doi: 10.3389/fphys.2013.00324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kamata H, Honda S, Maeda S, Chang L, Hirata H, Karin M. Reactive oxygen species promote TNFalpha-induced death and sustained JNK activation by inhibiting MAP kinase phosphatases. Cell. 2005;120:649–661. doi: 10.1016/j.cell.2004.12.041. [DOI] [PubMed] [Google Scholar]
- 44.Goldstein BJ, Mahadev K, Wu X. Redox paradox: insulin action is facilitated by insulin-stimulated reactive oxygen species with multiple potential signaling targets. Diabetes. 2005;54:311–321. doi: 10.2337/diabetes.54.2.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Otamiri T, Sjödahl R. Oxygen radicals: their role in selected gastrointestinal disorders. Dig Dis. 1991;9:133–141. doi: 10.1159/000171299. [DOI] [PubMed] [Google Scholar]
- 46.Vonlaufen A, Apte MV, Imhof BA, Frossard JL. The role of inflammatory and parenchymal cells in acute pancreatitis. J Pathol. 2007;213:239–248. doi: 10.1002/path.2231. [DOI] [PubMed] [Google Scholar]
- 47.Tsuji N, Watanabe N, Okamoto T, Niitsu Y. Specific interaction of pancreatic elastase and leucocytes to produce oxygen radicals and its implication in pancreatitis. Gut. 1994;35:1659–1664. doi: 10.1136/gut.35.11.1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dabrowski A, Konturek SJ, Konturek JW, Gabryelewicz A. Role of oxidative stress in the pathogenesis of caerulein-induced acute pancreatitis. Eur J Pharmacol. 1999;377:1–11. doi: 10.1016/s0014-2999(99)00421-5. [DOI] [PubMed] [Google Scholar]
- 49.Gukovskaya AS, Gukovsky I, Zaninovic V, Song M, Sandoval D, Gukovsky S, Pandol SJ. Pancreatic acinar cells produce, release, and respond to tumor necrosis factor-alpha. Role in regulating cell death and pancreatitis. J Clin Invest. 1997;100:1853–1862. doi: 10.1172/JCI119714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kimura K, Shimosegawa T, Sasano H, Abe R, Satoh A, Masamune A, Koizumi M, Nagura H, Toyota T. Endogenous glucocorticoids decrease the acinar cell sensitivity to apoptosis during cerulein pancreatitis in rats. Gastroenterology. 1998;114:372–381. doi: 10.1016/s0016-5085(98)70490-1. [DOI] [PubMed] [Google Scholar]
- 51.Kaiser AM, Saluja AK, Sengupta A, Saluja M, Steer ML. Relationship between severity, necrosis, and apoptosis in five models of experimental acute pancreatitis. Am J Physiol. 1995;269:C1295–C1304. doi: 10.1152/ajpcell.1995.269.5.C1295. [DOI] [PubMed] [Google Scholar]
- 52.Sata N, Klonowski-Stumpe H, Han B, Lüthen R, Häussinger D, Niederau C. Supraphysiologic concentrations of cerulein induce apoptosis in the rat pancreatic acinar cell line AR4-2J. Pancreas. 1999;19:76–82. doi: 10.1097/00006676-199907000-00012. [DOI] [PubMed] [Google Scholar]
- 53.Wang X, Yang C, Chai J, Shi Y, Xue D. Mechanisms of AIF-mediated apoptotic DNA degradation in Caenorhabditis elegans. Science. 2002;298:1587–1592. doi: 10.1126/science.1076194. [DOI] [PubMed] [Google Scholar]
- 54.Cregan SP, Dawson VL, Slack RS. Role of AIF in caspase-dependent and caspase-independent cell death. Oncogene. 2004;23:2785–2796. doi: 10.1038/sj.onc.1207517. [DOI] [PubMed] [Google Scholar]
- 55.Yu JH, Kim H, Kim KH. Calcium-dependent apoptotic gene expression in cerulein-treated AR42J cells. Ann N Y Acad Sci. 2003;1010:66–69. doi: 10.1196/annals.1299.009. [DOI] [PubMed] [Google Scholar]
- 56.Snook JH, Li J, Helmke BP, Guilford WH. Peroxynitrite inhibits myofibrillar protein function in an in vitro assay of motility. Free Radic Biol Med. 2008;44:14–23. doi: 10.1016/j.freeradbiomed.2007.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Crimi E, Ignarro LJ, Napoli C. Microcirculation and oxidative stress. Free Radic Res. 2007;41:1364–1375. doi: 10.1080/10715760701732830. [DOI] [PubMed] [Google Scholar]
- 58.Okumura N, Sakakibara A, Hayakawa T, Noda A. Pancreatic endocrine function in experimental pancreatolithiasis in dogs. Am J Gastroenterol. 1982;77:392–396. [PubMed] [Google Scholar]
- 59.Aho HJ, Nevalainen TJ, Havia VT, Heinonen RJ, Aho AJ. Human acute pancreatitis: a light and electron microscopic study. Acta Pathol Microbiol Immunol Scand A. 1982;90:367–373. [PubMed] [Google Scholar]
- 60.Uys CJ, Bank S, Marks IN. The pathology of chronic pancreatitis in Cape Town. Digestion. 1973;9:454–468. doi: 10.1159/000197474. [DOI] [PubMed] [Google Scholar]
- 61.Naito Y, Yoshikawa T, Tanigawa T, Sakurai K, Yamasaki K, Uchida M, Kondo M. Hydroxyl radical scavenging by rebamipide and related compounds: electron paramagnetic resonance study. Free Radic Biol Med. 1995;18:117–123. doi: 10.1016/0891-5849(94)00110-6. [DOI] [PubMed] [Google Scholar]
- 62.Ogino K, Hobara T, Ishiyama H, Yamasaki K, Kobayashi H, Izumi Y, Oka S. Antiulcer mechanism of action of rebamipide, a novel antiulcer compound, on diethyldithiocarbamate-induced antral gastric ulcers in rats. Eur J Pharmacol. 1992;212:9–13. doi: 10.1016/0014-2999(92)90065-c. [DOI] [PubMed] [Google Scholar]
- 63.Yu JH, Lim JW, Namkung W, Kim H, Kim KH. Suppression of cerulein-induced cytokine expression by antioxidants in pancreatic acinar cells. Lab Invest. 2002;82:1359–1368. doi: 10.1097/01.lab.0000032377.09626.c7. [DOI] [PubMed] [Google Scholar]
- 64.Park BK, Chung JB, Lee JH, Suh JH, Park SW, Song SY, Kim H, Kim KH, Kang JK. Role of oxygen free radicals in patients with acute pancreatitis. World J Gastroenterol. 2003;9:2266–2269. doi: 10.3748/wjg.v9.i10.2266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Verlaan M, Roelofs HM, van-Schaik A, Wanten GJ, Jansen JB, Peters WH, Drenth JP. Assessment of oxidative stress in chronic pancreatitis patients. World J Gastroenterol. 2006;12:5705–5710. doi: 10.3748/wjg.v12.i35.5705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yoo BM, Oh TY, Kim YB, Yeo M, Lee JS, Surh YJ, Ahn BO, Kim WH, Sohn S, Kim JH, et al. Novel antioxidant ameliorates the fibrosis and inflammation of cerulein-induced chronic pancreatitis in a mouse model. Pancreatology. 2005;5:165–176. doi: 10.1159/000085268. [DOI] [PubMed] [Google Scholar]
- 67.Bhardwaj P, Garg PK, Maulik SK, Saraya A, Tandon RK, Acharya SK. A randomized controlled trial of antioxidant supplementation for pain relief in patients with chronic pancreatitis. Gastroenterology. 2009;136:149–159.e2. doi: 10.1053/j.gastro.2008.09.028. [DOI] [PubMed] [Google Scholar]
- 68.Siriwardena AK, Mason JM, Balachandra S, Bagul A, Galloway S, Formela L, Hardman JG, Jamdar S. Randomised, double blind, placebo controlled trial of intravenous antioxidant (n-acetylcysteine, selenium, vitamin C) therapy in severe acute pancreatitis. Gut. 2007;56:1439–1444. doi: 10.1136/gut.2006.115873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Booth DM, Murphy JA, Mukherjee R, Awais M, Neoptolemos JP, Gerasimenko OV, Tepikin AV, Petersen OH, Sutton R, Criddle DN. Reactive oxygen species induced by bile acid induce apoptosis and protect against necrosis in pancreatic acinar cells. Gastroenterology. 2011;140:2116–2125. doi: 10.1053/j.gastro.2011.02.054. [DOI] [PubMed] [Google Scholar]
- 70.Apte MV, Haber PS, Applegate TL, Norton ID, McCaughan GW, Korsten MA, Pirola RC, Wilson JS. Periacinar stellate shaped cells in rat pancreas: identification, isolation, and culture. Gut. 1998;43:128–133. doi: 10.1136/gut.43.1.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bachem MG, Schünemann M, Ramadani M, Siech M, Beger H, Buck A, Zhou S, Schmid-Kotsas A, Adler G. Pancreatic carcinoma cells induce fibrosis by stimulating proliferation and matrix synthesis of stellate cells. Gastroenterology. 2005;128:907–921. doi: 10.1053/j.gastro.2004.12.036. [DOI] [PubMed] [Google Scholar]
- 72.Omary MB, Lugea A, Lowe AW, Pandol SJ. The pancreatic stellate cell: a star on the rise in pancreatic diseases. J Clin Invest. 2007;117:50–59. doi: 10.1172/JCI30082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Apte MV, Phillips PA, Fahmy RG, Darby SJ, Rodgers SC, McCaughan GW, Korsten MA, Pirola RC, Naidoo D, Wilson JS. Does alcohol directly stimulate pancreatic fibrogenesis? Studies with rat pancreatic stellate cells. Gastroenterology. 2000;118:780–794. doi: 10.1016/s0016-5085(00)70148-x. [DOI] [PubMed] [Google Scholar]