

Abstract
Air pollution exposure is associated with cardiovascular events triggered by clot formation. Endothelial activation and initiation of coagulation are pathophysiological mechanisms that could link inhaled air pollutants to vascular events. Here we investigated the underlying mechanisms of increased endothelial cell procoagulant activity following exposure to soluble components of ultrafine particles (soluble UF). Human coronary artery endothelial cells (HCAEC) were exposed to soluble UF and assessed for their ability to trigger procoagulant activity in platelet-free plasma. Exposed HCAEC triggered earlier thrombin generation and faster fibrin clot formation, which was abolished by an anti-tissue factor (TF) antibody, indicating TF-dependent effects. Soluble UF exposure increased TF mRNA expression without compensatory increases in key anticoagulant proteins. To identify early events that regulate TF expression, we measured endothelial H2O2 production following soluble UF exposure and identified the enzymatic source. Soluble UF exposure increased endothelial H2O2 production, and antioxidants attenuated UF-induced upregulation of TF, linking the procoagulant responses to reactive oxygen species (ROS) formation. Chemical inhibitors and RNA silencing showed that NOX-4, an important endothelial source of H2O2, was involved in UF-induced upregulation of TF mRNA. These data indicate that soluble UF exposure induces endothelial cell procoagulant activity, which involves de novo TF synthesis, ROS production, and the NOX-4 enzyme. These findings provide mechanistic insight into the adverse cardiovascular effects associated with air pollution exposure.
Keywords: air pollution, NADPH oxidases, reactive oxygen species, tissue factor, thrombin generation
Epidemiological studies reveal a strong association between air pollution exposure and increased cardiovascular (CV) morbidity and mortality (Pope et al., 2004, 2006). Exposure to air pollution can increase thrombus formation in vivo in humans (Lucking et al., 2008) and animal models (Nemmar et al., 2003), and may lead to thrombotic events such as acute myocardial infarctions (Nawrot et al., 2011; Peters et al., 2004) and venous thromboembolism (Baccarelli et al., 2008). One of the major components of air pollution is particulate matter (PM), which is categorized based on the diameter of the particles: coarse (10–2.5 μm), fine (2.5–0.1 μm), and ultrafine (UF; <0.1 μm). The United States Environmental Protection Agency currently monitors and regulates coarse and fine PM, but increasing evidence suggests that UF particles, which are not currently regulated, are particularly important mediators of CV effects of air pollution (Nel, 2005). Although UF particles comprise only a small portion of ambient PM by mass concentration, they constitute the majority of particle number and have a large surface area-to-mass ratio. UF particles can deposit in the alveolar region of the lung due to their small diameter (Kreyling et al., 2004). These extremely small particles are not efficiently cleared by alveolar macrophages (Gonzalez et al., 1996), and they may persist in the lung, increasing the potential to cause adverse effects. Furthermore, soluble components of UF particles (soluble UF) could enter the vasculature by traversing the alveolar-capillary barrier (Wallenborn et al., 2007). These soluble components, which include transition metals, induce oxidative stress and procoagulant activity in animal and human models (Araujo et al., 2008; Sorensen et al., 2005); however, the mechanisms behind these responses have not been delineated.
A causal link between vascular effects of PM exposure and procoagulant responses is suggested by the finding that soluble UF exposure increases tissue factor (TF) mRNA expression in human pulmonary artery endothelial cells (Karoly et al., 2007). TF is not highly expressed on endothelial cells under normal physiological conditions, but surface expression is induced by various stimuli, including inflammatory cytokines, endotoxin, and hypoxia (Chu, 2005). Altered expression and activity of TF is of particular interest because, once activated, this membrane-bound protein is the primary initiator of the extrinsic coagulation pathway, resulting in thrombin generation and thrombus formation. Elevated TF levels are associated with adverse CV effects including thrombosis and atherosclerosis (Mackman et al., 2007).
TF expression is redox-regulated, and TF mRNA and protein levels are modulated by reactive oxygen species (ROS) in vascular cells (Herkert and Gorlach, 2002). Endothelial cells produce ROS in response to several physiological and pathological stimuli, including exposure to UF particles (Mo et al., 2009). One major cellular source of ROS production is the NADPH oxidase (NOX) family of enzymes (Lassegue et al., 2012). Endothelial cells express mainly NOX-2 and NOX-4 isoforms with variable expression levels between endothelial cell types (Guzik et al., 2004). Nox enzymes produce basal physiological levels of ROS, and NOX activity is augmented upon agonist stimulation producing increased ROS levels that play a role in pathophysiological responses (Lassegue et al., 2012). For example, thrombin enhances TF expression and procoagulant activity in vascular smooth muscle cells (VSMC) by stimulating ROS production by NOX (Herkert et al., 2002). Furthermore, NOX-induced oxidative stress in vascular cells is involved in the pathogenesis of CV disorders (Madamanchi et al., 2005), potentially through these procoagulant mechanisms.
The objective of our study was to determine if soluble UF induces procoagulant responses in human endothelial cells and to define the mechanisms behind these effects. We show here that endothelial cells exposed to soluble UF induce faster onset of thrombin generation and fibrin clot formation via TF upregulation, which involves ROS production and the NOX-4 enzyme.
MATERIALS AND METHODS
Reagents and chemicals
Human coronary artery endothelial cells (HCAEC), endothelial growth medium (EGM-2), and EGM-2 Bullet Kit were obtained from Lonza (Walkersville, MD). All other chemicals and reagents were from Sigma Chemical Company (St. Louis, MO) unless otherwise stated.
Cell culture
HCAEC were cultured in EGM-2 media supplemented with the EGM-2 Bullet Kit. Cells were grown to confluence and used between passages 5–8.
UF particles
UF particles collected from February to May 2007 in Chapel Hill, NC were analyzed for chemical components (Table 1) by the Research Triangle Institute (Research Triangle Park, NC) as previously described (Becker et al., 2005). The water-soluble fraction of UF particles was obtained as previously described (Karoly et al., 2007).
TABLE 1. Components of UF PM (ng/mg) Collected in Chapel Hill, NC from February to May 2007.
| Component | ng/mg | Component | ng/mg |
|---|---|---|---|
| Aluminum | 855.0 | Nitrate | 44100.0 |
| Arsenic | 55.8 | Nitrite | 579.0 |
| Bromide | 1824.0 | Selenium | 76.5 |
| Chromium | 12.3 | Silicon | 3480.0 |
| Copper | 108.0 | Sulfur | 99300.0 |
| Iron | 624.0 | Titanium | 21.5 |
| Lead | 79.5 | Vanadium | 57.0 |
| Nickel | 34.2 | Zinc | 735.0 |
Calibrated automated thrombography assay
Platelet-free plasma (PFP) from healthy donors was prepared as previously described (Machlus et al., 2009). Cellular procoagulant activity was measured as previously described with alterations (Campbell et al., 2009). Briefly, HCAEC were cultured on 96-well plates and exposed to soluble UF for 6 h. Following exposure, cells and PFP were incubated with an inhibitory anti-human TF monoclonal inhibitory antibody or control mouse IgG for 15 min at 37°C. PFP with antibodies, MP reagent containing phospholipids (4μM; 60% phosphatidylcholine/20% phosphatidylethanolamine/20% phosphatidylserine; Diagnostica Stago, Parsippany, NJ), and a fluorogenic thrombin substrate (Z-Gly-Gly-Arg-AMC; Diagnostica Stago) were then added to the cells (Machlus et al., 2009). Reactions were calibrated against wells containing a thrombin calibrator (α2-macroglobulin/thrombin complex; Diagnostica Stago). Thrombin generation was measured using a Fluoroskan Ascent fluorometer (ThermoLabsystem, Helsinki, Finland). Thrombinoscope software version 3.0.0.29 (Thrombinoscope BV, Maastricht, Netherlands) was used to calculate lag time (first time point after the thrombin concentration exceeds one-sixth peak height), peak (greatest amount of thrombin generation at a single point in time), time to peak (time to reach the maximum peak height), and endogenous thrombin potential (ETP; total amount of thrombin generated during the test).
Turbidity assay
HCAEC were cultured and exposed to soluble UF as described above and fibrin clot formation measured as previously described (Gray et al., 2011). Clot formation onset (time to reach inflection point before turbidity increase) and Vmax (slope of the line fitted to maximum rate of turbidity increase) were calculated using Softmax Pro Software version 1.21 (Molecular Devices, Sunnyvale, CA).
Real-time quantitative PCR
Relative gene expression in HCAEC was obtained using quantitative RT-PCR as previously described (Karoly et al., 2007). Total RNA was isolated from HCAEC using an RNeasy kit (Qiagen, Valencia, CA) as per manufacturer's protocol. RNA was quantified using a Nanodrop 1000 Spectrophotometer (Thermo Scientific, Wilmington, DE). Oligonucleotide primer pairs and fluorescent probes for β-actin (forward, 5′-CCTGGCACCCAGCACAAT-3′; reverse, 5′-GCCGATCCACACGGAGTACT-3′; probe, 5′-ATCAAGATCATTGCTCCTCCTGAGCGC-3′) and TF (forward, 5′-TTGGCACGGGTCTTCTCCTA-3′; reverse, 5′-CGAGGTTTGTCTCCAGGTAAGG-3′; and probe, 5′-AGAACCGGTGCTCTCCACATTCCCTG-3′) were designed using Primer Express (Applied Biosystems, Foster City, CA) and obtained from Integrated DNA Technologies (Coralville, IA). Thrombomodulin, endothelial protein C receptor (EPCR), tissue factor pathway inhibitor (TFPI), GAPDH, and NOX-4 primer pairs and fluorescent probe sets were obtained as Taqman pre-developed assay reagents from Applied Biosystems. Standard curves generated from a serially diluted standard pool of cDNA prepared from cultured human endothelial cells exposed to 100 ng/ml TNF-α for 6 h were used to determine the relative abundance of mRNA levels, which were normalized to relative abundance of β-actin mRNA.
H2O2 measurement
Extracellular H2O2 release was measured using the Amplex Red reagent (10-acetyl-3,7-dihydroxyphenoxazine; Invitrogen, Carlsbad, CA) as previously described (Li et al., 2006). Following analysis, cellular protein was collected and measured using the Bio-Rad protein reagent (Bio-Rad, Richmond, CA) as per manufacturer's protocol.
Peroxy Green 1 (PG1; kindly provided by Dr Christopher Chang, University of California-Berkeley), a small-molecule fluorophore that is more responsive to H2O2 than other ROS (Miller et al., 2007), was used to measure intracellular H2O2 as previously described (Cheng et al., 2010).
siRNA transfection
HCAEC were transfected with 90nM concentration of NOX-4 or scrambled no. 5 siRNA using siPORT Amine transfection agent (Ambion, Austin, TX) as per manufacturer's protocol. Cells were used for experiments 48 h post-transfection.
Statistical analysis
Statistical analysis was performed using GraphPad Prism (GraphPad Software, San Diego, CA). A t-test or one-way ANOVA analysis followed by Bonferroni's post-hoc test was used to compare exposed cells with control cells. A p-value < 0.05 was considered statistically significant.
RESULTS
Soluble UF Cause HCAEC to Promote Earlier Onset of TF-Dependent Thrombin Generation and Fibrin Clot Formation
HCAEC were exposed to 10, 50, and 100 μg/ml soluble UF. We found that these doses are non-cytotoxic to the cells (Supplementary fig. 1). Six hours following exposure, cells were assessed for functional coagulation balance using two methods. We used the calibrated automated thrombography (CAT) assay to quantify thrombin generation (Figs. 1A–C) and the turbidity assay to measure the rate of fibrin clot formation (Figs. 1D–F) in PFP. The CAT assay showed that lag time (Fig. 1B) and time to peak (Fig. 1C) significantly shortened with increasing doses of soluble UF, indicating earlier onset of thrombin generation. Soluble UF exposure did not significantly affect thrombin peak or ETP (data not shown). In the turbidity assay, HCAEC exposure to soluble UF led to a significant shortening in the onset time to clot formation (Fig. 1E) and an increase in Vmax (Fig. 1F), indicating earlier onset and faster rate of fibrin clot formation. These effects on thrombin and fibrin were abolished by addition of an inhibitory anti-human TF antibody. These data suggest soluble UF increases endothelial procoagulant activity via a TF-dependent mechanism.
Fig. 1.

Soluble UF exposure leads to faster onset of TF-dependent thrombin generation and fibrin clot formation. Endothelial cell coagulation balance was assessed by measuring capacity of HCAEC to induce thrombin generation (A-C) and fibrin clot formation (D-F) in PFP following soluble UF exposure (0, 10, 50, and 100 μg/ml). Thrombin parameters lag time (B) and time to peak (C) and the fibrin clot parameters clot formation onset (E) and Vmax (F) were measured. Graphs are representative of three separate experiments with samples in triplicate. Error bars indicate ± SE. *p < 0.05, **p < 0.01, ***p < 0.001 compared with control.
Soluble UF Increases TF mRNA Expression in HCAEC
To characterize the cause of soluble UF-induced changes in HCAEC cellular procoagulant activity, we quantified mRNA expression of key procoagulant and anticoagulant proteins by RT-PCR following exposure for 6 and 24 h. Exposure to 50 and 100 μg/ml of soluble UF led to a significant (p < 0.001) 3.8-fold and 5.1-fold, respectively, increase of TF mRNA expression in HCAEC at 6 h, and increased expression was sustained at 24 h (Fig. 2A). Conversely, we found no significant changes in thrombomodulin, EPCR, or TFPI following soluble UF exposure (Figs. 2B–D). The upregulation of TF and lack of compensatory changes in the anticoagulant proteins provide a mechanistic rationale for the increased cellular procoagulant activity seen in Figure 1.
Fig. 2.

Soluble UF exposure leads to TF upregulation. (A-D) HCAEC were exposed to soluble UF (0, 10, 50, and 100 μg/ml) for 6 or 24 h. TF (A), thrombomodulin (B), EPCR (C), and TFPI (D) mRNA expression were quantified by RT-PCR. Error bars indicate ± SE of n = 3 separate experiments with samples in triplicate. ***p < 0.001 compared with control.
Soluble UF-Induced TF Upregulation Involves ROS
To characterize the events initiating increased TF production, we treated HCAEC with PEG-SOD and PEG-catalase prior to 6 h soluble UF exposure (50 μg/ml) and measured TF mRNA expression. These cell-permeable antioxidants significantly suppressed TF mRNA upregulation by soluble UF by 55% and 36%, respectively (p < 0.001, Fig. 3A). In addition, pretreatment with L-NAME, a nitric oxide synthase inhibitor, did not attenuate TF mRNA levels following exposure (Fig. 3B). These data indicate that ROS production is involved in increasing TF mRNA expression in soluble UF-treated HCAEC.
Fig. 3.
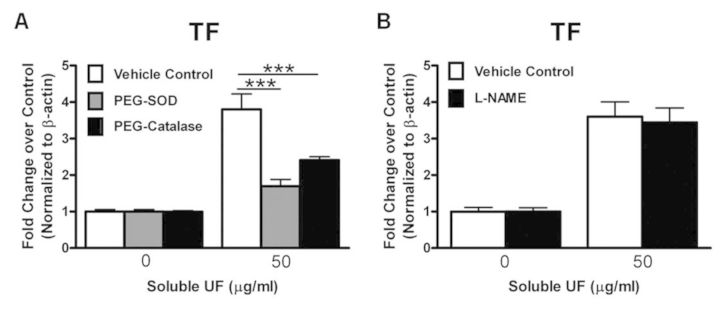
TF upregulation by soluble UF is ROS dependent. HCAEC were pretreated with 100 U/ml PEG-SOD or PEG-catalase for 1 h (A) or 1mM L-NAME for 30 min (B) prior to a 6 h exposure to 0 or 50 μg/ml soluble UF. TF mRNA expression was quantified by RT-PCR. Error bars indicate ± SE of n = 3 separate experiments with samples in triplicate. ***p < 0.001 compared with control.
Soluble UF Exposure Causes H2O2 Production in HCAEC
To characterize ROS production by endothelial cells, we measured extracellular and intracellular H2O2 production in soluble UF-treated HCAEC. Using the Amplex Red assay to quantify extracellular H2O2 release, we found an 11.8-fold (p < 0.05) and 19.7-fold (p < 0.001) increase in extracellular H2O2 levels following HCAEC exposure to 50 and 100 μg/ml soluble UF, respectively (Fig. 4A). Using the chemical indicator PG1, we observed a significant and progressive dose-dependent increase in intracellular H2O2 in cells exposed to 50 (21%, p < 0.05) and 100 μg/ml (32%, p < 0.001) (Fig. 4B). We then visualized the distribution of intracellular H2O2 production in individual living cells in real time using confocal microscopy. Soluble UF (100 μg/ml) induced intracellular H2O2 in HCAEC in a diffuse, cytoplasmic distribution as indicated by increased PG1 fluorescence intensity (Fig. 4C). Quantitative analysis of the confocal images indicated a significant increase (p < 0.001) in fold change over baseline in soluble UF-exposed HCAEC (Fig. 4D). These data indicate that soluble UF exposure results in immediate increases in extracellular and intracellular H2O2 production in HCAEC.
Fig. 4.

Soluble UF exposure induces rapid H2O2 generation by HCAEC. (A) HCAEC were exposed to soluble UF (0, 50, and 100 μg/ml) and extracellular H2O2 release was measured using the Amplex Red reagent. (B) HCAEC were exposed to soluble UF (0, 10, 50, and 100 μg/ml) for 60 min in the presence of the H2O2 indicator PG1. Fluorescence was measured with a fluorescent plate reader. Error bars indicate ± SE of n = 3 independent experiments with samples in triplicate. (C) PG1 fluorescence was detected by confocal microscopy. HCAEC were pre-incubated with PG1 for a baseline measurement, then exposed to 0 or 100 μg/ml soluble UF for 30 min. Representative images shown in a pseudo-colored scheme are from baseline and 30 min post-exposure. (D) Quantitative data from the confocal images were gathered by outlining individual cells and measuring the Relative Fluorrescense Units (RFU) using Nikon NIS-Elements software. Error bars indicate ± SE of n = 4 independent experiments. *p < 0.05, ***p < 0.001 compared with control.
Soluble UF-Induced TF mRNA Expression Involves the NOX-4 Enzyme
To determine the cellular source of soluble UF-induced H2O2 production, we measured extracellular H2O2 production by HCAEC in the presence of chemical inhibitors targeted towards the major sources of endothelial ROS production. DPI, a NOX inhibitor, reduced extracellular H2O2 production in soluble UF-exposed (50 μg/ml) HCAEC by 54% (p < 0.05) (Fig. 5A). Soluble UF-induced extracellular H2O2 levels were not affected by inhibitors of xanthine oxidases (allopurinol) or mitochondrial sources (KCN, rotenone), which are other important sources of endothelial ROS production (data not shown). These data indicate that NOX enzymes are likely the major source of soluble UF-induced H2O2 production. To determine if NOX enzymes mediate the increase in TF mRNA, we pretreated HCAEC with DPI followed by a 6 h soluble UF exposure (50 μg/ml). Pretreatment with the NOX inhibitor led to a 50% attenuation (p < 0.01) of the increased TF mRNA levels in exposed cells (Fig. 5B).
Fig. 5.

TF mRNA expression following soluble UF exposure requires ROS production and NOX-4. HCAEC pretreated with the NOX inhibitor DPI (50μM) for 30 min followed by exposure to 0 or 50 μg/ml soluble UF were used to measure (A) extracellular H2O2 and (B) TF mRNA expression. (C) HCAEC were transfected with NOX-4 or scramble siRNA for 48 h. mRNA expression of the target gene NOX-4 and non-target gene GAPDH were quantified by RT-PCR. (D) HCAEC were transfected with NOX-4 or scramble siRNA for 48 h and then exposed to 0 or 50 μg/ml soluble UF for 6 h. TF mRNA expression was quantified by RT-PCR. Error bars indicate ± SE of n = 3 separate experiments with samples in duplicate. *p < 0.05, **p < 0.01, and ***p < 0.001 compared with control.
Because HCAEC predominately express the NOX-4 isoform (Yoshida and Tsunawaki, 2008), we more specifically probed the role of NOX-4 in soluble UF-induced TF mRNA upregulation by transfecting HCAEC with NOX-4 siRNA. Transfection resulted in 84% knockdown of the target gene mRNA levels as compared with the scrambled siRNA control after 48 h (Fig. 5C). HCAEC were then transfected with NOX-4 or scrambled siRNA for 48 h prior to soluble UF exposure for 6 h. Compared with cells transfected with scrambled siRNA, NOX-4 silencing significantly (p < 0.001) attenuated soluble UF-induced TF mRNA upregulation by 56% (Fig. 5D). Together, these data indicate that following soluble UF exposure, ROS and the NOX-4 enzyme are involved in upregulating TF mRNA expression and endothelial cell-dependent procoagulant activity.
DISCUSSION
There is strong epidemiological evidence supporting a causal relationship between air pollution exposure and adverse CV health effects in humans that include clinical events mediated by thrombosis (Pope et al., 2004, 2006). An important but understudied area in this field is the effect of inhaled air pollutants on vascular cells via soluble components that can translocate into the vasculature. Here we report that the water-soluble components of UF PM induce a procoagulant phenotype in endothelial cells, supporting earlier onset of thrombin generation and faster fibrin clot formation, and that these activities are driven by TF upregulation. We further show that this increase in TF mRNA is regulated by increased ROS production and the NOX-4 enzyme, and can be attenuated by treatment of cells with antioxidants or by inhibiting NOX-4. These novel findings provide mechanistic insight into the enhanced thrombosis and endothelial dysfunction that might underlie increased risk for CV morbidity and mortality associated with air pollution exposure.
Our results add to a growing body of evidence linking air pollution exposure to thrombosis. Recent studies demonstrated hypercoagulable effects of air pollution on plasma thrombin generation in human and animal models (Emmerechts et al., 2012; Kilinc et al., 2011). Emmerechts et al. demonstrated that elevated levels of coarse PM shortened the lag time of thrombin generation in microvesicle-rich plasma obtained from diabetic individuals exposed to ambient air pollution (Emmerechts et al., 2012). Our results extend these observations to potentially pathologic effects of PM on cellular procoagulant activity. Specifically, using the CAT assay, we show that soluble UF exposure decreased lag time in a dose-dependent manner, which has been previously shown to be a sensitive reflection of TF activity (Ollivier et al., 2010). We did not find significant differences in ETP or peak thrombin; however, this is not surprising because these parameters are typically more associated with alterations in levels of soluble clotting factors (Machlus et al., 2009). In vivo, we anticipate that these observed changes in plasma (Emmerechts et al., 2012; Kilinc et al., 2011) and cellular procoagulant activity (endothelial cells and possibly platelets) may be additive or even synergistic, ultimately dysregulating multiple thrombin generation parameters simultaneously. Moreover, by disrupting endothelial cell function, soluble UF may also alter expression of endothelial-derived fibrinolytic enzymes (e.g., tissue plasminogen activator), with additive negative consequences for clot stability. Further study of these intriguing possibilities may reveal additional therapeutic targets for reducing deleterious effects of air pollution on clot quality.
TF expression is generally suppressed in endothelial cells under normal physiological conditions, but is induced by a variety of stimuli in vitro (Chu, 2005; Karoly et al., 2007) and possibly in vivo (Mackman et al., 2007). Activation of TF from injury or pathological conditions is typically balanced by expression of anticoagulant proteins such as TFPI (Crawley and Lane, 2008); however, our findings show lack of parallel upregulation of these anticoagulant proteins, resulting in a procoagulant environment that favors thrombin generation. The TF gene in endothelial cells contains binding sites for redox-sensitive transcription factors (Herkert and Gorlach, 2002). Accordingly, we found that in HCAEC, TF mRNA levels following soluble UF exposure involved the ROS molecules H2O2 and superoxide rather than NO. This finding supports previous studies showing attenuated TF mRNA expression in the presence of antioxidants following exposure to ionizing radiation and inflammatory cytokines (Szotowski et al., 2007).
ROS are implicated in the pathogenesis and progression of several CV diseases including atherosclerosis, hypertension, and diabetes (Madamanchi et al., 2005). Additionally, concentrations of 7-hydro-8-oxo-2’-deoxyguanosine in lymphocyte DNA correlated with elevated levels of water-soluble transition metals in fine PM in humans exposed to ambient air pollution (Sorensen et al., 2005). This measurement has been used as a biomarker to assess oxidative damage caused by increased ROS production and suggests air pollution exposure triggers oxidative stress. We showed that soluble UF components lead to significant increases in both extracellular and intracellular endothelial H2O2 production immediately following exposure. Our data, using the indicator PG1, indicate that ROS are initially formed intracellularly after soluble UF exposure. This premise is further supported by confocal microscopy, which showed a cytoplasmic distribution of H2O2.
NOX is an important source of ROS in vascular cells, and NOX enzymes have been implicated as a source of oxidative stress in numerous CV diseases (Lassegue et al., 2012). Although endothelial cells have several major sources of ROS production, NOX enzymes induced ROS production in mouse pulmonary microvascular endothelial cells exposed to whole UF particles (Mo et al., 2009). Our findings are important because we show that NOX-4 is the likely source of soluble UF-induced H2O2 production in human endothelial cells. The intracellular distribution of NOX-4 is broad and has been shown to be variably located in the plasma membrane, endoplasmic reticulum, mitochondria, and nucleus of endothelial cells (Lassegue et al., 2012). Our data further suggest that NOX-4-derived ROS produced following soluble UF exposure is cytoplasmic, but can escape to the extracellular space. This finding has important implications for paracrine signaling effects by cellular H2O2.
NOX-4 is involved in increasing TF mRNA expression in HCAEC by soluble UF. Other researchers have shown that ROS from NOX enzymes induce TF mRNA expression in VSMC after agonist stimulation by thrombin (Herkert et al., 2002). Our data extend this finding to endothelial cells after a more subtle challenge with soluble UF. NOX enzymes are important potential therapeutic targets for strategies to limit overproduction of ROS (Schramm et al., 2012) and our data reinforce the concept that inhibiting these enzymes is a potential approach to combat adverse CV effects from air pollution exposure.
Our study is novel in the use of primary HCAEC to model effects of soluble air pollution components. Because these endothelial cells contact blood immediately from the lungs, they seem likely to receive high levels of any soluble air pollution components that enter the vasculature. Although it is not known whether even the smallest PM can translocate from the lung into the vasculature and cause pathologic effects (Mills et al., 2006; Nemmar et al., 2002), water-soluble components of the particles appear in the vasculature following intratracheal instillation in rats (Wallenborn et al., 2007). Moreover, we previously determined that the insoluble fraction of UF PM does not induce significant adverse responses in HCAEC (Snow and Carraway, 2010). Our study strongly supports the potential for a pathogenic role for the water-soluble components of UF particles and implies that in endothelial cell culture systems, soluble UF exposure is more physiologically relevant than whole UF particle exposures. Furthermore, PM can cause cell death in vitro at high concentrations; however, we confirmed that the doses used in this study were non-cytotoxic, indicating that the procoagulant activity following soluble UF exposure was due to endothelial activation rather than cellular death.
Our study has a few limitations that are common to in vitro models. First, our study cannot clarify the uncertainty of whether endothelial TF is expressed in vivo in humans following PM exposure, even though TF expression does increase in vitro and in animals in response to PM (Sun et al., 2008). Furthermore, it has been shown that endothelial TF upregulation occurs in vascular disease in humans (Shet et al., 2003; Thiruvikraman et al., 1996), which is relevant to our postulate that air pollutants induce endothelial activation. Second, our data must be interpreted in light of the lack of current evidence for TF-initiated coagulation as a key early event in adverse CV responses to air pollution. Several negative studies on the role of coagulation considered acute exposures to larger air pollution particles. Yet, a recent study in diabetic individuals did find a trend towards early increased monocyte and platelet TF expression and TF+ microparticles following smaller UF particle exposure (Stewart et al., 2010), which suggests effects may differ between particles depending on size and associated components. Finally, in vitro studies are limited by the uncertainty in predicting PM component levels experienced by vascular cells after inhaling known particle concentrations. However, individuals exposed to indoor smoking, fire smoke, or indoor cook stoves can be transiently exposed to elevated UF particle concentrations on par with those used to generate soluble fractions for this study (Afshari et al., 2005; Baxter et al., 2010; Karoly et al., 2007). Therefore, we estimated levels that would emulate levels of particle components to which vascular cells could be exposed.
In summary, we have shown that exposure of HCAEC to soluble UF leads to increased production of intracellular ROS and activation of the NOX-4 enzyme that regulates TF mRNA. This activation of the extrinsic pathway results in faster onset of thrombin generation and fibrin clot formation. These novel findings support and extend important information regarding mechanism(s) by which PM exposure results in thrombosis and adverse CV health effects.
SUPPLEMENTARY DATA
Supplementary data are available online at http://toxsci.oxfordjournals.org/.
FUNDING
National Institutes of Health (CR833237 and T32ES007126 to S.J.S., R01HL094740 to A.S.W.).
Supplementary Material
Acknowledgments
We thank Lisa Dailey, Joleen Soukup, and Laura Gray for their excellent technical assistance in the execution of this study.
Disclaimer: The authors declare no competing financial interests. The research described in this article has been reviewed by the National Health and Environmental Effects Research Laboratory, U.S. EPA, and approved for publication. The contents of this article should not be construed to represent Agency policy nor does mention of trade names or commercial products constitute endorsement or recommendation for use.
REFERENCES
- Afshari A., Matson U., Ekberg L. E. Characterization of indoor sources of fine and ultrafine particles: A study conducted in a full-scale chamber. Indoor Air. 2005;15:141–150. doi: 10.1111/j.1600-0668.2005.00332.x. [DOI] [PubMed] [Google Scholar]
- Araujo J. A., Barajas B., Kleinman M., Wang X., Bennett B. J., Gong K. W., Navab M., Harkema J., Sioutas C., Lusis A. J., et al. Ambient particulate pollutants in the ultrafine range promote early atherosclerosis and systemic oxidative stress. Circ. Res. 2008;102:589–596. doi: 10.1161/CIRCRESAHA.107.164970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baccarelli A., Martinelli I., Zanobetti A., Grillo P., Hou L. F., Bertazzi P. A., Mannucci P. M., Schwartz J. Exposure to particulate air pollution and risk of deep vein thrombosis. Arch. Intern. Med. 2008;168:920–927. doi: 10.1001/archinte.168.9.920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baxter C. S., Ross C. S., Fabian T., Borgerson J. L., Shawon J., Gandhi P. D., Dalton J. M., Lockey J. E. Ultrafine particle exposure during fire suppression–is it an important contributory factor for coronary heart disease in firefighters? J. Occup. Environ. Med. 2010;52:791–796. doi: 10.1097/JOM.0b013e3181ed2c6e. [DOI] [PubMed] [Google Scholar]
- Becker S., Dailey L. A., Soukup J. M., Grambow S. C., Devlin R. B., Huang Y. C. Seasonal variations in air pollution particle-induced inflammatory mediator release and oxidative stress. Environ. Health Perspect. 2005;113:1032–1038. doi: 10.1289/ehp.7996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell R. A., Overmyer K. A., Selzman C. H., Sheridan B. C., Wolberg A. S. Contributions of extravascular and intravascular cells to fibrin network formation, structure, and stability. Blood. 2009;114:4886–4896. doi: 10.1182/blood-2009-06-228940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng W. Y., Tong H., Miller E. W., Chang C. J., Remington J., Zucker R. M., Bromberg P. A., Samet J. M., Hofer T. P. An integrated imaging approach to the study of oxidative stress generation by mitochondrial dysfunction in living cells. Environ. Health Perspect. 2010;118:902–908. doi: 10.1289/ehp.0901811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu A. J. Tissue factor mediates inflammation. Arch. Biochem. Biophys. 2005;440:123–132. doi: 10.1016/j.abb.2005.06.005. [DOI] [PubMed] [Google Scholar]
- Crawley J. T., Lane D. A. The haemostatic role of tissue factor pathway inhibitor. Arterioscler. Thromb. Vasc. Biol. 2008;28:233–242. doi: 10.1161/ATVBAHA.107.141606. [DOI] [PubMed] [Google Scholar]
- Emmerechts J., Jacobs L., Van Kerckhoven S., Loyen S., Mathieu C., Fierens F., Nemery B., Nawrot T. S., Hoylaerts M. F. Air pollution-associated procoagulant changes: The role of circulating microvesicles. J. Thromb. Haemost. 2012;10:96–106. doi: 10.1111/j.1538-7836.2011.04557.x. [DOI] [PubMed] [Google Scholar]
- Gonzalez O., Smith R. L., Goodman S. B. Effect of size, concentration, surface area, and volume of polymethylmethacrylate particles on human macrophages in vitro. J. Biomed. Mater. Res. 1996;30:463–473. doi: 10.1002/(SICI)1097-4636(199604)30:4<463::AID-JBM4>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- Gray L. D., Hussey M. A., Larson B. M., Machlus K. R., Campbell R. A., Koch G., Ezban M., Hedner U., Wolberg A. S. Recombinant factor viia analog nn1731 (v158d/e296v/m298q-fviia) enhances fibrin formation, structure and stability in lipidated hemophilic plasma. Thromb. Res. 2011;128:570–576. doi: 10.1016/j.thromres.2011.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzik T. J., Sadowski J., Kapelak B., Jopek A., Rudzinski P., Pillai R., Korbut R., Channon K. M. Systemic regulation of vascular nad(p)h oxidase activity and nox isoform expression in human arteries and veins. Arterioscler. Thromb. Vasc. Biol. 2004;24:1614–1620. doi: 10.1161/01.ATV.0000139011.94634.9d. [DOI] [PubMed] [Google Scholar]
- Herkert O., Diebold I., Brandes R. P., Hess J., Busse R., Gorlach A. Nadph oxidase mediates tissue factor-dependent surface procoagulant activity by thrombin in human vascular smooth muscle cells. Circulation. 2002;105:2030–2036. doi: 10.1161/01.cir.0000014611.28864.1e. [DOI] [PubMed] [Google Scholar]
- Herkert O., Gorlach A. Redox control of tissue factor expression in smooth muscle cells and other vascular cells. Methods Enzymol. 2002;352:220–231. doi: 10.1016/s0076-6879(02)52021-8. [DOI] [PubMed] [Google Scholar]
- Karoly E. D., Li Z., Dailey L. A., Hyseni X., Huang Y. C. Up-regulation of tissue factor in human pulmonary artery endothelial cells after ultrafine particle exposure. Environ. Health Perspect. 2007;115:535–540. doi: 10.1289/ehp.9556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilinc E., Van Oerle R., Borissoff J. I., Oschatz C., Gerlofs-Nijland M. E., Janssen N. A., Cassee F. R., Sandstrom T., Renne T., Ten Cate H., et al. Factor xii activation is essential to sustain the procoagulant effects of particulate matter. J. Thromb. Haemost. 2011;9:1359–1367. doi: 10.1111/j.1538-7836.2011.04280.x. [DOI] [PubMed] [Google Scholar]
- Kreyling W. G., Semmler M., Moller W. Dosimetry and toxicology of ultrafine particles. J. Aerosol. Med. 2004;17:140–152. doi: 10.1089/0894268041457147. [DOI] [PubMed] [Google Scholar]
- Lassegue B., San Martin A., Griendling K. K. Biochemistry, physiology, and pathophysiology of nadph oxidases in the cardiovascular system. Circ. Res. 2012;110:1364–1390. doi: 10.1161/CIRCRESAHA.111.243972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z., Hyseni X., Carter J. D., Soukup J. M., Dailey L. A., Huang Y. C. Pollutant particles enhanced h2o2 production from nad(p)h oxidase and mitochondria in human pulmonary artery endothelial cells. Am. J. Physiol. Cell Physiol. 2006;291:C357–365. doi: 10.1152/ajpcell.00365.2005. [DOI] [PubMed] [Google Scholar]
- Lucking A. J., Lundback M., Mills N. L., Faratian D., Barath S. L., Pourazar J., Cassee F. R., Donaldson K., Boon N. A., Badimon J. J., et al. Diesel exhaust inhalation increases thrombus formation in man. Eur. Heart J. 2008;29:3043–3051. doi: 10.1093/eurheartj/ehn464. [DOI] [PubMed] [Google Scholar]
- Machlus K. R., Colby E. A., Wu J. R., Koch G. G., Key N. S., Wolberg A. S. Effects of tissue factor, thrombomodulin and elevated clotting factor levels on thrombin generation in the calibrated automated thrombogram. Thromb. Haemost. 2009;102:936–944. doi: 10.1160/TH09-03-0180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackman N., Tilley R. E., Key N. S. Role of the extrinsic pathway of blood coagulation in hemostasis and thrombosis. Arterioscler. Thromb. Vasc. Biol. 2007;27:1687–1693. doi: 10.1161/ATVBAHA.107.141911. [DOI] [PubMed] [Google Scholar]
- Madamanchi N. R., Vendrov A., Runge M. S. Oxidative stress and vascular disease. Arterioscler. Thromb. Vasc. Biol. 2005;25:29–38. doi: 10.1161/01.ATV.0000150649.39934.13. [DOI] [PubMed] [Google Scholar]
- Miller E. W., Tulyathan O., Isacoff E. Y., Chang C. J. Molecular imaging of hydrogen peroxide produced for cell signaling. Nat. Chem. Biol. 2007;3:263–267. doi: 10.1038/nchembio871. [DOI] [PubMed] [Google Scholar]
- Mills N. L., Amin N., Robinson S. D., Anand A., Davies J., Patel D., de la Fuente J. M., Cassee F. R., Boon N. A., Macnee W., et al. Do inhaled carbon nanoparticles translocate directly into the circulation in humans. Am. J. Respir. Crit. Care Med. 2006;173:426–431. doi: 10.1164/rccm.200506-865OC. [DOI] [PubMed] [Google Scholar]
- Mo Y., Wan R., Chien S., Tollerud D. J., Zhang Q. Activation of endothelial cells after exposure to ambient ultrafine particles: The role of nadph oxidase. Toxicol. Appl. Pharmacol. 2009;236:183–193. doi: 10.1016/j.taap.2009.01.017. [DOI] [PubMed] [Google Scholar]
- Nawrot T. S., Perez L., Kunzli N., Munters E., Nemery B. Public health importance of triggers of myocardial infarction: A comparative risk assessment. Lancet. 2011;377:732–740. doi: 10.1016/S0140-6736(10)62296-9. [DOI] [PubMed] [Google Scholar]
- Nel A. Atmosphere. Air pollution-related illness: Effects of particles. Science. 2005;308:804–806. doi: 10.1126/science.1108752. [DOI] [PubMed] [Google Scholar]
- Nemmar A., Hoet P. H., Dinsdale D., Vermylen J., Hoylaerts M. F., Nemery B. Diesel exhaust particles in lung acutely enhance experimental peripheral thrombosis. Circulation. 2003;107:1202–1208. doi: 10.1161/01.cir.0000053568.13058.67. [DOI] [PubMed] [Google Scholar]
- Nemmar A., Hoet P. H., Vanquickenborne B., Dinsdale D., Thomeer M., Hoylaerts M. F., Vanbilloen H., Mortelmans L., Nemery B. Passage of inhaled particles into the blood circulation in humans. Circulation. 2002;105:411–414. doi: 10.1161/hc0402.104118. [DOI] [PubMed] [Google Scholar]
- Ollivier V., Wang J., Manly D., Machlus K. R., Wolberg A. S., Jandrot-Perrus M., Mackman N. Detection of endogenous tissue factor levels in plasma using the calibrated automated thrombogram assay. Thromb. Res. 2010;125:90–96. doi: 10.1016/j.thromres.2009.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters A., von Klot S., Heier M., Trentinaglia I., Hormann A., Wichmann H. E., Lowel H. Cooperative Health Research in the Region of Augsburg Study, G. Exposure to traffic and the onset of myocardial infarction. N. Engl. J. Med. 2004;351:1721–1730. doi: 10.1056/NEJMoa040203. [DOI] [PubMed] [Google Scholar]
- Pope C. A., III, Burnett R. T., Thurston G. D., Thun M. J., Calle E. E., Krewski D., Godleski J. J. Cardiovascular mortality and long-term exposure to particulate air pollution: Epidemiological evidence of general pathophysiological pathways of disease. Circulation. 2004;109:71–77. doi: 10.1161/01.CIR.0000108927.80044.7F. [DOI] [PubMed] [Google Scholar]
- Pope C. A., III, Muhlestein J. B., May H. T., Renlund D. G., Anderson J. L., Horne B. D. Ischemic heart disease events triggered by short-term exposure to fine particulate air pollution. Circulation. 2006;114:2443–2448. doi: 10.1161/CIRCULATIONAHA.106.636977. [DOI] [PubMed] [Google Scholar]
- Schramm A., Matusik P., Osmenda G., Guzik T. J. Targeting nadph oxidases in vascular pharmacology. Vascul. Pharmacol. 2012;56:216–231. doi: 10.1016/j.vph.2012.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shet A. S., Aras O., Gupta K., Hass M. J., Rausch D. J., Saba N., Koopmeiners L., Key N. S., Hebbel R. P. Sickle blood contains tissue factor-positive microparticles derived from endothelial cells and monocytes. Blood. 2003;102:2678–2683. doi: 10.1182/blood-2003-03-0693. [DOI] [PubMed] [Google Scholar]
- Snow S. J., Carraway M. S. Soluble components of ultrafine particulate matter stimulate endothelial hydrogen peroxide production. Toxicol. Sci. 2010;(Suppl. 114):313. [Google Scholar]
- Sorensen M., Schins R. P., Hertel O., Loft S. Transition metals in personal samples of pm2.5 and oxidative stress in human volunteers. Cancer Epidemiol. Biomarkers Prev. 2005;14:1340–1343. doi: 10.1158/1055-9965.EPI-04-0899. [DOI] [PubMed] [Google Scholar]
- Stewart J. C., Chalupa D. C., Devlin R. B., Frasier L. M., Huang L. S., Little E. L., Lee S. M., Phipps R. P., Pietropaoli A. P., Taubman M. B., et al. Vascular effects of ultrafine particles in persons with type 2 diabetes. Environ. Health Perspect. 2010;118:1692–1698. doi: 10.1289/ehp.1002237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Q., Yue P., Kirk R. I., Wang A., Moatti D., Jin X., Lu B., Schecter A. D., Lippmann M., Gordon T., et al. Ambient air particulate matter exposure and tissue factor expression in atherosclerosis. Inhal. Toxicol. 2008;20:127–137. doi: 10.1080/08958370701821482. [DOI] [PubMed] [Google Scholar]
- Szotowski B., Antoniak S., Goldin-Lang P., Tran Q. V., Pels K., Rosenthal P., Bogdanov V. Y., Borchert H. H., Schultheiss H. P., Rauch U. Antioxidative treatment inhibits the release of thrombogenic tissue factor from irradiation- and cytokine-induced endothelial cells. Cardiovasc. Res. 2007;73:806–812. doi: 10.1016/j.cardiores.2006.12.018. [DOI] [PubMed] [Google Scholar]
- Thiruvikraman S. V., Guha A., Roboz J., Taubman M. B., Nemerson Y., Fallon J. T. In situ localization of tissue factor in human atherosclerotic plaques by binding of digoxigenin-labeled factors viia and x. Lab. Invest. 1996;75:451–461. [PubMed] [Google Scholar]
- Wallenborn J. G., McGee J. K., Schladweiler M. C., Ledbetter A. D., Kodavanti U. P. Systemic translocation of particulate matter-associated metals following a single intratracheal instillation in rats. Toxicol. Sci. 2007;98:231–239. doi: 10.1093/toxsci/kfm088. [DOI] [PubMed] [Google Scholar]
- Yoshida L. S., Tsunawaki S. Expression of nadph oxidases and enhanced h(2)o(2)-generating activity in human coronary artery endothelial cells upon induction with tumor necrosis factor-alpha. Int. Immunopharmacol. 2008;8:1377–1385. doi: 10.1016/j.intimp.2008.05.004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.