

Abstract
Aims
Doxorubicin (DOX) is an effective anti-cancer therapeutic, but is associated with both acute and late-stage cardiotoxicity. Children are particularly sensitive to DOX-induced heart failure. Here, the impact of p53 inhibition on acute vs. late-stage DOX cardiotoxicity was examined in a juvenile model.
Methods and results
Two-week-old MHC-CB7 mice (which express dominant-interfering p53 in cardiomyocytes) and their non-transgenic (NON-TXG) littermates received weekly DOX injections for 5 weeks (25 mg/kg cumulative dose). One week after the last DOX treatment (acute stage), MHC-CB7 mice exhibited improved cardiac function and lower levels of cardiomyocyte apoptosis when compared with the NON-TXG mice. Surprisingly, by 13 weeks following the last DOX treatment (late stage), MHC-CB7 exhibited a progressive decrease in cardiac function and higher rates of cardiomyocyte apoptosis when compared with NON-TXG mice. p53 inhibition blocked transient DOX-induced STAT3 activation in MHC-CB7 mice, which was associated with enhanced induction of the DNA repair proteins Ku70 and Ku80. Mice with cardiomyocyte-restricted deletion of STAT3 exhibited worse cardiac function, higher levels of cardiomyocyte apoptosis, and a greater induction of Ku70 and Ku80 in response to DOX treatment during the acute stage when compared with control animals.
Conclusion
These data support a model wherein a p53-dependent cardioprotective pathway, mediated via STAT3 activation, mitigates DOX-induced myocardial stress during drug delivery. Furthermore, these data suggest an explanation as to how p53 inhibition can result in cardioprotection during drug treatment and, paradoxically, enhanced cardiotoxicity long after the cessation of drug treatment.
Keywords: Heart failure, Apoptosis, Myocytes
1. Introduction
Anthracyclines, as exemplified by doxorubicin (DOX), are widely used anti-cancer chemotherapeutics. Although highly efficacious, anthracyclines are also cardiotoxic. Anthracycline-induced cardiotoxicity can be observed during drug delivery (acute stage) as well as years after the termination of drug treatment (late stage). Acute stage cardiotoxicity is characterized by hypotension, tachycardia, arrhythmia, and transient depression of left ventricular function.1–4 Late-stage anthracycline cardiotoxicity is characterized by recalcitrant heart failure, the severity of which is directly related to cumulative drug exposure. Children are particularly sensitive to anthracycline-induced cardiotoxicity.5,6
It is generally thought that free radical-induced damage contributes to anthracycline-induced cardiotoxicity.7 However, numerous other cellular changes have been noted, including DNA damage, inhibition of protein synthesis, myofibre degeneration, transcriptional inhibition of myogenic programmes, and cardiomyocyte apoptosis.8–10 It has recently been reported that cardiomyocyte-restricted deletion of topoisomerase-II β (Top2β) reduced DOX-induced cardiotoxicity in mice.11 Interestingly, in the absence of Top2β, DOX-induced DNA damage was reduced, with a concomitant reduction in p53 induction. Despite progress in identifying cellular pathways perturbed by anthracyclines, it has proved difficult to determine the precise molecular aetiologies which give rise to acute vs. late-stage cardiotoxicity.
Many studies examining anthracycline cardiotoxicity utilized a limited number of high-dose DOX injections with terminal analyses shortly after the onset of treatment. Although such high-dose/short-duration studies provide insight into the mechanism of immediate drug toxicity, the clinical use of DOX entails the administration of multiple lower doses over an extended period of time, and the most challenging pathophysiological outcome is the recalcitrant heart failure that can occur long after the termination of treatment. Accordingly, several groups have developed long-term rodent models in an effort to better mimic the clinical problem.12–15 These studies have suggested that unique molecular mechanisms can contribute to acute vs. late-stage DOX cardiotoxicity.
It is well established, using high-dose/short-duration studies, that DOX treatment induces transient expression of the tumour suppressor p53, and furthermore that pharmacologic or genetic inhibition of p53 activity reduces DOX-induced cardiomyocyte apoptosis with a concomitant improvement in cardiac function.15–17 In this study, MHC-CB7 transgenic mice (which express dominant-interfering p53 in cardiomyocytes)18 were employed to examine the impact of p53 inhibition on acute vs. late-stage DOX-induced cardiotoxicity using a juvenile mouse model of chronic DOX-induced cardiac dysfunction. Although inhibition of p53 activity was protective during acute cardiotoxicity, a progressive reduction in systolic function, and comparatively higher rates of cardiomyocyte apoptosis, were observed during late-stage DOX cardiotoxicity in MHC-CB7 mice when compared with their non-transgenic littermates. Molecular analyses revealed a transient induction of STAT3 activity in DOX-treated NON-TXG mice, but not in DOX-treated MHC-CB7 mice. A potential mechanisms as to how p53 inhibition can promote cardioprotection during drug treatment and, paradoxically, cardiotoxicity long after the cessation of drug treatment is discussed.
2. Methods
2.1. Mice
This study utilized MHC-CB7 mice18 (n = 72), and their non-transgenic littermates (n = 70). The MHC-CB7 transgene utilizes the cardiomyocyte-restricted alpha-cardiac myosin heavy chain promoter19 to target expression of the p53 CB7 allele, which harbours an arginine to proline substitution at amino acid residue #193 that results in a dominant-interfering phenotype by blocking p53-dependent transcription.20,21 Mice were maintained in a DBA/2J genetic background. Mice with cardiac-restricted STAT3 deletion (STAT3-CKO)22 were also utilized (n = 16 STAT3-CKO mice and 17 wild-type controls). These animals (maintained in a C57Bl/6J genetic background) carried a modified STAT3 allele wherein loxP sites were inserted between exons 17 and 18 and between exons 20 and 21.22 These animals also carried a transgene encoding Cre recombinase under the transcriptional regulation of the myosin heavy chain promoter23 to drive cardiomyocyte-restricted STAT3 deletion. The wild-type controls lacked the Cre-encoding transgene, and were either heterozygous or homozygous for the floxed STAT3 allele. Numbers of mice used to generate each dataset are indicated in Supplementary material online, Tables S1–S4.
2.2. DOX cardiotoxicity model
A juvenile model featuring aspects of acute and chronic DOX cardiotoxicity was employed.15 Mice received a total of 25 mg/kg of DOX (five intra-peritoneal injections of 5 mg/kg in saline, given at 1 week intervals beginning at 2 weeks of age, Sigma, St Louis, MO, USA; control mice received saline injections). To avoid local tissue damage, a different region of the peritoneal cavity was injected at each time point, and no overt inflammation or damage was apparent at the injection sites. One cohort of mice was sacrificed 1 week after the final DOX injection (acute stage). A second cohort was sacrificed 13 weeks after the final DOX injection (late stage). Mice were sacrificed via cervical dislocation under isofluorane anaesthesia (1.5%), and hearts were immediately harvested. All animal protocols were approved by the Indiana University School of Medicine Institutional Animal Care and Use Committee.
2.3. Echocardiography
Mice were lightly anaesthetized with 1.5% isofluorane until their heart rate stabilized at 400–500 bpm. Two-dimensional images were obtained using a high-resolution Micro-Ultrasound system (Vevo 770, VisualSonics Inc., Toronto, Canada) equipped with a 40-MHz mechanical scan probe. Fractional shortening (FS, calculated from short-axis images) and ejection fraction (EF, calculated from parasternal long axis images) were calculated using the Vevo Analysis software (version 2.2.3) as described previously.24
2.4. Western blot analysis
Proteins were extracted and quantitated using the Coomassie Blue method (Pierce, Rockford, IL, USA) as described previously.18 Samples were solubilized in sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis (PAGE) loading buffer for 5 min at 95°C and resolved on 7 or 10% SDS–PAGE gels.25 Fractionated proteins were then electro-transferred from the gel to nitrocellulose (Amersham) filters in Towbin buffer at 200-mA constant current and analysed by western blotting as described.26 The filters were stained with 0.1% naphthol blue–black in 45% methanol, 10% acetic acid to assess the efficiency of transfer. Antibodies used recognized p53 (#PC-35, EMD Chemicals, Gibbstown, NJ, USA), PARP1 (#9542, Cell Signaling Technology, Danvers, MA, USA), Actin (#sc-1616-R, Santa Cruz Biotechnology), P-STAT3[Tyr705] (#9145S, Cell Signaling Technology), total STAT3 (#9139S, Cell Signaling Technology), Ku70 (#sc-365766, Santa Cruz Biotechnology), and Ku80 (#2180S, Cell Signaling Technology). Signal was visualized by the ECL method according to the manufacturer's protocol (Amersham). Western signal was digitized and quantitated using ImageJ software.
2.5. Histology
Hearts were harvested, cryoprotected in 30% sucrose, embedded and sectioned at 10 μm using standard techniques.27,28 To quantitate cardiomyocyte apoptosis, four transverse sections from each heart, sampled from the midpoint between the apex and base, were post-fixed in 4% paraformaldehyde and screened for anti-activated caspase-3 immune reactivity (antibody #G7481, Promega, Madison, WI, USA), followed by a horseradish peroxidase-conjugated secondary antibody; signal was visualized with a diaminobenzidine reaction, as described previously.18 TUNEL analyses were performed on adjacent sections using the ApopTag Apoptosis Detection kit according to the manufacturer's protocol (Chemicon International, Billerica, MA, USA).
2.6. Statistic analysis
All values are presented as mean ± SEM. Statistical significance was determined by Student's t-test (for two group comparison for quantification of western blot signals for p-STAT3/STAT3 α- and β-subunits) or two-way ANOVA (Holm–Sidak method) for multi-group comparison. P < 0.05 was considered significant.
3. Results
3.1 Inhibition of p53 activity does not prevent late-stage DOX-induced cardiac dysfunction in a juvenile cardiotoxicity model
We have previously shown, using a chronic juvenile mouse model, that DOX treatment results in low levels of cardiomyocyte apoptosis during drug delivery, but comparatively higher levels of apoptosis long after drug withdrawal.15 To elucidate the role of p53 in this process, 14-day-old MHC-CB7 mice and their NON-TXG littermates were given weekly DOX injections for a total of 5 weeks. One week after the last injection, half of the animals were sacrificed and their hearts were harvested for cell and molecular analyses (acute stage). The remaining animals were allowed to recover in the absence of DOX for an additional 12 weeks prior to sacrifice (late stage). Echocardiography was performed to monitor cardiac function throughout the study. In agreement with previous findings,15 both FS (Figure 1) and EF (see Supplementary material online, Table S1) progressively deteriorated in DOX-treated NON-TXG juvenile mice during the acute stage when compared with the saline-treated NON-TXG controls, and remained stable albeit at a suppressed level throughout the late stage. In contrast, DOX had little impact on FS and EF in MHC-CB7 mice during the acute stage, consistent with a cardioprotective role for p53 inhibition. However, systolic function deteriorated in the DOX-treated MHC-CB7 mice during the late stage (Figure 1 and Supplementary material online, Table S1).
Figure 1.

Cardiac function in saline- and DOX-treated juvenile mice in the acute and late stages. (A) FS in saline- and DOX-treated NON-TXG and MHC-CB7 mice in the acute and late stages. X-axis indicates the age of the mice at analysis; vertical arrows indicate the age at DOX injection. *P < 0.05 for DOX-treated NON-TXG vs. saline-treated NON-TXG mice, †P < 0.05 for DOX-treated NON-TXG vs. DOX-treated MHC-CB7 mice, ‡P < 0.05 for DOX-treated MHC-CB7 vs. saline-treated MHC-CB7 mice. (B) Representative short-axis echocardiograms from saline- and DOX-treated NON-TXG and MHC-CB7 mice in the acute and late stages.
3.2. Inhibition of p53 results in higher levels of late-stage DOX-induced cardiomyocyte apoptosis
Activated caspase-3 immune reactivity was used to quantitate cardiomyocyte apoptosis. Since activated caspase-3 immune reactivity is present in the cytoplasm, cardiomyocytes at early stages of apoptosis can be identified based on cell size and shape (Figure 2A).18 In agreement with previous results,15 cardiomyocyte-activated caspase-3 immune reactivity was induced in hearts from DOX-treated NON-TXG juvenile mice during acute toxicity, and the level was approximately four-fold higher during the late stage (Figure 2B). There was no induction of cardiomyocyte-activated caspase-3 immune reactivity during the acute stage in DOX-treated MHC-CB7 juvenile mice when compared with saline-treated animals, again supporting the notion that p53 inhibition can be cardioprotective. Surprisingly, a very high level of cardiomyocyte-activated caspase-3 immune reactivity was present in late-stage MHC-CB7 hearts, exceeding that seen in the late-stage NON-TXG mice (Figure 2B). Similar results were obtained using TUNEL analysis, with >2-fold more TUNEL-positive nuclei in late-stage DOX-treated MHC-CB7 hearts when compared with late-stage DOX-treated NON-TXG hearts (see Supplementary material online, Table S2, for the cardiac attributes for these animals).
Figure 2.
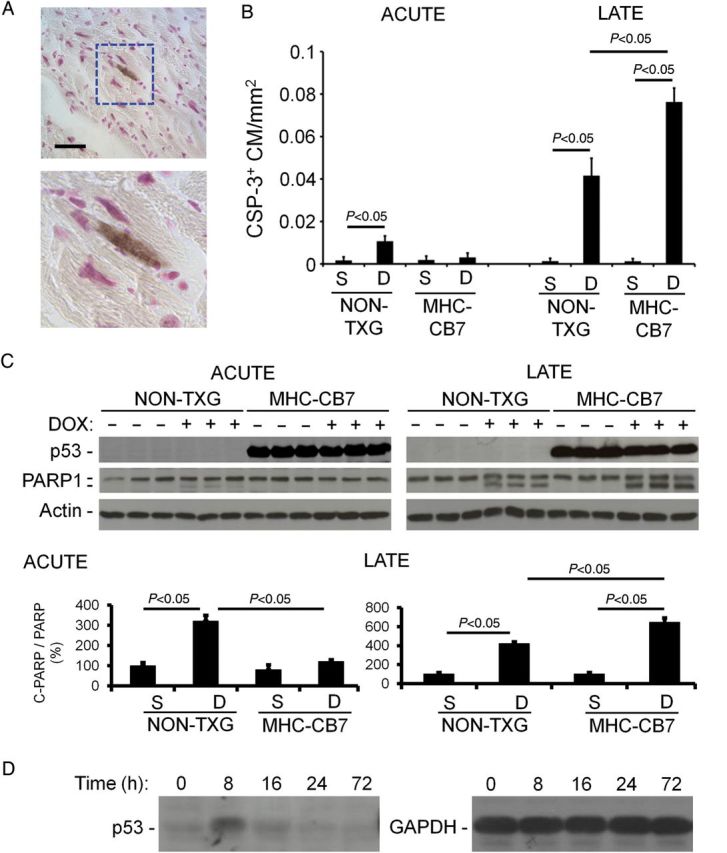
Cardiomyocyte apoptosis in saline- and DOX-treated NON-TXG and MHC-CB7 mice in the acute and late stages. (A) Representative activated caspase-3 immune reactive cardiomyocyte from a DOX-treated MHC-CB7 mouse (bar = 50 µm). (B) Quantitation of activated caspase-3 immune reactive cardiomyocytes (CSP-3+ CM) per mm2 in hearts from saline- and DOX-treated NON-TXG and MHC-CB7 mice in the acute and late stages. (C) Western blot analysis of cleaved PARP1 accumulation during the early and late stages in NON-TXG and MHC-CB7 mice; densitometric quantitation of the PARP1 signal shown. (D) p53 protein (left panel) is induced in juvenile NON-TXG hearts following DOX treatment (single injection, 5 mg/kg; time indicates the number of hours post-injection when the hearts were harvested); right panel shows a GAPDH western blot from the same transfer to confirm equal loading.
PARP1 is a caspase-3 substrate, and accumulation of cleaved PARP provides another marker for apoptosis.29 Western blots revealed the presence of cleaved PARP1 in hearts from DOX-treated NON-TXG mice, but not in NON-TXG saline-treated controls, during the acute stage (Figure 2C). Even higher levels of cleaved PARP1 were observed in DOX-treated NON-TXG hearts during the late stage, in agreement with the observed increase in activated caspase-3 immune reactivity and TUNEL signal (above). No cleaved PARP1 was detected in hearts from acute-stage DOX-treated MHC-CB7 mice, but very high levels were present in late-stage hearts (exceeding those seen in the late-stage NON-TXG hearts, Figure 2C). Western blot analyses demonstrated transient induction of endogenous p53 in response to DOX treatment (Figure 2D). The observation that p53 inhibition blocks acute stage apoptosis, but potentiates late-stage apoptosis, indicates a fundamental difference in the underlying mechanisms of DOX-induced cardiomyocyte death at these two stages.
3.3. Inhibition of p53 activity blocks STAT3 activation during DOX treatment
The presence of higher levels of cardiomyocyte apoptosis in MHC-CB7 vs. NON-TXG mice during late-stage DOX cardiotoxicity was somewhat surprising, and suggests the presence of a p53-dependent cardioprotective activity. Since DOX treatment results in only transient p53 induction (Figure 2), it is likely that this cardoprotective activity occurs during the acute stage. Indeed, such an activity has been hypothesized by others.30 If true, one would anticipate that the cardioprotective pathway would be present during DOX treatment in NON-TXG mice (i.e. in the acute stage and not in the late stage), and would be completely absent in DOX-treated MHC-CB7 mice. Western blot screens were performed to identify such an activity. Signal Transducers and Activators of Transcription 3 (STAT3) is a transcription factor implicated in the regulation of adaptive growth and survival in cardiomyocytes.31–33 A marked increase in the level of phosphorylated STAT3 at tyrosine residue 705 (P-STAT3[tyr705]) was observed during the acute stage in NON-TXG mice, but not in MHC-CB7 mice (Figure 3). Phosphorylation at tyrosine residue 705 facilitates STAT3 dimerization, and consequently, pathway activation.34 P-STAT3[tyr705] levels in DOX-treated NON-TXG mice returned to base-line in the late stage. In contrast, no overt changes in STAT3 phosphorylation at serine residue 727 (P-STAT3[ser727], another regulatory residue)35 was observed in DOX-treated NON-TXG or MHC-CB7 mice (Figure 3).
Figure 3.

p53 inhibition blocks transient, DOX-induced P-STAT3[tyr705] phosphorylation in the acute stage, and enhances Ku70 and Ku80 expression in the acute and late stages. Densitometric quantitation of the western blot signals is shown. Note that the STAT3-β isoform lacks the serine 727 residue, and hence is not detected in the anti-P-STAT3[ser727] immune blot.
3.4. STAT3 activity mitigates DOX-induced Ku expression
Given that STAT3 is cardioprotective, one would anticipate that markers indicative of cardiac stress would also be differentially expressed in NON-TXG vs. MHC-CB7 DOX-treated hearts. Since DOX cardiotoxicity is thought to result at least in part from reactive oxygen species-induced stress, and since reactive oxygen species are known to damage DNA, western blot screens for changes in the expression of proteins associated with DNA damage were performed. Ku (a heterodimer protein complex comprising Ku70 and Ku80) is induced in response to DNA damage; Ku binds to double-stranded DNA breaks and facilitates non-homologous end joining.36 Interestingly, it has also been suggested that Ku70 functions as a transcriptional co-factor in response to oxidative stress.37,38 DOX treatment resulted in an induction of Ku70 and Ku80 in NON-TXG mice during the acute stage; the induction of Ku70 and Ku80 was even greater in MHC-CB7 hearts. Ku70 and Ku80 expression remained elevated in late-stage DOX-treated NON-TXG hearts; once again, levels were higher in the DOX-treated MHC-CB7 hearts (Figure 3).
To directly evaluate whether STAT3 activity mitigates DOX-induced Ku expression, mice with cardiomyocyte-restricted deletion of STAT3 (STAT3-CKO) and their wild-type (WT) controls were given weekly DOX injections for a total of 5 weeks, and 1 week after the last injection, the animals were sacrificed and their hearts were harvested for cell and molecular analyses. Cardiac function was monitored over the course of the experiment. DOX treatment resulted in a rapid decrease in cardiac function (FS and EF) in the WT mice during the acute stage (Figure 4A and B). The impact of DOX treatment on cardiac function was even more pronounced in the STAT3-CKO mice. Activated caspase-3 immune reactivity revealed an increased level of cardiomyocyte-activated caspase-3 immune reactivity in STAT3-CKO hearts when compared with WT hearts (Figure 4C). Similar results were obtained using TUNEL analysis, with >2-fold more TUNEL-positive nuclei in DOX-treated STAT3-CKO hearts when compared with DOX-treated WT hearts (see Supplementary material online, Table S4, for the cardiac attributes of these animals). The level of cleaved PARP1 was greater in acute-stage STAT3-CKO hearts when compared with WT hearts, further supporting higher rates of cardiomyocyte apoptosis in these animals (Figure 5). Western blot analysis also revealed that DOX treatment resulted in enhanced induction of Ku70 and Ku80 in STAT3-CKO hearts when compared with WT hearts (Figure 5). Collectively, these data demonstrate that the loss of STAT3 renders the heart more susceptible to DOX-induced stress, and that STAT3 activity mitigates DOX-induced Ku expression.
Figure 4.

Cardiomyocyte-restricted STAT3 deletion exacerbates acute-stage DOX cardiotoxicity. (A) FS in saline- and DOX-treated WT and STAT3-CKO mice. X-axis indicates the age of the mice at analysis; vertical arrows indicate the age at DOX injection. *P < 0.05 for DOX-treated WT vs. saline-treated WT mice, †P < 0.05 for DOX-treated WT vs. DOX-treated STAT3-CKO mice, ‡P < 0.05 for DOX-treated STAT3-CKO vs. saline-treated STAT3-CKO mice. (B) Representative short-axis echocardiograms from saline- and DOX-treated WT and STAT3-CKO mice. (C) Quantitation of activated caspase-3 immune reactive cardiomyocytes (CSP+ CM) per mm2 in hearts from saline- and DOX-treated NON-TXG and STAT3-CKO mice in the acute stage.
Figure 5.

Western blot analysis of protein expression in saline- and DOX-treated WT and STAT3-CKO mice. Densitometric quantitation of the western blot signals is also shown.
4. Discussion
The data presented here, utilizing juvenile mice with cardiomyocyte-restricted expression of a dominant-interfering p53 variant, demonstrated that p53 inhibition blocked acute cardiotoxicity when DOX was delivered at low doses over a 5-week period. The surprising finding was that inhibition of p53 activity resulted in greater levels of cardiomyocyte apoptosis long (3 months) after the termination of DOX treatment, when compared with DOX-treated NON-TXG mice. p53 inhibition blocked transient P-STAT3[thr705] phosphorylation during the acute stage, rendering the myocardium more sensitive to DOX-induced stress (as evidenced by the hyper-induction of acute stage Ku levels). This increase in DOX-induced stress during drug treatment was associated with a greater rate of cardiomyocyte dropout during the late stage, and progressive late-stage cardiac dysfunction. Collectively, these data suggest an explanation as to how p53 inhibition can result in cardioprotection during drug treatment and, paradoxically, cardiotoxicity long after the cessation of drug treatment.
STAT3 has previously been implicated in modulating DOX-induced cardiotoxicity in high-dose/short-duration studies. For example, transgenic mice overexpressing STAT3 in cardiomyocytes exhibited enhanced survival,39 whereas STAT3-CKO mice exhibited reduced cardiac function (Figure 4),22 when compared with their respective DOX-treated controls. Moreover, numerous studies have shown a correlation between STAT3 activation and detoxification of reactive oxygen species.40–44 The transient induction of P-STAT3[thr705] levels during the acute stage in NON-TXG mice, but not MHC-CB7 mice, makes it a prime candidate for the proposed p53-dependent cardioprotective activity. Although P-STAT3[ser727] is also present in the heart, the absence of dynamic regulation during DOX treatment indicates that changes in phosphorylation at this residue are not responsible for the p53-dependent cardioprotective activity proposed here. Given the observation that phospho-mimetic mutants at this residue can impact cardiomyocyte survival,35 the combined loss P-STAT[thr705] and P-STAT3[ser727] in STAT3-CKO mice readily explains the relative decrease in cardiac function and increase in cardiomyocyte apoptosis following DOX treatment, when compared with DOX-treated WT mice.
The mechanistic link between the induction of p53 activity and the activation of STAT3 is presently not clear. Although the absence of previous work describing a link between p53 induction and JAK (the canonical upstream regulator of STAT3) pathway activation argues against direct activation, one cannot rule out the possibility that, as of yet, an unidentified p53-dependent protein kinase mediates P-STAT3[tyr705] phosphorylation during DOX treatment. It is also possible that the activation occurs via an indirect mechanism. For example, it has previously been shown using a high-dose/short-duration model that DOX treatment resulted in cardiac atrophy via a p53-dependent mechanism, and that expression of the MHC-CB7 transgene effectively blocked DOX-mediated atrophy.45 Inhibition of p53 activity also blocked DOX-induced cardiac atrophy during acute-stage cardiotoxicity in the current study (see Supplementary material online, Table S2). In other studies, atrophic cardiac remodelling following heterotopic heart transplantation resulted in STAT3 activation.46 Thus, the absence of STAT3 activation in the MHC-CB7 mice may result from the absence of DOX-induced cardiac atrophy. It should be noted that enforced expression of p53 inhibited STAT3 phosphorylation in prostate cancer cell lines,47 indicating that STAT3 can be negatively regulated by p53. On the other hand, both p53 induction48 and STAT3 activation49 are observed during autophagy, leading to the suggestion that the pathways function in parallel to regulate increased autophagic activity.50 Thus, the impact of p53 on STAT3 activity is dependent upon both the cell type and the physiological pathway under analysis.
In the absence of STAT3 activation (due to inhibition of p53 in the MHC-CB7 mice or lineage-specific gene deletion in the STAT3-CKO mice), a hyper-induction of Ku70 and 80 was observed following DOX treatment. The absence of increased apoptosis during the acute stage in MHC-CB7 mice argues that Ku expression is not simply tracking cardiomyocyte death. Additionally, the absence of Ku expression in saline-treated MHC-CB7 hearts indicates that transgene expression per se is not responsible for its induction. It is also unlikely that Ku is a downstream effector of STAT3, as both Ku70 and Ku80 were hyper-induced in DOX-treated STAT3-CKO mice. Although Ku70 has been reported to encode anti-apoptotic activity (by binding BAX and blocking its translocation to the mitochondria),51 this likely is not the case in the results presented here given the elevated late-stage cardiomyocyte apoptosis seen in MHC-CB7 mice. Rather, it is more likely that the accumulation of Ku reflects the relative level of myocardial stress (given its role as a transcription co-factor) and/or the relative level of DNA damage (given its role in DNA repair) following DOX treatment.
As mentioned above, cardiomyocyte-restricted deletion of Topoisomerase-II β (Top2β) reduced DOX-induced cardiotoxicity in mice by blocking DNA damaged-induced p53 activation.11 This work is noteworthy in that it suggested that reactive oxygen species generation was secondary to a p53-mediated reduction of the transcription of genes required for mitochondrial biosynthesis and oxidative phosphorylation (leading to a concomitant decrease in mitochondrial function). However, since expression of the CB7 protein blocks p53-mediated transcriptional activity,21 this mechanism cannot underlie the late-stage cardiotoxicity observed in the MHC-CB7 mice in the current study.
In summary, the data presented here indicate that p53 inhibition is beneficial during acute-stage DOX cardiotoxicity, but detrimental during late-stage cardiotoxicity. Furthermore, the data suggest a working model that could explain in part how changes in protein activity during drug treatment in the acute stage can result in more severe cardiotoxicity long after the termination of drug treatment. Direct proof of the proposed mechanism will require the generation of additional transgenic animals with the conditional expression of the relevant proteins (as for example conditional expression of the CB7 protein during the acute stage only, and/or conditional expression of a constitutively active STAT3 during the acute stage in MHC-CB7 mice). Nonetheless, the data presented indicate that interventions to ameliorate DOX cardiotoxicity could have a rather unexpected negative impact. These results further underscore the complex nature of anthracycline cardiotoxicity.
Supplementary material
Supplementary Material is available at Cardiovascular Research online.
Funding
This work was supported by the National Institutes of Health (HL109205 to L.J.F.) and an American Cancer Society postdoctoral fellowship (121101 to W.Z.).
Supplementary Material
Acknowledgements
We thank Dorothy Field for technical support and Dr Michael Rubart for assistance with the statistical analyses.
Conflict of interest: none declared.
References
- 1.Bristow MR, Billingham ME, Mason JW, Daniels JR. Clinical spectrum of anthracycline antibiotic cardiotoxicity. Cancer Treat Rep. 1978;62:873–879. [PubMed] [Google Scholar]
- 2.Lefrak EA, Pitha J, Rosenheim S, Gottlieb JA. A clinicopathologic analysis of adriamycin cardiotoxicity. Cancer. 1973;32:302–314. doi: 10.1002/1097-0142(197308)32:2<302::aid-cncr2820320205>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 3.Lipshultz SE, Alvarez JA, Scully RE. Anthracycline associated cardiotoxicity in survivors of childhood cancer. Heart. 2008;94:525–533. doi: 10.1136/hrt.2007.136093. [DOI] [PubMed] [Google Scholar]
- 4.Singal PK, Iliskovic N. Doxorubicin-induced cardiomyopathy. N Engl J Med. 1998;339:900–905. doi: 10.1056/NEJM199809243391307. [DOI] [PubMed] [Google Scholar]
- 5.Lipshultz SE, Lipsitz SR, Sallan SE, Dalton VM, Mone SM, Gelber RD, Colan SD. Chronic progressive cardiac dysfunction years after doxorubicin therapy for childhood acute lymphoblastic leukemia. J Clin Oncol. 2005;23:2629–2636. doi: 10.1200/JCO.2005.12.121. [DOI] [PubMed] [Google Scholar]
- 6.Scully RE, Lipshultz SE. Anthracycline cardiotoxicity in long-term survivors of childhood cancer. Cardiovasc Toxicol. 2007;7:122–128. doi: 10.1007/s12012-007-0006-4. [DOI] [PubMed] [Google Scholar]
- 7.Zhou S, Starkov A, Froberg MK, Leino RL, Wallace KB. Cumulative and irreversible cardiac mitochondrial dysfunction induced by doxorubicin. Cancer Res. 2001;61:771–777. [PubMed] [Google Scholar]
- 8.Carvalho FS, Burgeiro A, Garcia R, Moreno AJ, Carvalho RA, Oliveira PJ. Doxorubicin-induced cardiotoxicity: from bioenergetic failure and cell death to cardiomyopathy. Med Res Rev. 2014;34:106–135. doi: 10.1002/med.21280. [DOI] [PubMed] [Google Scholar]
- 9.Octavia Y, Tocchetti CG, Gabrielson KL, Janssens S, Crijns HJ, Moens AL. Doxorubicin-induced cardiomyopathy: from molecular mechanisms to therapeutic strategies. J Mol Cell Cardiol. 2012;52:1213–1225. doi: 10.1016/j.yjmcc.2012.03.006. [DOI] [PubMed] [Google Scholar]
- 10.Volkova M, Russell R., 3rd Anthracycline cardiotoxicity: prevalence, pathogenesis and treatment. Curr Cardiol Rev. 2011;7:214–220. doi: 10.2174/157340311799960645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang S, Liu X, Bawa-Khalfe T, Lu LS, Lyu YL, Liu LF, Yeh ET. Identification of the molecular basis of doxorubicin-induced cardiotoxicity. Nat Med. 2012;18:1639–1642. doi: 10.1038/nm.2919. [DOI] [PubMed] [Google Scholar]
- 12.Delgado RM, 3rd, Nawar MA, Zewail AM, Kar B, Vaughn WK, Wu KK, Aleksic N, Sivasubramanian N, McKay K, Mann DL, Willerson JT. Cyclooxygenase-2 inhibitor treatment improves left ventricular function and mortality in a murine model of doxorubicin-induced heart failure. Circulation. 2004;109:1428–1433. doi: 10.1161/01.CIR.0000121354.34067.48. [DOI] [PubMed] [Google Scholar]
- 13.Lebrecht D, Geist A, Ketelsen UP, Haberstroh J, Setzer B, Walker UA. Dexrazoxane prevents doxorubicin-induced long-term cardiotoxicity and protects myocardial mitochondria from genetic and functional lesions in rats. Br J Pharmacol. 2007;151:771–778. doi: 10.1038/sj.bjp.0707294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lebrecht D, Setzer B, Ketelsen UP, Haberstroh J, Walker UA. Time-dependent and tissue-specific accumulation of mtDNA and respiratory chain defects in chronic doxorubicin cardiomyopathy. Circulation. 2003;108:2423–2429. doi: 10.1161/01.CIR.0000093196.59829.DF. [DOI] [PubMed] [Google Scholar]
- 15.Zhu W, Shou W, Payne RM, Caldwell R, Field LJ. A mouse model for juvenile doxorubicin-induced cardiac dysfunction. Pediatr Res. 2008;64:488–494. doi: 10.1203/PDR.0b013e318184d732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu X, Chua CC, Gao J, Chen Z, Landy CL, Hamdy R, Chua BH. Pifithrin-alpha protects against doxorubicin-induced apoptosis and acute cardiotoxicity in mice. Am J Physiol Heart Circ Physiol. 2004;286:H933–H939. doi: 10.1152/ajpheart.00759.2003. [DOI] [PubMed] [Google Scholar]
- 17.Shizukuda Y, Matoba S, Mian OY, Nguyen T, Hwang PM. Targeted disruption of p53 attenuates doxorubicin-induced cardiac toxicity in mice. Mol Cell Biochem. 2005;273:25–32. doi: 10.1007/s11010-005-5905-8. [DOI] [PubMed] [Google Scholar]
- 18.Nakajima H, Nakajima HO, Tsai SC, Field LJ. Expression of mutant p193 and p53 permits cardiomyocyte cell cycle reentry after myocardial infarction in transgenic mice. Circ Res. 2004;94:1606–1614. doi: 10.1161/01.RES.0000132279.99249.f4. [DOI] [PubMed] [Google Scholar]
- 19.Gulick J, Subramaniam A, Neumann J, Robbins J. Isolation and characterization of the mouse cardiac myosin heavy chain genes. J Biol Chem. 1991;266:9180–9185. [PubMed] [Google Scholar]
- 20.Munroe DG, Peacock JW, Benchimol S. Inactivation of the cellular p53 gene is a common feature of Friend virus-induced erythroleukemia: relationship of inactivation to dominant transforming alleles. Mol Cell Biol. 1990;10:3307–3313. doi: 10.1128/mcb.10.7.3307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Timmers C, Sharma N, Opavsky R, Maiti B, Wu L, Wu J, Orringer D, Trikha P, Saavedra HI, Leone G. E2f1, E2f2, and E2f3 control E2F target expression and cellular proliferation via a p53-dependent negative feedback loop. Mol Cell Biol. 2007;27:65–78. doi: 10.1128/MCB.02147-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jacoby JJ, Kalinowski A, Liu MG, Zhang SS, Gao Q, Chai GX, Ji L, Iwamoto Y, Li E, Schneider M, Russell KS, Fu XY. Cardiomyocyte-restricted knockout of STAT3 results in higher sensitivity to inflammation, cardiac fibrosis, and heart failure with advanced age. Proc Natl Acad Sci USA. 2003;100:12929–12934. doi: 10.1073/pnas.2134694100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Agah R, Frenkel PA, French BA, Michael LH, Overbeek PA, Schneider MD. Gene recombination in postmitotic cells. Targeted expression of Cre recombinase provokes cardiac-restricted, site-specific rearrangement in adult ventricular muscle in vivo. J Clin Invest. 1997;100:169–179. doi: 10.1172/JCI119509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shen WH, Chen Z, Shi S, Chen H, Zhu W, Penner A, Bu G, Li W, Boyle DW, Rubart M, Field LJ, Abraham R, Liechty EA, Shou W. Cardiac restricted overexpression of kinase-dead mammalian target of rapamycin (mTOR) mutant impairs the mTOR-mediated signaling and cardiac function. J Biol Chem. 2008;283:13842–13849. doi: 10.1074/jbc.M801510200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 26.Towbin H, Staehelin T, Gordon J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc Natl Acad Sci USA. 1979;76:4350–4354. doi: 10.1073/pnas.76.9.4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bullock GR, Petrusz P. Techniques in Immunocytochemistry. New York, NY: Academic Press; 1982. [Google Scholar]
- 28.Junqueira LCU, Carneiro J, Kelley RO. Basic Histology. Norwalk, Conn: Appleton & Lange; 1992. [Google Scholar]
- 29.Boulares AH, Yakovlev AG, Ivanova V, Stoica BA, Wang G, Iyer S, Smulson M. Role of poly(ADP-ribose) polymerase (PARP) cleavage in apoptosis. Caspase 3-resistant PARP mutant increases rates of apoptosis in transfected cells. J Biol Chem. 1999;274:22932–22940. doi: 10.1074/jbc.274.33.22932. [DOI] [PubMed] [Google Scholar]
- 30.Nithipongvanitch R, Ittarat W, Velez JM, Zhao R, St Clair DK, Oberley TD. Evidence for p53 as guardian of the cardiomyocyte mitochondrial genome following acute adriamycin treatment. J Histochem Cytochem. 2007;55:629–639. doi: 10.1369/jhc.6A7146.2007. [DOI] [PubMed] [Google Scholar]
- 31.Fischer P, Hilfiker-Kleiner D. Survival pathways in hypertrophy and heart failure: the gp130-STAT axis. Basic Res Cardiol. 2007;102:393–411. doi: 10.1007/s00395-007-0674-z. [DOI] [PubMed] [Google Scholar]
- 32.Hilfiker-Kleiner D, Hilfiker A, Drexler H. Many good reasons to have STAT3 in the heart. Pharmacol Ther. 2005;107:131–137. doi: 10.1016/j.pharmthera.2005.02.003. [DOI] [PubMed] [Google Scholar]
- 33.Booz GW, Day JN, Baker KM. Interplay between the cardiac renin angiotensin system and JAK-STAT signaling: role in cardiac hypertrophy, ischemia/reperfusion dysfunction, and heart failure. J Mol Cell Cardiol. 2002;34:1443–1453. doi: 10.1006/jmcc.2002.2076. [DOI] [PubMed] [Google Scholar]
- 34.Schindler C, Darnell JE., Jr Transcriptional responses to polypeptide ligands: the JAK-STAT pathway. Annu Rev Biochem. 1995;64:621–651. doi: 10.1146/annurev.bi.64.070195.003201. [DOI] [PubMed] [Google Scholar]
- 35.Shen Y, La Perle KM, Levy DE, Darnell JE., Jr Reduced STAT3 activity in mice mimics clinical disease syndromes. Biochem Biophys Res Commun. 2005;330:305–309. doi: 10.1016/j.bbrc.2005.02.154. [DOI] [PubMed] [Google Scholar]
- 36.Lee SH, Kim CH. DNA-dependent protein kinase complex: a multifunctional protein in DNA repair and damage checkpoint. Mol Cells. 2002;13:159–166. [PubMed] [Google Scholar]
- 37.Antoniali G, Lirussi L, D'Ambrosio C, Dal Piaz F, Vascotto C, Casarano E, Marasco D, Scaloni A, Fogolari F, Tell G. SIRT1 gene expression upon genotoxic damage is regulated by APE1 through nCaRE-promoter elements. Mol Biol Cell. 2014;25:532–547. doi: 10.1091/mbc.E13-05-0286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brenkman AB, van den Broek NJ, de Keizer PL, van Gent DC, Burgering BM. The DNA damage repair protein Ku70 interacts with FOXO4 to coordinate a conserved cellular stress response. Faseb J. 2010;24:4271–4280. doi: 10.1096/fj.10-158717. [DOI] [PubMed] [Google Scholar]
- 39.Kunisada K, Negoro S, Tone E, Funamoto M, Osugi T, Yamada S, Okabe M, Kishimoto T, Yamauchi-Takihara K. Signal transducer and activator of transcription 3 in the heart transduces not only a hypertrophic signal but a protective signal against doxorubicin-induced cardiomyopathy. Proc Natl Acad Sci USA. 2000;97:315–319. doi: 10.1073/pnas.97.1.315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Negoro S, Kunisada K, Fujio Y, Funamoto M, Darville MI, Eizirik DL, Osugi T, Izumi M, Oshima Y, Nakaoka Y, Hirota H, Kishimoto T, Yamauchi-Takihara K. Activation of signal transducer and activator of transcription 3 protects cardiomyocytes from hypoxia/reoxygenation-induced oxidative stress through the upregulation of manganese superoxide dismutase. Circulation. 2001;104:979–981. doi: 10.1161/hc3401.095947. [DOI] [PubMed] [Google Scholar]
- 41.Terui K, Enosawa S, Haga S, Zhang HQ, Kuroda H, Kouchi K, Matsunaga T, Yoshida H, Engelhardt JF, Irani K, Ohnuma N, Ozaki M. Stat3 confers resistance against hypoxia/reoxygenation-induced oxidative injury in hepatocytes through upregulation of Mn-SOD. J Hepatol. 2004;41:957–965. doi: 10.1016/j.jhep.2004.08.019. [DOI] [PubMed] [Google Scholar]
- 42.Jung JE, Kim GS, Narasimhan P, Song YS, Chan PH. Regulation of Mn-superoxide dismutase activity and neuroprotection by STAT3 in mice after cerebral ischemia. J Neurosci. 2009;29:7003–7014. doi: 10.1523/JNEUROSCI.1110-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lapp DW, Zhang SS, Barnstable CJ. Stat3 mediates LIF-induced protection of astrocytes against toxic ROS by upregulating the UPC2 mRNA pool. Glia. 2014;62:159–170. doi: 10.1002/glia.22594. [DOI] [PubMed] [Google Scholar]
- 44.Boengler K, Ungefug E, Heusch G, Schulz R. The STAT3 inhibitor Stattic impairs cardiomyocyte mitochondrial function through increased reactive oxygen species formation. Curr Pharm Des. 2013;19:6890–6895. doi: 10.2174/138161281939131127115940. [DOI] [PubMed] [Google Scholar]
- 45.Zhu W, Soonpaa MH, Chen H, Shen W, Payne RM, Liechty EA, Caldwell RL, Shou W, Field LJ. Acute doxorubicin cardiotoxicity is associated with p53-induced inhibition of the mammalian target of rapamycin pathway. Circulation. 2009;119:99–106. doi: 10.1161/CIRCULATIONAHA.108.799700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sharma S, Ying J, Razeghi P, Stepkowski S, Taegtmeyer H. Atrophic remodeling of the transplanted rat heart. Cardiology. 2006;105:128–136. doi: 10.1159/000090550. [DOI] [PubMed] [Google Scholar]
- 47.Lin J, Tang H, Jin X, Jia G, Hsieh JT. p53 regulates Stat3 phosphorylation and DNA binding activity in human prostate cancer cells expressing constitutively active Stat3. Oncogene. 2002;21:3082–3088. doi: 10.1038/sj.onc.1205426. [DOI] [PubMed] [Google Scholar]
- 48.Crighton D, Wilkinson S, O'Prey J, Syed N, Smith P, Harrison PR, Gasco M, Garrone O, Crook T, Ryan KM. DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell. 2006;126:121–134. doi: 10.1016/j.cell.2006.05.034. [DOI] [PubMed] [Google Scholar]
- 49.Shen S, Niso-Santano M, Adjemian S, Takehara T, Malik SA, Minoux H, Souquere S, Marino G, Lachkar S, Senovilla L, Galluzzi L, Kepp O, Pierron G, Maiuri MC, Hikita H, Kroemer R, Kroemer G. Cytoplasmic STAT3 represses autophagy by inhibiting PKR activity. Mol Cell. 2012;48:667–680. doi: 10.1016/j.molcel.2012.09.013. [DOI] [PubMed] [Google Scholar]
- 50.Pietrocola F, Izzo V, Niso-Santano M, Vacchelli E, Galluzzi L, Maiuri MC, Kroemer G. Regulation of autophagy by stress-responsive transcription factors. Semin Cancer Biol. 2013;23:310–322. doi: 10.1016/j.semcancer.2013.05.008. [DOI] [PubMed] [Google Scholar]
- 51.Sawada M, Sun W, Hayes P, Leskov K, Boothman DA, Matsuyama S. Ku70 suppresses the apoptotic translocation of Bax to mitochondria. Nat Cell Biol. 2003;5:320–329. doi: 10.1038/ncb950. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.