

Abstract
Fistulae represent an important complication in patient suffering from Crohn’s disease (CD). Cumulative incidence of fistula formation in CD patients is 17%-50% and about one third of patients suffer from recurring fistulae formation. Medical treatment options often fail and also surgery frequently is not successful. Available data indicate that CD-associated fistulae originate from an epithelial defect that may be caused by ongoing inflammation. Having undergone epithelial to mesenchymal transition (EMT), intestinal epithelial cells (IEC) penetrate into deeper layers of the mucosa and the gut wall causing localized tissue damage formation of a tube like structure and finally a connection to other organs or the body surface. EMT of IEC may be initially aimed to improve wound repair mechanisms since “conventional” wound healing mechanisms, such as migration of fibroblasts, are impaired in CD patients. EMT also enhances activation of matrix remodelling enzymes such as matrix metalloproteinase (MMP)-3 and MMP-9 causing further tissue damage and inflammation. Finally, soluble mediators like TNF and interleukin-13 further induce their own expression in an autocrine manner and enhance expression of molecules associated with cell invasiveness aggravating the process. Additionally, pathogen-associated molecular patterns also seem to play a role for induction of EMT and fistula development. Though current knowledge suggests a number of therapeutic options, new and more effective therapeutic approaches are urgently needed for patients suffering from CD-associated fistulae. A better understanding of the pathophysiology of fistula formation, however, is a prerequisite for the development of more efficacious medical anti-fistula treatments.
Keywords: Crohn’s disease, Fistula, Tumor necrosis factor, Interleukin-13, Transforming growth factor, Epithelial to mesenchymal transition
Core tip: Fistulae represent an important complication in Crohn’s disease (CD) patients. CD-associated fistulae originate from an epithelial defect due to destructive inflammation. Having undergone epithelial-to-mesenchymal transition (EMT), intestinal epithelial cells (IEC) penetrate into deeper layers of the gut wall causing further tissue damage finally forming connections to other organs or the body surface. EMT of IEC results in activation of matrix remodelling enzymes. Soluble mediators like TNF and IL-13 induce their own expression and expression of molecules associated with cell invasiveness. A better understanding of the pathophysiology of fistula formation is a prerequisite for developing more efficacious medical anti-fistula treatments.
INTRODUCTION
Crohn’s disease (CD) and ulcerative colitis are the two main forms of inflammatory bowel diseases (IBD) and are characterized by chronic intestinal inflammation. An important complication of CD is the formation of fistulae. This frequent clinical problem often causes a severely impaired quality of life in the affected patients. According to population-based studies, the cumulative incidence of fistula formation in CD patients is 17%-50%[1-3]. About 35% of patients suffer from at least one fistula during their disease course[2]. After 10 years of disease, the cumulative incidence for fistulas is reported to be up to 33% and after 20 years it is about 50%[2]. About one third of patients suffer from recurring fistulae[2]. In roughly 10% of the CD patients fistulae are the initial disease presentation and may precede the manifestation of intestinal disease by several years[1]. Therapeutic options are limited: Medical options include antibiotics, immunosuppressives (such as azathioprine or cyclosporine) and anti-TNF antibodies. Their clinical effect is often limited and despite medical treatment more than one third of patients suffers from recurring fistulae[4]. However, also surgical option do not always provide a definitive solution and permanent fistula closure can only be achieved in about 34% of CD patients[5].
MORPHOLOGICAL CHARACTERISATION OF CD FISTULAE
CD fistulae are found perianal in the majority of cases (54%), as well as entero-enteric (24%), recto-vaginal (9%) or in other locations, such as entero-cutaneous or entero-vesical[2]. On a histomorphologic level, CD-associated fistulae reveal a central fissure that penetrates through the lamina propria and muscularis mucosa into deeper layers of the underlying gut wall. In general, fistulae are lined by granulation tissue consisting of histiocytes and a dense network of tender capillaries. The lumen is frequently filled up by nuclear debris, erythrocytes as well as neutrophils and lymphocytes indicating non-specific acute or chronic inflammation. In more than 80% there are signs of moderate to severe acute inflammation. In CD fistulae, the wall of the fistulae is frequently infiltrated by CD45RO+ positive T-cells, followed by a small band of CD68+ positive macrophages and finally a dense infiltrate of CD20+ B-cells. This is in contrast to non-CD fistulae, where often an intense infiltration by CD68+ macrophages, but only a few CD20+ B-lymphocytes and CD45RO+ T-lymphocytes are observed[6].
Independent of the inflammatory infiltrate about one fourth of CD fistulae feature a lining epithelium central in the fistulising inflammation. Depending on the fistula location, this lining epithelium consists of flattened epithelial cells of the small intestine or colon without goblet cells or of a narrow squamous epithelium in cutaneous or perianal fistulae. The cells reveal tight junctions and a basement membrane. In “non-epithelialized” fistulae some areas are lined with myofibroblast-like cells, so-called transitional cells (TC). The region where the mucosal epithelial cells transform into the TC is called transitional zone. The TC are connected by gap junctions to each other and in certain areas a new basement membrane develops between TC and the underlying granulation tissue. The TC and the new basement membrane are connected by fibronexus. However, in adjacent areas there are also disordered myofibroblasts showing no gap junctions and a fragmented basement membrane[6].
ALTERED MIGRATORY POTENTIAL OF COLONIC LAMINA PROPRIA FIBROBLASTS
On a functional level, the migratory potential of colonic lamina propria fibroblasts (CLPF) in the intestine of CD patients is clearly less than in non-IBD or UC patients. Of note, mucosal fibroblasts derived from CD-associated fistulae reveal an even further reduced migratory potential what might contribute to decreased wound healing potential in this disease[7,8]. The decrease in the migratory potential of the CLPF can be induced by pro-inflammatory cytokines, such as TNF or IFN and is a persistent functional change since it is not reversible even after removing the cytokines. Additionally, it is accompanied by a decreased expression and phosphorylation, meaning activation, of the focal adhesion kinase (FAK)[7,9] which is a central regulator of cell migration[10]. Fibronectin is also an essential factor for the induction of migration of CLPF. In CD fistulae, the ED-A and ED-B isoforms of fibronectin are almost absent[11]. This might also critically contribute to the reduced migratory potential of CD fistula CLPF, since the ED-A subunit is usually increased during wound healing and is an important inducer of fibroblast migration[12]. A further stimulator of CLPF migration in the intestine is galectin-3 which is secreted by colonic epithelial cells[13,14]. Though galectin-3 is able to induce the migration of CLPF derived from CD fistulae, its expression is clearly decreased in CD fistulae[14]. These observations might explain the reduced mesenchyme-mediated wound healing potential in patients with CD fistulae. Other mechanisms have to step in to repair the epithelial barrier. Increased IEC migration is a mechanism aiming to replace the malfunctioning fibroblast migration, to improve wound repair and to restore intestinal barrier function. Thus, the migration of epithelial towards the defect might be induced as part of a compensatory mechanism, since the migratory potential of CD fistula CLPF is critically impaired[15].
A KEY ROLE FOR EPITHELIAL TO MESENCHYMAL TRANSITION
In order to migrate, IEC must undergo a conversion into mesenchymal-like cells so-called myofibroblasts. This process is called epithelial-to-mesenchymal transition. During EMT, differentiated IEC characterized by strong intercellular junctions and cell polarity lose their epithelial phenotype and acquire a mesenchymal differentiation featuring reduced cell-cell contacts and a fibroblast-like morphology and function[16,17]. EMT plays an important role during embryogenesis and organ development, but also for tissue remodelling and wound healing[16-18]. However, recent studies also suggested that EMT is critically involved in pathologic conditions, such as cancer growth and fibrosis[16,17]. While TGF is the most potent inducer of EMT in vivo[19], there are further markers for the onset of EMT, such as decreased expression of E-cadherin and β-catenin, translocation of membrane-bound-catenin into the cytosol or the nucleus and increased expression of β6-integrin[16,17,20,21].
Available data strongly suggest that EMT might also be critically involved in the formation of the TC layer covering the fistula tracts of CD patients. TGF-1 and TGF-2 expression are both upregulated in fistula lining cells as compared to regular IEC[22]. E-cadherin is involved in the formation of intercellular zonulae adherentes and is found at the lateral cell membrane at the cell-cell contact sites of normal IEC. In the TCs lining the CD fistula tract not only a decreased expression, but also redistribution of membranous E-cadherin is detectable. This change in localization of E-cadherin can especially be observed in the transition zone[22]. In TC-catenin expression is diffuse, much weaker and found in the cytoplasm and nuclei whereas it is located in the lateral cell membrane of normal mucosal IEC[22]. Six integrin expression normally is restricted to epithelial cells during embryonic development and organogenesis. Its re-induction indicates an important role of this molecule during intestinal EMT[20,21]. In contrast to normal IEC, TC localised in the transitional zones feature a strong staining pattern for 6-integrin[22]. IEC as well as mesenchymal-like myofibroblast-like TC expressed both of the epithelial markers, cytokeratin (CK) 8 and CK 20[22]. These staining patterns of the TC strongly suggest an epithelial origin of these myofibroblast-like cells that show characteristic features of EMT.
A further event that is characteristic to EMT, is the nuclear expression of the transcription factors SNAIL1 and SLUG (SNAIL2) which are activated by TGF and are involved in the down-regulation of E-cadherin[16,23,24]. Interestingly, there are different expression pattern for SNAIL1 and SLUG in CD fistulae. While SNAIL1 is heavily expressed in the nuclei of the TC lining the fistula tracts indicating transcriptionally active protein, SLUG is mainly detected in cells around the fistula tract and only to a very limited level in the fistula tract lining TC[25]. CLPF from CD patients with fistulae express higher SLUG mRNA levels than CLPF from patients without fistulae[26]. Expression of fibroblast growth factors 1 and 2 that are also associated with increased SNAIL1 expression, reduced E-cadherin expression and the onset of EMT are detectable in the tissue layers below the fistula tract and in the fistula-associated inflammatory infiltrates[25]. A number of EMT characteristic events are detectable in and around fistula tracts clearly supporting a role for EMT in the pathogenesis of CD-associated fistulae. The detection of epithelial markers in submucosal myofibroblast-like cells further supports this hypothesis and demonstrates the transformation of former IEC into these mesenchymal-like cells.
INVOLVEMENT OF MATRIX REMODELLING MOLECULES
The intercellular matrix is constantly remodelled by a number of enzymes that degrade all components of the extracellular matrix (ECM), namely the matrix metalloproteinases (MMP). Increased MMP activity finally results in immune-mediated tissue injury and is associated with a number of pathologies, such as cancer growth and CD[27-29]. The importance of MMPs for the development of CD is highlighted by the fact that in the murine DSS-induced colitis model, targeted deletion of MMP-9 has a protective effect[30,31] while mice overexpressing MMP-9 in the intestinal epithelium develop more severe colitis when compared to wild type animals[32]. Further, addition of MMP-3 caused extensive tissue injury in an ex vivo human fetal model of intestinal inflammation and tissue injury was effectively blocked by inhibiting MMP activity[33]. The physiological inhibitors of MMPs are the tissue inhibitors of MMP (TIMP) which are also secreted by the MMP producing cells[27].
In CD fistulae, strong MMP-3 expression is observed independent of the stage of inflammation. MMP3 mRNA and protein expression is detected in mononuclear cells and fibroblasts[34]. Inactive and active MMP-9 is expressed around CD fistulae and mRNA and protein levels are found in granulocytes and fibroblasts[34,35]. The activated isoform of MMP-13 is present in the supernatant of untreated CD fistula CLPF, but is almost absent in the supernatant of non-fistula CLPF. MMP-13 protein expression also is clearly detectable in mononuclear cells around CD fistulae[26,35]. In contrast, expression of MMP-1 and MMP-7 is only weak around CD fistulae, MMP-10 is not detectable and MMP-2 protein is equally expressed in fistula and control tissue. Activated MMP-2 is only detectable in CD fistulae[34]. Protein levels of TIMP-1, TIMP-2 and TIMP-3 are low around CD fistulae[34]. These observations suggest a critical role for matrix remodelling enzymes in fistula pathogenesis.
MOLECULES ASSOCIATED WITH CELL MIGRATION AND INVASION
The published data on fistula pathogenesis strongly suggest that fistula originate from an epithelial injury and due to defective wound healing mechanisms IEC re-differentiate into mesenchymal-like cells acquiring migratory potential. In line with this assumption, molecules associated with IEC migration, such as 6-integrin, Ets-1 transcription factor and the Wnt-inhibitor Dickkopf-homolog 1 (DKK-1) are detected in CD fistulae.
While regular mucosal epithelial cells do not express 6-integrin, TC located in the transitional zone are strongly positive for β6-integrin staining[22] and fistula CLPF express higher 6-integrin mRNA levels than CLPF from control or CD patients without fistulae[26]. These observations are of particular interest, since 6-integrin expression has been associated with migration, invasion, metastasis and shortened survival in certain carcinomas, such as colorectal cancer, head-and-neck cancer, breast cancer or squamous cell cancer[20,36-39]. Up-regulated β6-integrin is associated with increased levels of EMT[20,38-41], represents a receptor for fibronectin and tenascin what might be important for cell migration, regulates secretion and activation of MMP-9[42,43] and mediates TGF activation and adhesion what has also been implicated in increased survival, progression and metastases of various tumours[44,45]. Additionally, 6-integrin can induce its own expression in an autocrine manner[46]. On a transcriptional level, 6-integrin is regulated by Ets-1 transcription factor[20]. While tissue samples from control and IBD-patients in remission display only low expression levels of Ets-1 protein, a strong staining signal is detected in tissue samples derived from patients with active inflammation and in TC along the fistula tract[47] providing further support for the regulatory effect of Ets-1 on 6-integrin expression.
The Wnt-inhibitor DKK-1 represents an important factor involved in the regulation of cell migration. Loss of DKK-1 has been associated with progression of certain types of carcinomas, such as CRC[48]. The secreted glycoprotein is capable to block IEC migration, is a potent antagonist of the canonical Wnt/β-catenin signalling pathway and has been implicated to act as a mediator of inflammation[49,50]. In the intestinal tissue of non-IBD control patients, Dkk-1 staining is very weak, while tissue samples from CD-patients with a perianal fistulae reveal strong DKK-1 staining in TC lining the fistula tracts and patients with active CD also exhibit considerable DKK-1 expression in inflammatory infiltrates[51].
TGF
TGF is the most important inducer of EMT[19] and expression of TGF-1 and TGF-2 is higher in TC than in normal IEC[22]. TGF induces the mRNA expression of interleukin-13 (IL-13) in the HT-29 IEC spheroid model of EMT and the secretion of IL-13 from fistula CLPF, but not from CLPF from non-IBD control patients or CD patients without fistulae[26]. The effect of TGF on IL-13 expression in IEC is mediated via -catenin and DKK-1[51]. In HT-29 IEC, TGF treatment induces DKK-1 levels and this effect is inhibited by knock-down of -catenin. Interestingly, knock-down of either-catenin or DKK-1 prevents the TGF-induced increase in IL-13 expression[51]. These observations fit to the nuclear staining pattern of -catenin in TC. Increased TGF levels in TC induce nuclear accumulation, meaning transcriptional activation, of -catenin what results in enhanced expression of DKK-1 and IL-13. The TGF-induced up-regulation of DKK-1 might serve hereby as a negative feed-back mechanism to control TGF-mediated effects. However, IL-13 decreases the expression of DKK-1 in IEC and fistula CLPF[51] what finally dis-ruptures this regulatory mechanism and might result in uncontrolled secretion of IL-13.
IL-13
IL-13 has been implicated in the pathogenesis of tissue fibrosis, such as in the lung or liver, and, in this context, induces the secretion of TGF[52-54]. It is mainly secreted by Th2-cells and its alpha 1 receptor (IL-13R1) is the signal transducing receptor while the alpha 2 receptor (IL-13R2) acts as a decoy receptor[55,56]. In TC lining the fistula tracts as well as in deformed crypts adjacent to the fistula, IL-13 and IL-13R1 are heavily expressed, while they are almost absent in the intestine of non-IBD patients, UC patients and CD patients without fistula regardless their inflammation status[26]. These observations suggest that IL-13 expression and associated effects might be induced in CLPF and fistula-associated IEC in a positive feedback mechanism. On a functional level, IL-13 induces the expression of SLUG and 6-integrin in HT29 cells grown as monolayers or spheroids. Interestingly the IL-13 induced 6-integrin expression is mediated, at least in part, via SLUG transcription factor and SLUG expression is sensitive to anti-IL-13 antibody treatment. However, in contrast to TGF, IL-13 treatment is not sufficient to induce EMT in the HT29-IEC spheroid model[26].
TNF
TNF has been demonstrated to induce EMT in IEC and is able to induce the expression of TGF[47,57,58]. Similar to IL-13, strong staining for TNF and its receptor, TNF-receptor I (TNF-RI) is detected in TC lining the fistula tracts as well as in IEC of adjacent crypts in CD patients. TNF is also expressed in fistula surrounding immune cells[25]. This observation further supports the hypothesis that TNF, similar to IL-13, induces its expression in an autocrine manner. Correlating, TNF induces its own expression in IEC and fistula CLPF in vitro[47,57]. In IEC and CLPF, TNF stimulates the expression of 6-integrin and Ets-1 transcription factor and knock-down of Ets-1 results in diminished 6-integrin levels in response to TNF. Of note, while TNF induces TGF and EMT in IEC, it is not sufficient to stimulate IL-13 neither in IEC nor in fistula CLPF[47]. These observations suggest that TNF, which is naturally present in acutely and chronically inflamed intestinal tissue, acts via two different pathways: TNF induces EMT on the one hand by its own, on the other had via TGF, as part of a wound healing mechanism. However, aberrant responses of IEC and/or CLPF to TGF then result in increased expression of IL-13. IL-13, similar to TNF, then stimulates its own expression via a positive feedback mechanism what finally causes the expression of molecules being associated with cell migration and invasiveness, such as Ets-1 and 6-integrin. TNF-induced effects can be effectively blocked by administration of an anti-TNF antibody in vitro[47], what might also explain, at least in part, the beneficial effect of anti-TNF antibodies for fistula treatment in CD patients.
PATHOGEN-ASSOCIATED MOLECULAR PATTERN
Polymorphisms within the nucleotide oligomerization domain 2 (NOD2) gene are associated with a fistulizing disease course of CD[59]. The bacterial wall component and pathogen-associated molecular pattern (PAMP), muramyl-dipeptide, is the natural ligand for NOD2 and following activation of NOD2, immune cells produce pro-inflammatory cytokines, such as TNF[60]. MDP treatment induces the expression of genes being associated with EMT as well as with cell invasiveness, such as TGF, SNAIL1, IL-13 and 6-Integrin, in IEC. While in non-fistula CLPF, MDP significantly induced mRNA expression of Ets-1, 6-Integrin, TNF, SNAIL1 and TGF, in fistula CLPF MDP treatment only induces mRNA levels of Ets-1 and TGF[47]. Since fistula CLPF express high levels of TNF and IL-13 via an autocrine mechanism, it might be that exogenous stimulation of these cells, i.e., by MDP, is not sufficient to further induce TNF or IL-13 levels in these cells. Interestingly, lipopolysaccharide (LPS) does not induce any of the fistula-associated molecules in either IEC or CLPF pointing towards a specific role for the MDP-NOD2 axis in fistula pathogenesis. These observations suggest that distinct PAMPs might play a critical role for fistula pathogenesis by inducing EMT and genes being associated with EMT and cell invasiveness what makes the use of antibiotics in fistula treatment plausible.
CONCLUSION
Taken together, available data demonstrate that CD-associated fistulae originate from an epithelial defect that occurs during chronic inflammation. Having undergone EMT, IEC penetrate into deeper tissue layers causing tissue damage and a connection to other organs or the body surface. EMT of IEC is part of a wound repair mechanism as inflammation causes ongoing tissue damage and conventional wound healing mechanisms, such as migration of fibroblasts, are impaired. The expression of EMT-associated molecules results in enhanced activation of matrix remodelling enzymes such as MMP-3 and MMP-9 causing further tissue damage and inflammation. Finally, soluble mediators such as TNF and IL-13 promote their own expression in an autocrine manner and enhance expression of molecules being associated with cell invasiveness. Subsequently, fistula formation and “growth” is constantly promoted and further supported by the presence of EMT-inducers, such as TGF, and PAMPs (Figure 1). Though current knowledge suggests a number of therapeutic options, new and more effective therapeutic approaches are urgently needed for patients suffering from CD-associated fistulae.
Figure 1.
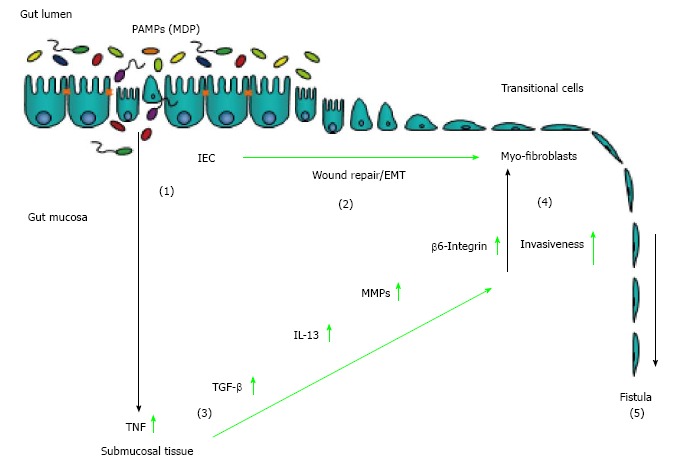
Pathogenesis of Crohn’s disease-associated fistulae. An epithelial barrier defect favours the invasion of pathogen-associated pattern (PAMPs) into the gut mucosa (1). On the one hand, for wound healing purposes, intestinal epithelial cells undergo epithelial-to-mesenchymal transition (2). On the other hand, presence of PAMPs induced an inflammatory reaction resulting in increased secretion of TNF (3). TNF is able to induce secretion of TGF as well as to induce EMT and expression of molecules associated with cell invasiveness, such as 6-integrin. TGF-induced IL-13 and elevated activation of matrix remodelling MMPs critically contribute to invasive cell growth (4). Finally, EMT, MMP over-activation and elevated expression of invasive molecules contribute to the development of fistulae (5).
Footnotes
Supported by A grant from Fonds zur Förderung des akademischen Nachwuchses (FAN) of the Zürcher Universitätsverein (ZUNIV) to MS; by a research grant from the Swiss Philanthropy Foundation to MS and GR; by a research credit from the University of Zurich to MS; by research grants from the Swiss National Science Foundation (SNF) to MS, No. 314730-146204, GR, No. 310030-120312, and the Swiss IBD Cohort, No. 3347CO-108792; and by the Zurich Center for Integrative Human Physiology (ZIHP) of the University of Zurich
P- Reviewer: Keshteli AH, Kellermayer R, Mullin JM S- Editor: Ji FF L- Editor: A E- Editor: Lu YJ
References
- 1.Hellers G, Bergstrand O, Ewerth S, Holmström B. Occurrence and outcome after primary treatment of anal fistulae in Crohn’s disease. Gut. 1980;21:525–527. doi: 10.1136/gut.21.6.525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schwartz DA, Loftus EV, Tremaine WJ, Panaccione R, Harmsen WS, Zinsmeister AR, Sandborn WJ. The natural history of fistulizing Crohn’s disease in Olmsted County, Minnesota. Gastroenterology. 2002;122:875–880. doi: 10.1053/gast.2002.32362. [DOI] [PubMed] [Google Scholar]
- 3.Solomon MJ. Fistulae and abscesses in symptomatic perianal Crohn’s disease. Int J Colorectal Dis. 1996;11:222–226. doi: 10.1007/s003840050051. [DOI] [PubMed] [Google Scholar]
- 4.Nielsen OH, Rogler G, Hahnloser D, Thomsen OØ. Diagnosis and management of fistulizing Crohn‘s disease. Nat Clin Pract Gastroenterol Hepatol. 2009;6:92–106. doi: 10.1038/ncpgasthep1340. [DOI] [PubMed] [Google Scholar]
- 5.Loftus EV, Schoenfeld P, Sandborn WJ. The epidemiology and natural history of Crohn’s disease in population-based patient cohorts from North America: a systematic review. Aliment Pharmacol Ther. 2002;16:51–60. doi: 10.1046/j.1365-2036.2002.01140.x. [DOI] [PubMed] [Google Scholar]
- 6.Bataille F, Klebl F, Rümmele P, Schroeder J, Farkas S, Wild PJ, Fürst A, Hofstädter F, Schölmerich J, Herfarth H, et al. Morphological characterisation of Crohn’s disease fistulae. Gut. 2004;53:1314–1321. doi: 10.1136/gut.2003.038208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Leeb SN, Vogl D, Gunckel M, Kiessling S, Falk W, Göke M, Schölmerich J, Gelbmann CM, Rogler G. Reduced migration of fibroblasts in inflammatory bowel disease: role of inflammatory mediators and focal adhesion kinase. Gastroenterology. 2003;125:1341–1354. doi: 10.1016/j.gastro.2003.07.004. [DOI] [PubMed] [Google Scholar]
- 8.Leeb SN, Vogl D, Falk W, Schölmerich J, Rogler G, Gelbmann CM. Regulation of migration of human colonic myofibroblasts. Growth Factors. 2002;20:81–91. doi: 10.1080/08977190290031941. [DOI] [PubMed] [Google Scholar]
- 9.Meier JK, Scharl M, Miller SN, Brenmoehl J, Hausmann M, Kellermeier S, Schölmerich J, Rogler G. Specific differences in migratory function of myofibroblasts isolated from Crohn’s disease fistulae and strictures. Inflamm Bowel Dis. 2011;17:202–212. doi: 10.1002/ibd.21344. [DOI] [PubMed] [Google Scholar]
- 10.Schlaepfer DD, Hauck CR, Sieg DJ. Signaling through focal adhesion kinase. Prog Biophys Mol Biol. 1999;71:435–478. doi: 10.1016/s0079-6107(98)00052-2. [DOI] [PubMed] [Google Scholar]
- 11.Brenmoehl J, Lang M, Hausmann M, Leeb SN, Falk W, Schölmerich J, Göke M, Rogler G. Evidence for a differential expression of fibronectin splice forms ED-A and ED-B in Crohn’s disease (CD) mucosa. Int J Colorectal Dis. 2007;22:611–623. doi: 10.1007/s00384-006-0188-4. [DOI] [PubMed] [Google Scholar]
- 12.Ffrench-Constant C, Van de Water L, Dvorak HF, Hynes RO. Reappearance of an embryonic pattern of fibronectin splicing during wound healing in the adult rat. J Cell Biol. 1989;109:903–914. doi: 10.1083/jcb.109.2.903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lippert E, Falk W, Bataille F, Kaehne T, Naumann M, Goeke M, Herfarth H, Schoelmerich J, Rogler G. Soluble galectin-3 is a strong, colonic epithelial-cell-derived, lamina propria fibroblast-stimulating factor. Gut. 2007;56:43–51. doi: 10.1136/gut.2005.081646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lippert E, Gunckel M, Brenmoehl J, Bataille F, Falk W, Scholmerich J, Obermeier F, Rogler G. Regulation of galectin-3 function in mucosal fibroblasts: potential role in mucosal inflammation. Clin Exp Immunol. 2008;152:285–297. doi: 10.1111/j.1365-2249.2008.03618.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dignass AU. Mechanisms and modulation of intestinal epithelial repair. Inflamm Bowel Dis. 2001;7:68–77. doi: 10.1097/00054725-200102000-00014. [DOI] [PubMed] [Google Scholar]
- 16.Kalluri R, Neilson EG. Epithelial-mesenchymal transition and its implications for fibrosis. J Clin Invest. 2003;112:1776–1784. doi: 10.1172/JCI20530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee JM, Dedhar S, Kalluri R, Thompson EW. The epithelial-mesenchymal transition: new insights in signaling, development, and disease. J Cell Biol. 2006;172:973–981. doi: 10.1083/jcb.200601018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Arias AM. Epithelial mesenchymal interactions in cancer and development. Cell. 2001;105:425–431. doi: 10.1016/s0092-8674(01)00365-8. [DOI] [PubMed] [Google Scholar]
- 19.Zavadil J, Böttinger EP. TGF-beta and epithelial-to-mesenchymal transitions. Oncogene. 2005;24:5764–5774. doi: 10.1038/sj.onc.1208927. [DOI] [PubMed] [Google Scholar]
- 20.Bates RC, Bellovin DI, Brown C, Maynard E, Wu B, Kawakatsu H, Sheppard D, Oettgen P, Mercurio AM. Transcriptional activation of integrin beta6 during the epithelial-mesenchymal transition defines a novel prognostic indicator of aggressive colon carcinoma. J Clin Invest. 2005;115:339–347. doi: 10.1172/JCI23183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bates RC. Colorectal cancer progression: integrin alphavbeta6 and the epithelial-mesenchymal transition (EMT) Cell Cycle. 2005;4:1350–1352. doi: 10.4161/cc.4.10.2053. [DOI] [PubMed] [Google Scholar]
- 22.Bataille F, Rohrmeier C, Bates R, Weber A, Rieder F, Brenmoehl J, Strauch U, Farkas S, Fürst A, Hofstädter F, et al. Evidence for a role of epithelial mesenchymal transition during pathogenesis of fistulae in Crohn’s disease. Inflamm Bowel Dis. 2008;14:1514–1527. doi: 10.1002/ibd.20590. [DOI] [PubMed] [Google Scholar]
- 23.Peinado H, Quintanilla M, Cano A. Transforming growth factor beta-1 induces snail transcription factor in epithelial cell lines: mechanisms for epithelial mesenchymal transitions. J Biol Chem. 2003;278:21113–21123. doi: 10.1074/jbc.M211304200. [DOI] [PubMed] [Google Scholar]
- 24.Zheng G, Lyons JG, Tan TK, Wang Y, Hsu TT, Min D, Succar L, Rangan GK, Hu M, Henderson BR, et al. Disruption of E-cadherin by matrix metalloproteinase directly mediates epithelial-mesenchymal transition downstream of transforming growth factor-beta1 in renal tubular epithelial cells. Am J Pathol. 2009;175:580–591. doi: 10.2353/ajpath.2009.080983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Scharl M, Weber A, Fürst A, Farkas S, Jehle E, Pesch T, Kellermeier S, Fried M, Rogler G. Potential role for SNAIL family transcription factors in the etiology of Crohn’s disease-associated fistulae. Inflamm Bowel Dis. 2011;17:1907–1916. doi: 10.1002/ibd.21555. [DOI] [PubMed] [Google Scholar]
- 26.Scharl M, Frei S, Pesch T, Kellermeier S, Arikkat J, Frei P, Fried M, Weber A, Jehle E, Rühl A, et al. Interleukin-13 and transforming growth factor β synergise in the pathogenesis of human intestinal fistulae. Gut. 2013;62:63–72. doi: 10.1136/gutjnl-2011-300498. [DOI] [PubMed] [Google Scholar]
- 27.Nelson AR, Fingleton B, Rothenberg ML, Matrisian LM. Matrix metalloproteinases: biologic activity and clinical implications. J Clin Oncol. 2000;18:1135–1149. doi: 10.1200/JCO.2000.18.5.1135. [DOI] [PubMed] [Google Scholar]
- 28.Baugh MD, Evans GS, Hollander AP, Davies DR, Perry MJ, Lobo AJ, Taylor CJ. Expression of matrix metalloproteases in inflammatory bowel disease. Ann N Y Acad Sci. 1998;859:249–253. doi: 10.1111/j.1749-6632.1998.tb11139.x. [DOI] [PubMed] [Google Scholar]
- 29.von Lampe B, Barthel B, Coupland SE, Riecken EO, Rosewicz S. Differential expression of matrix metalloproteinases and their tissue inhibitors in colon mucosa of patients with inflammatory bowel disease. Gut. 2000;47:63–73. doi: 10.1136/gut.47.1.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Castaneda FE, Walia B, Vijay-Kumar M, Patel NR, Roser S, Kolachala VL, Rojas M, Wang L, Oprea G, Garg P, et al. Targeted deletion of metalloproteinase 9 attenuates experimental colitis in mice: central role of epithelial-derived MMP. Gastroenterology. 2005;129:1991–2008. doi: 10.1053/j.gastro.2005.09.017. [DOI] [PubMed] [Google Scholar]
- 31.Santana A, Medina C, Paz-Cabrera MC, Díaz-Gonzalez F, Farré E, Salas A, Radomski MW, Quintero E. Attenuation of dextran sodium sulphate induced colitis in matrix metalloproteinase-9 deficient mice. World J Gastroenterol. 2006;12:6464–6472. doi: 10.3748/wjg.v12.i40.6464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu H, Patel NR, Walter L, Ingersoll S, Sitaraman SV, Garg P. Constitutive expression of MMP9 in intestinal epithelium worsens murine acute colitis and is associated with increased levels of proinflammatory cytokine Kc. Am J Physiol Gastrointest Liver Physiol. 2013;304:G793–G803. doi: 10.1152/ajpgi.00249.2012. [DOI] [PubMed] [Google Scholar]
- 33.Pender SL, Tickle SP, Docherty AJ, Howie D, Wathen NC, MacDonald TT. A major role for matrix metalloproteinases in T cell injury in the gut. J Immunol. 1997;158:1582–1590. [PubMed] [Google Scholar]
- 34.Kirkegaard T, Hansen A, Bruun E, Brynskov J. Expression and localisation of matrix metalloproteinases and their natural inhibitors in fistulae of patients with Crohn’s disease. Gut. 2004;53:701–709. doi: 10.1136/gut.2003.017442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Frei SM LS, Jehle EC, Fried M, Rogler G, Scharl M. Expression of Interleukins 22 and 33, Matrix Metalloproteinases 9 and 13, Mast Cell Markers and Hypoxia-Inducible Factor 1α in Crohn’s Disease Associated Fistulae Gastroenterology. DDW. 2013;144(5, Supplement 1):S441–S442. [Google Scholar]
- 36.Hazelbag S, Kenter GG, Gorter A, Dreef EJ, Koopman LA, Violette SM, Weinreb PH, Fleuren GJ. Overexpression of the alpha v beta 6 integrin in cervical squamous cell carcinoma is a prognostic factor for decreased survival. J Pathol. 2007;212:316–324. doi: 10.1002/path.2168. [DOI] [PubMed] [Google Scholar]
- 37.Arihiro K, Kaneko M, Fujii S, Inai K, Yokosaki Y. Significance of alpha 9 beta 1 and alpha v beta 6 integrin expression in breast carcinoma. Breast Cancer. 2000;7:19–26. doi: 10.1007/BF02967183. [DOI] [PubMed] [Google Scholar]
- 38.Li X, Yang Y, Hu Y, Dang D, Regezi J, Schmidt BL, Atakilit A, Chen B, Ellis D, Ramos DM. Alphavbeta6-Fyn signaling promotes oral cancer progression. J Biol Chem. 2003;278:41646–41653. doi: 10.1074/jbc.M306274200. [DOI] [PubMed] [Google Scholar]
- 39.Thomas GJ, Lewis MP, Hart IR, Marshall JF, Speight PM. AlphaVbeta6 integrin promotes invasion of squamous carcinoma cells through up-regulation of matrix metalloproteinase-9. Int J Cancer. 2001;92:641–650. doi: 10.1002/1097-0215(20010601)92:5<641::aid-ijc1243>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 40.Thomas GJ, Lewis MP, Whawell SA, Russell A, Sheppard D, Hart IR, Speight PM, Marshall JF. Expression of the alphavbeta6 integrin promotes migration and invasion in squamous carcinoma cells. J Invest Dermatol. 2001;117:67–73. doi: 10.1046/j.0022-202x.2001.01379.x. [DOI] [PubMed] [Google Scholar]
- 41.Thomas GJ, Poomsawat S, Lewis MP, Hart IR, Speight PM, Marshall JF. alpha v beta 6 Integrin upregulates matrix metalloproteinase 9 and promotes migration of normal oral keratinocytes. J Invest Dermatol. 2001;116:898–904. doi: 10.1046/j.1523-1747.2001.01352.x. [DOI] [PubMed] [Google Scholar]
- 42.Agrez M, Gu X, Turton J, Meldrum C, Niu J, Antalis T, Howard EW. The alpha v beta 6 integrin induces gelatinase B secretion in colon cancer cells. Int J Cancer. 1999;81:90–97. doi: 10.1002/(sici)1097-0215(19990331)81:1<90::aid-ijc16>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- 43.Agrez MV, Bates RC, Mitchell D, Wilson N, Ferguson N, Anseline P, Sheppard D. Multiplicity of fibronectin-binding alpha V integrin receptors in colorectal cancer. Br J Cancer. 1996;73:887–892. doi: 10.1038/bjc.1996.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Weinacker A, Chen A, Agrez M, Cone RI, Nishimura S, Wayner E, Pytela R, Sheppard D. Role of the integrin alpha v beta 6 in cell attachment to fibronectin. Heterologous expression of intact and secreted forms of the receptor. J Biol Chem. 1994;269:6940–6948. [PubMed] [Google Scholar]
- 45.Munger JS, Huang X, Kawakatsu H, Griffiths MJ, Dalton SL, Wu J, Pittet JF, Kaminski N, Garat C, Matthay MA, et al. The integrin alpha v beta 6 binds and activates latent TGF beta 1: a mechanism for regulating pulmonary inflammation and fibrosis. Cell. 1999;96:319–328. doi: 10.1016/s0092-8674(00)80545-0. [DOI] [PubMed] [Google Scholar]
- 46.Niu J, Gu X, Ahmed N, Andrews S, Turton J, Bates R, Agrez M. The alphaVbeta6 integrin regulates its own expression with cell crowding: implications for tumour progression. Int J Cancer. 2001;92:40–48. doi: 10.1002/1097-0215(200102)9999:9999<::aid-ijc1157>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 47.Frei SM, Pesch T, Lang S, Weber A, Jehle E, Vavricka SR, Fried M, Rogler G, Scharl M. A role for tumor necrosis factor and bacterial antigens in the pathogenesis of Crohn’s disease-associated fistulae. Inflamm Bowel Dis. 2013;19:2878–2887. doi: 10.1097/01.MIB.0000435760.82705.23. [DOI] [PubMed] [Google Scholar]
- 48.Aguilera O, Fraga MF, Ballestar E, Paz MF, Herranz M, Espada J, García JM, Muñoz A, Esteller M, González-Sancho JM. Epigenetic inactivation of the Wnt antagonist DICKKOPF-1 (DKK-1) gene in human colorectal cancer. Oncogene. 2006;25:4116–4121. doi: 10.1038/sj.onc.1209439. [DOI] [PubMed] [Google Scholar]
- 49.Koch S, Capaldo CT, Samarin S, Nava P, Neumaier I, Skerra A, Sacks DB, Parkos CA, Nusrat A. Dkk-1 inhibits intestinal epithelial cell migration by attenuating directional polarization of leading edge cells. Mol Biol Cell. 2009;20:4816–4825. doi: 10.1091/mbc.E09-05-0415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Niehrs C. Function and biological roles of the Dickkopf family of Wnt modulators. Oncogene. 2006;25:7469–7481. doi: 10.1038/sj.onc.1210054. [DOI] [PubMed] [Google Scholar]
- 51.Frei SM HC, Pesch T, Lang S, Weber A, Jehle E, Rhl A, Fried M, Rogler G, Scharl M. The Role for Dickkopf-Homolog-1 in the Pathogenesis of Crohn’s Disease-Associated Fistulae. PLoS One. 2013:In press. doi: 10.1371/journal.pone.0078882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhu Z, Homer RJ, Wang Z, Chen Q, Geba GP, Wang J, Zhang Y, Elias JA. Pulmonary expression of interleukin-13 causes inflammation, mucus hypersecretion, subepithelial fibrosis, physiologic abnormalities, and eotaxin production. J Clin Invest. 1999;103:779–788. doi: 10.1172/JCI5909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lee CG, Homer RJ, Zhu Z, Lanone S, Wang X, Koteliansky V, Shipley JM, Gotwals P, Noble P, Chen Q, et al. Interleukin-13 induces tissue fibrosis by selectively stimulating and activating transforming growth factor beta(1) J Exp Med. 2001;194:809–821. doi: 10.1084/jem.194.6.809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chiaramonte MG, Donaldson DD, Cheever AW, Wynn TA. An IL-13 inhibitor blocks the development of hepatic fibrosis during a T-helper type 2-dominated inflammatory response. J Clin Invest. 1999;104:777–785. doi: 10.1172/JCI7325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wynn TA. IL-13 effector functions. Annu Rev Immunol. 2003;21:425–456. doi: 10.1146/annurev.immunol.21.120601.141142. [DOI] [PubMed] [Google Scholar]
- 56.Donaldson DD, Whitters MJ, Fitz LJ, Neben TY, Finnerty H, Henderson SL, O’Hara RM, Beier DR, Turner KJ, Wood CR, et al. The murine IL-13 receptor alpha 2: molecular cloning, characterization, and comparison with murine IL-13 receptor alpha 1. J Immunol. 1998;161:2317–2324. [PubMed] [Google Scholar]
- 57.Bates RC, Mercurio AM. Tumor necrosis factor-alpha stimulates the epithelial-to-mesenchymal transition of human colonic organoids. Mol Biol Cell. 2003;14:1790–1800. doi: 10.1091/mbc.E02-09-0583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sullivan DE, Ferris M, Pociask D, Brody AR. Tumor necrosis factor-alpha induces transforming growth factor-beta1 expression in lung fibroblasts through the extracellular signal-regulated kinase pathway. Am J Respir Cell Mol Biol. 2005;32:342–349. doi: 10.1165/rcmb.2004-0288OC. [DOI] [PubMed] [Google Scholar]
- 59.Radlmayr M, Török HP, Martin K, Folwaczny C. The c-insertion mutation of the NOD2 gene is associated with fistulizing and fibrostenotic phenotypes in Crohn’s disease. Gastroenterology. 2002;122:2091–2092. doi: 10.1053/gast.2002.34020. [DOI] [PubMed] [Google Scholar]
- 60.Xavier RJ, Podolsky DK. Unravelling the pathogenesis of inflammatory bowel disease. Nature. 2007;448:427–434. doi: 10.1038/nature06005. [DOI] [PubMed] [Google Scholar]