

Abstract
K-ras mutations are associated with smoking-induced lung cancer and poor clinical outcomes. In mice, K-ras mutations are sufficient to induce lung tumors, which require phosphoinoside-3-kinase (PI3K) and further downstream, mammalian target of rapamycin (mTOR) activation. However, the roles of individual Akt isoforms that link PI3K and mTOR are unknown. Here, we show that deletion of Akt1 but not Akt2 or Akt3 prevents lung tumorigenesis in a tobacco carcinogen-induced model and a genetic model. Akt1 deletion prevented tumor initiation as well as tumor progression, coincident with decreased Akt signaling in tumor tissues. In contrast, deletion of Akt3 increased tumor multiplicity in the carcinogen model and increased tumor size in the genetic model. Fibroblasts lacking Akt1 are resistant to transformation by mutant K-ras and stimulation by epidermal growth factor. Human lung cancer cells with mutant K-ras and diminished Akt1 levels fail to grow in vivo. These data suggest that Akt1 is the primary Akt isoform activated by mutant K-ras in lung tumors, and that Akt3 may oppose Akt1 in lung tumorigenesis and lung tumor progression. Given that Akt inhibitors in clinical development as cancer therapeutics are not isoform selective, these studies support specific targeting of Akt1 to mitigate the effects of mutant K-ras in lung cancer.
Keywords: Akt1, Akt3, K-ras, lung
Introduction
Lung cancer is the leading cause of cancer death in the United States with nearly 160 000 yearly deaths. The overall 5-year survival rate is 15%, largely due to the long-term ineffectiveness of current therapies and frequent late stage at time of diagnoses (Jemal et al., 2010). Most lung cancers are linked to smoking, and genetic changes associated with these cancers have been attributed to tobacco carcinogens. Mutations in K-ras are one of the most important genetic changes induced by smoking. K-ras mutations constitutively activate its GTPase activity and are found in 20–30% of human lung adenocarcinomas and in other cancers as well (Friday and Adjei, 2005). GTPases are notoriously difficult drug targets. Previous attempts at targeting K-ras, such as through inhibition of farnesyltransferase (Macdonald et al., 2005), have failed.
Mutant K-ras activates several signaling cascades, including the serine threonine kinase Akt. In addition to K-ras, Akt is activated by many growth factor and cytokine receptors. Akt activation can lead to increased proliferation, escape from cell cycle arrest, inhibition of apoptosis and altered metabolism (Gonzalez and McGraw, 2009). Akt activation is one of the most frequent molecular alterations in cancer, and is observed in more than 70% of non-small cell lung cancers (NSCLC) (Tsurutani et al., 2006), as well as many other tumor types (Steelman et al., 2008). In NSCLC, Akt activation is associated with decreased survival (Tsurutani et al., 2006). The importance of mutant K-ras and Akt activation is recapitulated in mouse models of lung cancer. In genetically engineered mice, K-ras mutations cause high-multiplicity lung tumors (Johnson et al., 2001), and in A/J mice treated with 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK), a tobacco-specific carcinogen, lung tumors show Akt pathway activation and K-ras mutation (Belinsky et al., 1989; West et al., 2004).
Akt is a multigene family comprised of three isoforms with more than 75% identity in humans. The phosphorylation sites used to ascertain Akt activation are conserved between isoforms (for example T308 and S473 in Akt1 and T309 and S474 in Akt2). Akt isoforms are differentially expressed in tissues (Dummler et al., 2006), and recent evidence suggests that they may have different substrate specificities as well (Manning and Cantley, 2007). Mice with genetic deletion of individual isoforms manifest different phenotypes, suggesting unique developmental functions for each (Cho et al., 2001a, b; Easton et al., 2005). Isoform specific mutations have also been reported in human cancers. For example, activating mutations in Akt1 occur in multiple tumor types (Carpten et al., 2007; Malanga et al., 2008; Dannemann et al., 2009; Ricarte-Filho et al., 2009; Shoji et al., 2009; Zilberman et al., 2009; Askham et al., 2010), and Akt3 mutation, amplification and over-expression have been linked specifically to melanoma (Madhunapantula and Robertson, 2009).
As Akt is activated in a high percentage of tumors, inhibitors of Akt may have therapeutic value. Clinical trials with Akt inhibitors have shown modest efficacy and variable toxicities (Gills and Dennis, 2009). Although these inhibitors vary in the mechanisms by which they inhibit Akt, they were typically chosen for development without regard for isoform specificity or for inhibition of multiple Akt isoforms. We hypothesized that identification of specific roles for individual Akt family member(s) in lung tumor formation and/or maintenance could assist the development of more specific inhibitors with potentially better therapeutic potential and fewer side effects.
In the current study, genetically engineered mice deficient in individual Akt isoforms were used to determine which isoform(s) were required for mutant K-ras-mediated lung tumorigenesis. Only deletion of Akt1 prevented lung tumors in both carcinogen-driven and genetic mutant K-ras models. Akt1 deletion also prevented tumor progression in vivo and prevented transformation by mutant K-ras in vitro. In contrast to Akt1 deletion, deletion of Akt3 increased tumor initiation and progression in the carcinogen and genetic models, respectively, suggesting that the marginal activity of Akt inhibitors in clinical trials might be due to inhibition of isoforms that have opposing effects. Akt1 may therefore be an important and specific target for therapy of human lung cancers with mutant K-ras.
Results
Genetically engineered mice that lack individual Akt isoforms were obtained in a lung tumor–resistant C57BL/6 background. Therefore, they were backcrossed twice to A/J mice and the progeny were selected for homozygosity of the A/J K-ras allele that confers lung tumor susceptibility. In this mixed background, homozygous deletion of Akt1 led to decreased NNK-induced lung tumor incidence whereas heterozygous deletion of Akt1 had an intermediate affect (Figure 1a). Tumor multiplicity was decreased by 94% in Akt1−/− compared with Akt1+/+ littermates (Figure 1b). Akt1+/+ mice in a pure A/J background had many more NNK-induced tumors than the mixed background, but a similar trend of decreased tumor multiplicity with decreased Akt1 was observed (Figure 1c). In the NNK model, loss of Akt1 did not affect tumor size (data not shown).
Figure 1.
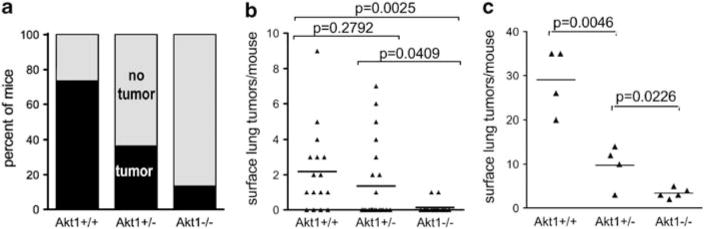
Akt1 is required for NNK-induced lung tumors in mice. Mice were treated with NNK as described in Materials and methods. (a) Incidence of lung tumors in mixed background mice with varying Akt1 (w2-test for trend, P=0.0008). (b) Lung tumor multiplicity in mice with a mixed AB6 genetic background. (c) Lung tumor multiplicity in mice with eight backcrosses to A/J. Each point represents one mouse; lines are at means.
Akt2−/− or Akt2+/− mice developed 80–90% fewer tumors than their Akt2+/+ littermates in response to NNK (Supplementary Table 1), but tumor multiplicity did not decrease with loss of Akt2 in two other mutant K-ras lung mouse models and is likely related to a linked polymorphism that affects NNK metabolism (described below). Deletion of Akt3 had the opposite effect as the deletion of Akt1 or Akt2, leading to more than double the number of lung tumors as Akt3+/+ littermates (Supplementary Table 1).
Both spontaneous and carcinogen-induced lung tumors that develop in A/J mice nearly always carry mutations in the K-ras gene (Belinsky et al., 1989), and expression of a mutant K-ras transgene in the lung is sufficient for lung tumorigenesis (Johnson et al., 2001). As K-ras mutations activate phosphoinoside-3-kinase (PI3K), which can activate all Akt isoforms, our results suggested that activation of Akt1 specifically is required for efficient K-ras-induced lung tumorigenesis. To further address this issue, K-rasLA2/+ (latent activatible allele) mice (Johnson et al., 2001) were crossed with Akt1−/−, Akt2−/− and Akt3−/− mice. Mice that were in a mixed A/J and C57BL/6 background developed spontaneous tumors with much shorter latencies than in the NNK model, with microscopic tumors observed as early as 2 weeks (data not shown). As in the NNK model, only Akt1 deletion significantly reduced lung tumor multiplicity in the K-rasLA2/+ model (Figure 2a, Supplementary Table 1). Akt1−/−/K-rasLA2/+ mice maintained lower lung tumor multiplicity through 16 weeks of age, at which time Akt1+/−/K-rasLA2/+ mice showed decreased tumor number as well (Figure 2a). Between 5 and 16 weeks, lung tumors in Akt1+/+/K-rasLA2/+ mice increased in size by an average of eightfold (Figure 2b) but tumor growth was significantly attenuated in both Akt1−/−/K-rasLA2/+ and Akt1+/−/K-rasLA2/+ mice (Figures 2b and c). Histological evaluation of lung lesions indicated that 8.5% (10/118) of tumors from Akt1+/+/K-rasLA2/+ mice were adenocarcinomas, whereas no adenocarcinomas (0/29) were observed in Akt1−/− K-rasLA2/+ mice. All other tumors were classified as adenomas (data not shown). These data suggest that Akt1 is required for tumor progression as well as initiation. Deletion of either Akt2 or Akt3 had no effect on lung tumor multiplicity (Figures 3a and b, left panels), but Akt3 deletion led to a nearly threefold increase in tumor volume at 4 weeks (Figure 3a, right panel).
Figure 2.
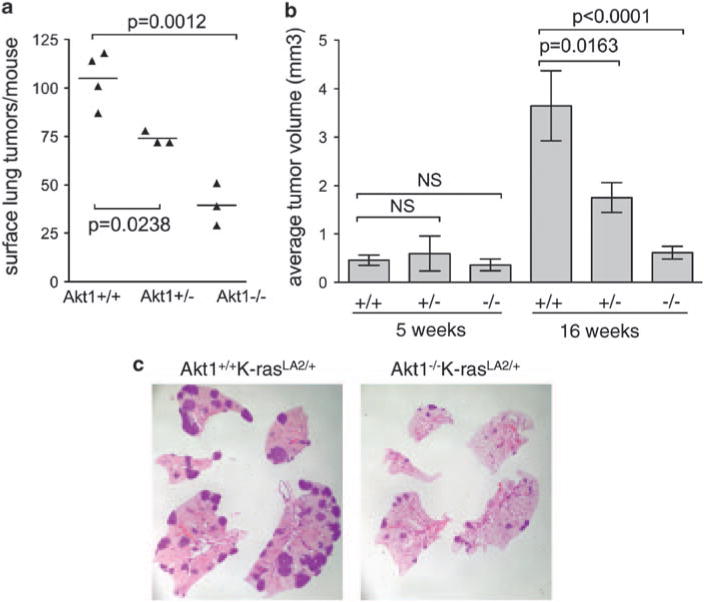
Akt1 is required for initiation and progression of lung tumors induced by mutant K-ras. Mice harbored a latent activatible mutant K-ras (K-rasLA2/+). (a) Lung tumor multiplicity at 16 weeks. Each point represents one mouse; lines are at means. (b) Lung tumor volume increases with time in +/+ but not Akt1−/− mice. Bars represent mean tumor volume±s.e.m. (c) Representative 16 week-old K-rasLA2/+ whole mounts of lungs stained with hematoxylin and eosin. Normal lung appears pink and tumors appear dark purple. Notice decreased number and size of lung tumors in the Akt1−/−/K-rasLA2/+ lungs. Overall size of lungs (and mice) are decreased in Akt1−/− mice, which has been described previously.
Figure 3.
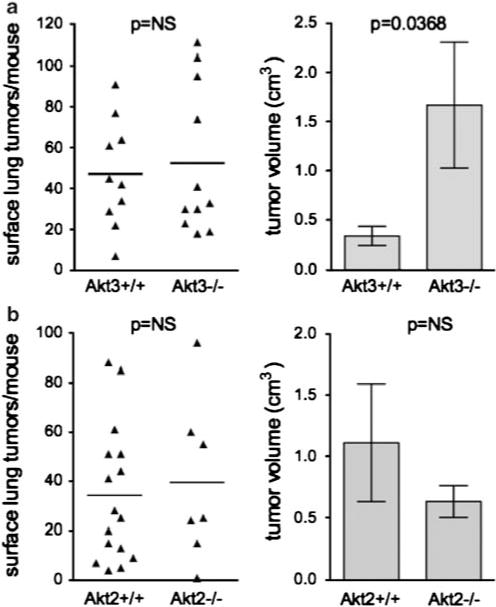
Akt2 or Akt3 deletion has no effect on lung tumor multiplicity, but Akt3 deletion increases K-rasLA2 lung tumor size. (a) Effects of Akt3 loss on lung tumor multiplicity and size. (b) Effects of Akt2 loss on lung tumor multiplicity and size. Data for Akt3 mice were at 5 weeks of age. Data for Akt2 mice were at 8 weeks of age. In the left panels, each point represents one mouse; lines are at means. In the right panels, bar graphs represent average tumor volume±s.e.m.
The discrepant role for Akt2 in the NNK model and K-rasLA2/+ model raised the possibility that loss of Akt2 specifically affected the response to NNK. To address this issue, mice were treated with urethane, which also induces K-ras mutant lung tumors (Lin et al., 1998). Akt2 deletion did not lead to fewer lung tumors in this model (Supplementary Table 1). Therefore, Akt2 was not required in two of the three mutant K-ras-mediated lung tumor models, suggesting that Akt2 may be specifically required for the response to NNK rather than generally required for mutant K-ras-mediated lung tumorigenesis.
To determine whether Akt1 deletion prevents Akt pathway activation by mutant K-ras, Akt signaling was assessed in comparably sized tumors from Akt1+/+ and Akt1−/− mice. Akt1+/+/K-rasLA2/+ tumors had increased activation of Akt, ERK, and targets downstream of mammalian target of rapamycin (mTOR) relative to normal lung (Figure 4a). However, total Akt levels, activation of Akt and propagation to downstream targets were attenuated in Akt1−/− tumors relative to Akt1+/+ tumors (Figure 4b). In particular, loss of Akt1 decreased mTOR activation by mutant K-ras, because p-S6, p4E-BP1, total 4E-BP1 and total eIF4E were diminished in Akt1−/− tumors (Figure 4). Decreased Akt activation in tumor cells from Akt1−/− tumors was also evident by loss of nuclear staining for pS-473 (Figure 4c). As total levels of Akt were decreased in Akt1−/− tumors without compensatory increases in levels of Akt2 or Akt3, this suggested that decreased total Akt could be a mechanism for attenuation of Akt signaling in tumors.
Figure 4.

Decreased total Akt, active Akt and downstream signaling in lung tumors from Akt1−/− K-rasLA2/+ mice. (a) Lysates were prepared from individual tumors that were approximately 0.8–1.3 mm in diameter, or whole lung without tumors (NL). (b) Quantitation of western blots from (a) and others (12–14 tumors each). (c) Decreased nuclear pS473-Akt staining (by immunohistochemistry, brown color) in K-rasLA2/+ tumors lacking Akt1.
To assess whether loss of total and active Akt was specific for Akt1−/−/K-rasLA2/+ lung tumors or was a general feature of lung tissues, Akt signaling was evaluated in lungs from mice lacking Akt1, 2 or 3 but had no K-ras mutation (Supplementary Figure 1). Although Akt1−/− lungs showed significantly less total Akt, they did not show a relative loss of Akt activation or downstream signaling, which was in contrast to the decreased signaling observed in tumors. Loss of Akt2 or Akt3 appeared to decrease Akt activation in lungs without affecting mTOR, even though total Akt varied between mice lacking Akt2 or Akt3. These data suggest that loss of Akt signaling was specific for K-ras-induced lung tumors in Akt1−/− mice.
Cells with mutant K-ras have increased responsiveness to epidermal growth factor (EGF) (Guerra et al., 2003). Even in the presence of wild-type K-ras, EGF signals through epidermal growth factor receptor, K-ras and PI3K to activate Akt (Cully and Downward, 2008). Therefore, EGF-induced Akt signaling was investigated in mouse embryo fibroblasts (MEF) lacking Akt1, 2 or 3. In wild-type cells, EGF treatment led to a time- and dose-dependent increase in activated Akt. This activation was attenuated in Akt1−/− MEF but not in Akt2−/− or Akt3−/− MEF (Figure 5). Akt1−/− MEF required approximately 10-fold higher doses of EGF for detectable Akt activation (Figure 5b). ERK activation did not differ between any of the genotypes. These data show that Akt1 preferentially controls the cellular response to exogenous EGF.
Figure 5.
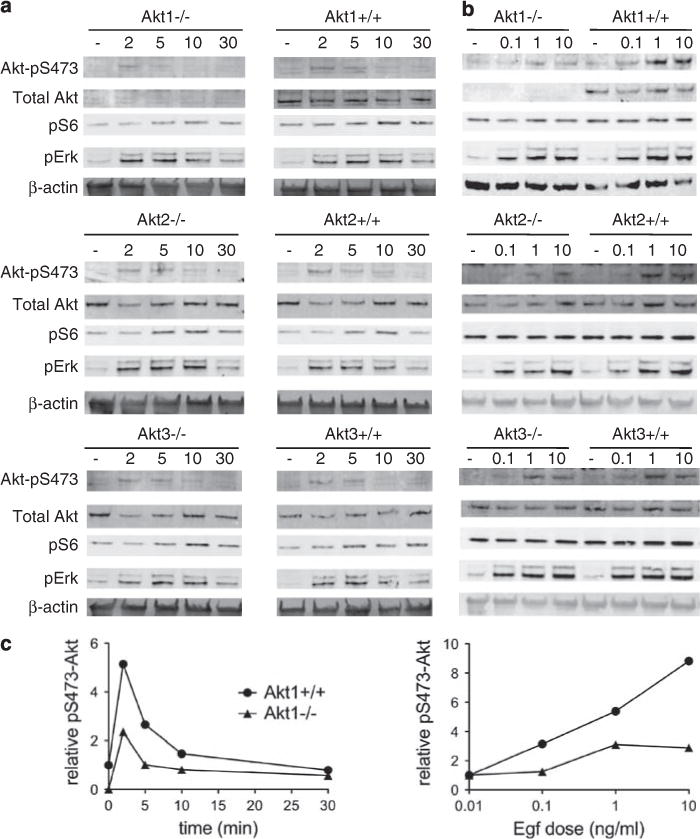
Akt1 but not Akt2 or Akt3 is required for efficient Akt activation by the EGF pathway. (a) Time course (in minutes) with 10 ng/ml EGF. (b) Dose response (nM) at 5 min. Samples for (a) were run on the same gel and intervening lanes have been removed. (c) Quantitation of pS473 signal for Akt1+/+ which and Akt1−/− blots. All experiments were done with two independent clones, gave similar results.
Immortalized cells can be transformed by Ras family members, and loss of Akt1 can inhibit transformation caused by mutant H-ras in combination with other oncogenes (Skeen et al., 2006). As mutant K-ras is the driving mutation for lung tumors in the mouse models used here, primary MEFs were immortalized with dominant negative p53 followed by transformation with mutant K-ras (G12V). Immortalized (p53DD) and immortalized/transformed (p53DD + K-rasG12V) MEF cell lines were generated successfully regardless of Akt1 status. Introduction of mutant K-ras transformed immortalized Akt1+/+/p53DD MEF, but had no noticeable effect on Akt1−/−/p53DD-immortalized MEF (Figure 6a). Akt1+/+/p53DD + K-rasG12V MEF formed large, dense colonies that exhibited loss of contact inhibition, whereas Akt1−/−/p53DD + K-rasG12V colonies retained the contact inhibition characteristic of the immortalized cells (Figure 6a, right panels). Akt1+/+/p53DD + K-rasG12V MEF formed colonies in soft agar but Akt1−/−/p53DD + K-rasG12V MEF did not (data not shown). These results were replicated in MEF lacking p53, which are already immortal. MEF lacking both Akt1 and p53 showed a lower level of transformation by mutant K-ras, and this effect was reversed by overexpression of Akt1 (Figure 6b). These data suggest that Akt1 deletion substantially diminishes transformation by mutant K-ras. In contrast, deletion of Akt2 or Akt3 did not affect transformation by mutant K-ras (data not shown).
Figure 6.
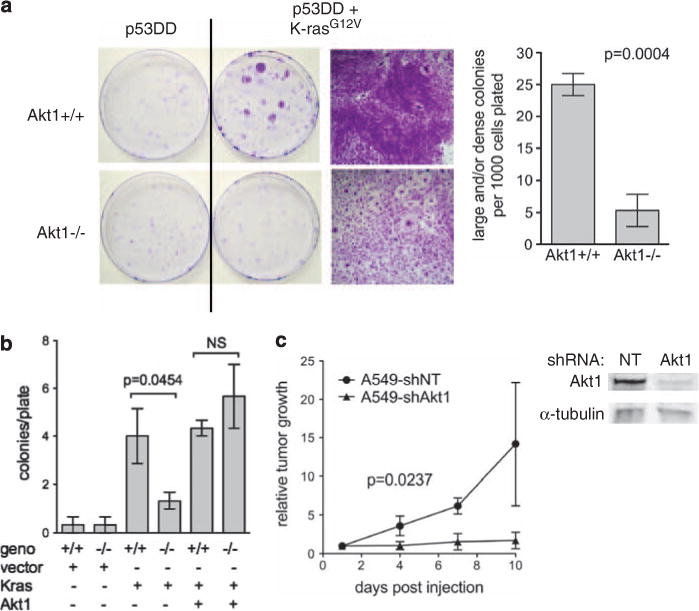
Akt1 is required for mutant K-ras-mediated transformation of immortalized MEF in vitro. (a) Primary MEF were infected with retroviruses expressing dominant-negative p53 (P53DD) and/or K-rasG12V. After hygromycin and/or puromycin selection, 1000 cells were plated in 10 cm dishes. Fourteen days after plating, cells were fixed with methanol/acetone and stained with crystal violet. Representative plates are shown, as well as high magnification of a transformed colony for Akt1+/+ and a typical colony for Akt1−/−. Quantitation of transformed foci is indicated on the right. (b) The p53−/− MEF were infected with retroviruses expressing K-rasG12V and/or Akt1 or empty vector. Colonies were plated and counted as above. (c) Akt1 knockdown in human K-ras-mutant lung tumor cells inhibits growth in xenografts. Tumor growth was measured by luminescence at injection sites. Five mice were analyzed. A549shNT includes two parental A549 and three A549shNT. A549 expressing a non-targeting (NT) shRNA and parental A549 had indistinguishable growth rates. Western blot showing Akt1 knockdown in the same cells in vitro.
The K-ras mutant A549 lung adenocarcinoma cell line was used as a model to test whether Akt1 is important in human lung tumor growth. Expression of individual Akt family members was decreased with short hairpin (sh)RNA, and tumor growth was monitored by imaging for the integrated luciferase. In xenografts, knockdown of Akt1 by 90–95% completely prevented tumor growth (Figure 6c). Visible tumors from parental A549 and A549shNT were apparent two weeks after injection of 1 × 105 cells, whereas tumors from A549-shAkt1 were not detectable for up to 40 days. Although the luciferase signal could still be detected from A549-shAkt1 injected cells at 40 days, no tumors were found at necropsy. Knockdown of Akt2 by 95% did not affect tumor growth (data not shown). Akt1 and Akt2 levels in A549 were similar whereas Akt3 levels were nearly undetectable when compared with a standard curve of recombinant proteins (data not shown).
Discussion
Akt activation is commonly observed in human tumors and is frequently associated with poor prognosis (Cicenas, 2008). However, commercially available antibodies used to ascertain Akt activation recognize all three isoforms, which obscures the importance of individual isoforms and highlights the need for relevant preclinical studies that distinguish roles for individual isoforms. Our studies show that Akt1 alone is required for efficient lung tumorigenesis as well as lung tumor progression by mutant K-ras. This was demonstrated in both a carcinogen-driven and a genetic model of mutant K-ras-induced lung tumors in mice.
The roles of Akt2 and Akt3 differed from that of Akt1. Akt2 was most variable in that loss of Akt2 decreased lung tumors in the NNK model but had no effect in the LA2 model, and increased tumors in the urethane model. Akt2 is on mouse chromosome 7 and is within 0.01 cM from multiple cytochrome p450 genes (NCBI Mapviewer). Given that these carcinogen experiments were done in mixed background strains, it is possible that Akt2 is very tightly linked to a gene(s) involved in metabolism of NNK and/or urethane, and that these linked genes differ between the Akt2+/+ and Akt2−/− littermates. However, as Akt2 is not required for tumor formation in the LA2 and urethane models, Akt2 most likely is dispensable for K-ras mutation-induced lung tumors in mice. However, lung tumor size was somewhat smaller in the Akt2−/−/K-rasLA2 mice compared with Akt2+/+/K-rasLA2 littermates (Figure 3b), although this difference was not significant. Previous studies have shown that dual deletion of Akt2 and Akt3 can recapitulate the small size of Akt1−/− mice (Dummler et al., 2006), suggesting that for some phenotypes, total levels of Akt expression are more important than expression of individual isoforms. In contrast, deletion of Akt1 was found to have effects on glucose metabolism in opposition to those found in mice lacking Akt2 (Buzzi et al., 2010), suggesting that other phenotypes are critically dependent on isoform. Consequently, a role for Akt2 in lung tumorigenesis cannot be discounted entirely, but any effect is minimal in the presence of normal levels of Akt1.
Akt3 may have a surprising role as a tumor suppressor in mutant K-ras-driven lung tumorigenesis. Akt3 deletion more than doubled lung tumor multiplicity in the NNK model and increased tumor size in the LA2 model, suggesting that Akt3 may oppose Akt1 in lung tumors. Despite these provocative results in lung tumorigenesis, it is likely that any tumor suppressor function of Akt3 is tissue-specific because Akt3 acts as an oncogene in melanoma (Robertson, 2005; Davies et al., 2008).
Loss of Akt1 could decrease lung tumorigenesis through several mechanisms that were identified in vivo and in vitro. First, loss of Akt1 interrupts the propagation of signaling in tumors to critical downstream targets such as mTOR, which is required for tumor formation (Granville et al., 2007). Second, the lack of nuclear, active Akt in Akt1−/− lung tumors suggests that subcellular localization of Akt1 may be important for tumorigenesis. This is consistent with the ability of the tumor suppressor Pml to reduce nuclear Akt and inhibit tumorigenesis in PTEN+/− mice (Trotman et al., 2006). Third, loss of Akt1 could diminish the response to exogenous growth factors. in vitro, Akt1−/− MEF but not Akt2−/− or Akt3−/− MEF was resistant to stimulation with EGF, suggesting that Akt2 and Akt3 do not contribute substantially to the activation of total Akt through this pathway. Fourth, loss of Akt1 could inhibit transformation of lung epithelial cells by mutant K-ras because immortalized Akt1−/− MEF were resistant to transformation by mutant K-ras. This is consistent with previous studies showing that mutant H-ras also had decreased transforming ability in Akt1−/− MEF (Chen et al., 2006).
The requirement for Akt1 emphasizes the primacy of the PI3K/Akt/mTOR pathway over other pathways such as the MEK/ERK pathway in mutant K-ras-induced lung tumorigenesis. A mutation in the p110a subunit of PI3K that prevents binding to K-ras decreases lung tumor multiplicity by 95% in the K-rasLA2 model and also decreases tumor size, which supports an essential role for PI3K signaling downstream of K-ras (Gupta et al., 2007). Activation of PI3K generates phosphoinositides that lead to activation of Akt, but it is not known which isoform(s) is responsive. Akt1 deletion decreases lung tumor multiplicity in the K-rasLA2 model and prevents progression of tumors that do form. As Akt2 or Akt3 deletion did not decrease tumor multiplicity or size in this model, it is likely that PI3K signals primarily through the Akt1 isoform in these K-ras-induced lung tumors. In the A/J NNK model of mouse lung tumorigenesis, treatment with the mTOR inhibitor, rapamycin, decreased tumor multiplicity by 90%, similar to what was observed for Akt1−/− mice (Granville et al., 2007). Although mTOR inhibition can lead to feedback activation of Akt in some cell types, rapamycin did not increase Akt activation in these studies. Collectively, these results suggest that Akt1 is an important regulator of mTOR activity in K-ras mutant lung tumors, and provide evidence that Akt1 may be a critical missing link between PI3K and mTOR in mouse lung tumorigenesis.
The reliance of mutant K-ras on Akt1 to induce lung tumors is consistent with previously published data wherein other tumor types also require Akt1. In PTEN+/− mice, deletion of Akt1 decreased tumors of the endometrium, prostate, thyroid and adrenal glands and decreased intestinal polyps (Chen et al., 2006). In multiple studies, deletion of Akt1 decreased mammary tumorigenesis by ErbB2 and other oncogenes (Ju et al., 2007; Maroulakou et al., 2007). Collectively, there is mounting evidence that Akt1 may be the most important isoform in both tumorigenesis and tumor progression, and is a potential target for therapeutic intervention in multiple tumor types.
Loss of Akt1 also occurs in human NSCLC in the setting of gross chromosomal abnormalities. A recent study described deletions of chromosome 14q32 in more than 20% of early stage NSCLC cases that were associated with increased survival. Although this study correlated deletion with decreased Hsp90 expression, these deletions also encompass Akt1, however, Akt1 levels were not investigated in this study (Gallegos Ruiz et al., 2008). Allelic loss of this region containing Akt1 could lead to decreased tumor growth and prolongation of survival, similar to what was observed for mice deficient in Akt1.
Identification of Akt1 as the only Akt isoform required for mutant K-ras-mediated lung tumor initiation and progression validates Akt1 as a potential therapeutic target for human NSCLC. The fact that Akt1+/− mice had decreased tumor multiplicity and decreased tumor progression implies that incomplete inhibition of Akt1 in lung tumors might prove therapeutically efficacious. In support of this, Akt1 shRNA delivery in nanoparticles prevented nascent urethane-induced lung tumor formation by 35% with just a 30% decrease in Akt1 (Jiang et al., 2009). This treatment also decreased both lung tumor number and tumor size in the K-rasLA1 transgenic model (Xu et al., 2008). The potential opposing roles of Akt1 and Akt3 in lung tumorigenesis suggest that development of non-selective Akt inhibitors may not be successful. The development of specific Akt1 inhibitors for lung cancer might improve efficacy and diminish toxicity, especially because inhibition of Akt2 is thought to be responsible for the hyperglycemia and hyperinsulinemia observed with non-selective Akt inhibitors.
Materials and methods
Mice
NIH is an Association for Assessment and Accreditation of Laboratory Animal Care-accredited animal facility and all experiments were done under an approved NCI animal study protocol. Mice were housed in Plexiglas cages and given autoclaved NIH 31 diet and water ad libidum. Lung tumor susceptibility was introduced into Akt1−/− (Cho et al., 2001b), Akt2−/− (Cho et al., 2001a) and Akt3−/− (Easton et al., 2005) mice through mating with A/J mice and selection of mice harboring two copies of the A/J K-ras allele (Hollander et al., 2008), which has been shown to be the major determinant of lung tumor susceptibility in this strain (Lin et al., 1998). For this study, breeder pairs of +/− mice from each strain were used to generate mice for treatment. Mice were genotyped by PCR on tail DNA (Hollander et al., 1999). As a mixed genetic background was used, littermates consisting of +/+, +/− and −/− mice for each strain were injected with three weekly doses of 100 mg/kg of NNK starting at 6 weeks of age. Akt1+/− mice were also backcrossed for eight generations to A/J mice and then treated with NNK as above. Sixteen weeks after the 1st NNK injection, mice were killed by cervical dislocation and lungs were inflated with 10% neutral buffered formalin. For urethane (ethyl carbamate)-induced carcinogenesis, progeny from Akt2+/− crosses were injected intraperitoneally with a single dose of 1000 mg/kg urethane. Lungs were taken as above, 16 weeks after dosing.
Akt1−/− mice were mated with K-rasLA2/+ mice (Johnson et al., 2001) and all tumorigenesis studies were done in a mixed A/J and C57BL/6 strain background. Akt1+//K-rasLA2/+ mice were mated with Akt1+/− mice to generate Akt1−/−/K-rasLA2/+, Akt1+//K-rasLA2/+ and Akt1+/+/K-rasLA2/+ mice for this study. The same breeding scheme was used for Akt2−/− and Akt3−/− mice. Mice were killed at 4–16 weeks of age by cervical dislocation and the lungs inflated with 10% neutral buffered formalin.
For all mice, lung lobes were separated and visible surface lung tumors were counted and measured using a dissecting microscope. This method has been shown to be a reasonable measure of total tumor number (Witschi, 1981; John, 2001), and particularly suited for high-volume studies. Tissues were processed to paraffin, sectioned on slides and stained with hematoxylin and eosin for histopathology analysis. For some mice, lungs were inflated after cervical dislocation with ice cold phosphate-buffered saline and tumors were dissected from normal lung tissue under a dissecting microscope to create lysates for western blotting.
Nude mice were injected with 1 × 105 cells per injection site, subcutaneously. The following day and at 3–4 day intervals thereafter, mice were imaged for luciferase activity on an IVIS imager (Caliper Lifesciences, Hopkinton, MA, USA). Mice were injected intraperitoneally with 200 μl of 15 mg/ml luciferin, anesthetized with isofluorane and imaged until the luminescent signal began to decrease. Maximum signal was noted for each injection site. Visible tumors were excised from mice and weighed 39–40 days after tumor cell injection.
Cell culture and lentivirus production
MEF were prepared from E13.5 embryos. Cells were maintained in Dulbecco’s modified Eagle medium with 10% fetal bovine serum. For all experiments, two independent −/− MEF clones (each from a single embryo) were tested for each genotype along with MEF from +/+ littermates. For treatment with EGF, cells were transferred to medium with 0.5% fetal bovine serum 4 h before treatment.
For introduction of dominant negative p53 and activated K-ras into MEF, Phoenix Eco packaging cells (Swift et al., 2001) were transfected with pBabe-hygro-p53DD (Addgene plasmid 9058, Cambridge, MA, USA) (Hahn et al., 2002), pBabe-puro-K-rasG12V (Addgene plasmid 12544) (Khosravi-Far et al., 1996) or pBabe-puroL-Akt1 (Addgene plasmid 9011) (DiRenzo et al., 2002) retroviral plasmids. Virus-containing medium was collected and used for infection of 2 × 105 cells as described previously (Bulavin et al., 2003). Two days after infection, cells were selected for 1–2 weeks in 300 μg/ml of hygromycin or 4 μg/ml of puromycin. For each MEF clone, several hundred drug-resistant colonies were pooled for further use. For colony formation, 1000 cells were plated per 10 cm dish, fixed after 14 days and stained with crystal violet. For soft agar colony formation, 1 × 104 cells were plated as described previously (Bulavin et al., 2003).
A549 and 293FT cells were grown in RPMI 1640 with 10% fetal bovine serum. A549 was infected with lentivirus expressing a green fluorescent protein–luciferase (gfp–luc) fusion protein (pSICO-pol2-luc/gfp). As there is no drug-selection for this construct, gfp positive cells were selected by fluorescence-activated cell sorting (FACS Vantage SE cell sorter, BD Biosciences, San Jose, CA, USA). These cells were used for all subsequent shRNA experiments. The shRNA vectors used were from Sigma-Aldrich (St Louis, MO, USA) unless otherwise noted: Akt1, NM_005163.1-628s1c1; Akt2, NM_001626.2-1509s1c1; Akt3, NM_005465.3-671s1c1;and non-targeting, pLKO-scr (Addgene plasmid 1864) were used. For transfection to generate lentivirus, 8 × 105 293FT cells were plated in 25 cm2 flasks. The following day, cells were transfected using 6 μl Lipofectamine 2000 (Life Technologies), 1 mg lentiviral shRNA plasmid, 750 ng of psPAX2 (Addgene plasmid 12260) and 250 ng of pMD2.G (Addgene plasmid 12259). Growth medium was replaced after 16 h at 37 °C and lentivirus-containing medium was obtained at 48 and 72 h post transfection. A549 was plated at 2 × 105 cells per 35 mm well and infected with lentiviral particles the following day at a multiplicity of infection of 0.1, which had been validated as the optimal multiplicity of infection for knockdown. After 2 days, medium was changed and cells were selected for 3–4 days in 2 μg/ml puromycin, split to a 75 cm2 flask and selected for a further 4–6 days, until reaching confluence when they were cryopreserved as designated in passage 2. Knockdown of specific shRNA targets was verified by western blotting.
Western blotting
Antibodies used were from Cell Signaling Technology unless otherwise indicated. Primary antibodies used were Akt1 (2967), Akt2 (2964), Akt3 (3788), pS473-Akt (9271), pT308-Akt (3780), pS235/236-S6 (2211), 4E-BP1 (9452), eIF4E (9742), pT202/204-ERK1/2 (4377), α-tubulin (2125), b-actin (CP-01, EMD Biosciences, San Diego, CA, USA). Cells were lysed in situ with 125 mM Tris, pH 8, 20% glycerol, 4% sodium dodecyl sulfate and sonicated to shear DNA. Individual tumors were microdissected from lungs and vortexed at high speed for 5 min in cell lysis buffer above with silicon carbide sharp particles (Biospec Products, Bartlesville, OK, USA). For lung extracts, the left lobe was homogenized in 300 mM NaCl, 50 mM Tris, pH 8, 0.1% Triton with protease inhibitors and phosphatase inhibitor cocktails (Sigma-Aldrich) using a dounce homogenizer. Samples were incubated on ice for 30 min, spun at 13 kr.p.m. for 10 min and the supernatant was collected. Lysates or extracts were incubated at 95 1C for 5 min with 5% β-mercaptoethanol and loaded onto 4–12% acrylamide XT gels (Bio-Rad, Hercules, CA, USA). Proteins were transferred to polyvinylidene difluoride membranes and incubated with primary antibodies (1:1000 dilution) overnight in 1% bovine serum albumin. Secondary antibodies were conjugated to IRdye and blots were imaged and the signal was quantified on an Odyssey infrared scanner (Li-Cor Biosciences, Lincoln, NE, USA).
Immunohistochemistry and immunofluorescence
Paraffin sections were rehydrated in a series of xylene and ethanol washes. Antigen retrieval was carried out using preheated target retrieval solution (pH 6.0) (Dako, Carpinteria, CA, USA) in a pressure cooker. Vectastain Elite ABC kits (Vector Laboratories, Burlingame, CA, USA) were used according to manufacturer’s instructions for blocking, dilution of primary antibody and labeling. Primary antibody directed at pS473-Akt (Cell Signaling) was incubated with sections overnight at 4 1C. The 3,3-diaminobenzidine was prepared fresh from tablets (Sigma-Aldrich).
Statistical methods
Prism software (GraphPad Software, La Jolla, CA, USA) was used for all statistical analysis. For tumor number analysis, an unpaired t-test was used with Welch’s correction for those samples with unequal variances. For incidence analysis, a contingency table was used with w2-test for trend. For analysis of colony formation and western blot signals, t-tests were used. For xenografts growth, two way analysis of variance was used.
Supplementary Material
Acknowledgments
This project was funded through the NIH intramural research program.
Footnotes
Conflict of interest
The authors declare no conflict of interest.
Supplementary Information accompanies the paper on the Oncogene website (http://www.nature.com/onc)
References
- Askham JM, Platt F, Chambers PA, Snowden H, Taylor CF, Knowles MA. AKT1 mutations in bladder cancer: identification of a novel oncogenic mutation that can co-operate with E17K. Oncogene. 2010;29:150–155. doi: 10.1038/onc.2009.315. [DOI] [PubMed] [Google Scholar]
- Belinsky SA, Devereux TR, Maronpot RR, Stoner GD, Anderson MW. Relationship between the formation of promutagenic adducts and the activation of the K-ras protooncogene in lung tumors from A/J mice treated with nitrosamines. Cancer Res. 1989;49:5305–5311. [PubMed] [Google Scholar]
- Bulavin D, Kovalsky O, Hollander M, Fornace AJ. Loss of oncogenic H-ras-induced cell cycle arrest and p38 mitogen-activated protein kinase activation by disruption of Gadd45a. Mol Cell Biol. 2003;23:3859–3871. doi: 10.1128/MCB.23.11.3859-3871.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buzzi F, Xu L, Zuellig RA, Boller SB, Spinas GA, Hynx D, et al. Differential effects of protein kinase B/Akt isoforms on glucose homeostasis and islet mass. Mol Cell Biol. 2010;30:601–612. doi: 10.1128/MCB.00719-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpten J, Faber A, Horn C, Donoho G, Briggs S, Robbins C, et al. A transforming mutation in the pleckstrin homology domain of AKT1 in cancer. Nature. 2007;448:439–444. doi: 10.1038/nature05933. [DOI] [PubMed] [Google Scholar]
- Chen M, Xu P, Peng X, Chen W, Guzman G, Yang X, et al. The deficiency of Akt1 is sufficient to suppress tumor development in Pten+/− mice. Genes Dev. 2006;20:1569–1574. doi: 10.1101/gad.1395006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho H, Mu J, Kim J, Thorvaldsen J, Chu Q, Crenshaw Er, et al. Insulin resistance and a diabetes mellitus-like syndrome in mice lacking the protein kinase Akt2 (PKB beta) Science. 2001a;292:1728–1731. doi: 10.1126/science.292.5522.1728. [DOI] [PubMed] [Google Scholar]
- Cho H, Thorvaldsen J, Chu Q, Feng F, Birnbaum M. Akt1/PKBalpha is required for normal growth but dispensable for maintenance of glucose homeostasis in mice. J Biol Chem. 2001b;276:38349–38352. doi: 10.1074/jbc.C100462200. [DOI] [PubMed] [Google Scholar]
- Cicenas J. The potential role of Akt phosphorylation in human cancers. Int J Biol Markers. 2008;23:1–9. doi: 10.1177/172460080802300101. [DOI] [PubMed] [Google Scholar]
- Cully M, Downward J. SnapShot: Ras signaling. Cell. 2008;133:1292–1292 e1291. doi: 10.1016/j.cell.2008.06.020. [DOI] [PubMed] [Google Scholar]
- Dannemann N, Hart JR, Ueno L, Vogt PK. Phosphatidylinositol 4,5-bisphosphate-specific AKT1 is oncogenic. Int J Cancer. 2009;127:239–244. doi: 10.1002/ijc.25012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies M, Stemke-Hale K, Tellez C, Calderone T, Deng W, Prieto V, et al. A novel AKT3 mutation in melanoma tumours and cell lines. Br J Cancer. 2008;99:1265–1268. doi: 10.1038/sj.bjc.6604637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiRenzo J, Signoretti S, Nakamura N, Rivera-Gonzalez R, Sellers W, Loda M, et al. Growth factor requirements and basal phenotype of an immortalized mammary epithelial cell line. Cancer Res. 2002;62:89–98. [PubMed] [Google Scholar]
- Dummler B, Tschopp O, Hynx D, Yang Z, Dirnhofer S, Hemmings B. Life with a single isoform of Akt: mice lacking Akt2 and Akt3 are viable but display impaired glucose homeostasis and growth deficiencies. Mol Cell Biol. 2006;26:8042–8051. doi: 10.1128/MCB.00722-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Easton R, Cho H, Roovers K, Shineman D, Mizrahi M, Forman M, et al. Role for Akt3/protein kinase Bgamma in attainment of normal brain size. Mol Cell Biol. 2005;25:1869–1878. doi: 10.1128/MCB.25.5.1869-1878.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friday B, Adjei A. K-ras as a target for cancer therapy. Biochim Biophys Acta. 2005;1756:127–144. doi: 10.1016/j.bbcan.2005.08.001. [DOI] [PubMed] [Google Scholar]
- Gallegos Ruiz M, Floor K, Roepman P, Rodriguez J, Meijer G, Mooi W, et al. Integration of gene dosage and gene expression in non-small cell lung cancer, identification of HSP90 as potential target. PLoS One. 2008;3:e0001722. doi: 10.1371/journal.pone.0001722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gills J, Dennis P. Perifosine: update on a novel Akt inhibitor. Curr Oncol Rep. 2009;11:102–110. doi: 10.1007/s11912-009-0016-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez E, McGraw T. The Akt kinases: isoform specificity in metabolism and cancer. Cell Cycle. 2009;8:2502–2508. doi: 10.4161/cc.8.16.9335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granville CA, Warfel N, Tsurutani J, Hollander MC, Robertson M, Fox SD, et al. Identification of a highly effective rapamycin schedule that markedly reduces the size, multiplicity, and pheno-typic progression of tobacco carcinogen-induced murine lung tumors. Clin Cancer Res. 2007;13:2281–2289. doi: 10.1158/1078-0432.CCR-06-2570. [DOI] [PubMed] [Google Scholar]
- Guerra C, Mijimolle N, Dhawahir A, Dubus P, Barradas M, Serrano M, et al. Tumor induction by an endogenous K-ras oncogene is highly dependent on cellular context. Cancer Cell. 2003;4:111–120. doi: 10.1016/s1535-6108(03)00191-0. [DOI] [PubMed] [Google Scholar]
- Gupta S, Ramjaun A, Haiko P, Wang Y, Warne P, Nicke B, et al. Binding of ras to phosphoinositide 3-kinase p110alpha is required for ras-driven tumorigenesis in mice. Cell. 2007;129:957–968. doi: 10.1016/j.cell.2007.03.051. [DOI] [PubMed] [Google Scholar]
- Hahn W, Dessain S, Brooks M, King J, Elenbaas B, Sabatini D, et al. Enumeration of the simian virus 40 early region elements necessary for human cell transformation. Mol Cell Biol. 2002;22:2111–2123. doi: 10.1128/MCB.22.7.2111-2123.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollander M, Sheikh M, Bulavin D, Lundgren K, Augeri-Henmueller L, Shehee R, et al. Genomic instability in Gadd45a-deficient mice. Nat Genet. 1999;23:176–184. doi: 10.1038/13802. [DOI] [PubMed] [Google Scholar]
- Hollander MC, Balogh AR, Liwanag J, Han W, Linnoila RI, Anver MR, et al. Strain-specific spontaneous and NNK-mediated tumor-igenesis in Pten+/− mice. Neoplasia. 2008;10:866–872. doi: 10.1593/neo.08406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jemal A, Siegel R, Xu J, Ward E. Cancer statistics, 2010. CA Cancer J Clin. 2010;60:277–300. doi: 10.3322/caac.20073. [DOI] [PubMed] [Google Scholar]
- Jiang HL, Xu CX, Kim YK, Arote R, Jere D, Lim HT, et al. The suppression of lung tumorigenesis by aerosol-delivered folate-chitosan-graft-polyethylenimine/Akt1 shRNA complexes through the Akt signaling pathway. Biomaterials. 2009;30:5844–5852. doi: 10.1016/j.biomaterials.2009.07.017. [DOI] [PubMed] [Google Scholar]
- John LC. Assessment of murine lung tumour development: a comparison of two techniques. Br J Biomed Sci. 2001;58:159–163. [PubMed] [Google Scholar]
- Johnson L, Mercer K, Greenbaum D, Bronson R, Crowley D, Tuveson D, et al. Somatic activation of the K-ras oncogene causes early onset lung cancer in mice. Nature. 2001;410:1111–1116. doi: 10.1038/35074129. [DOI] [PubMed] [Google Scholar]
- Ju X, Katiyar S, Wang C, Liu M, Jiao X, Li S, et al. Akt1 governs breast cancer progression in vivo. Proc Natl Acad Sci USA. 2007;104:7438–7443. doi: 10.1073/pnas.0605874104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khosravi-Far R, White M, Westwick J, Solski P, Chrzanowska-Wodnicka M, Van Aelst L, et al. Oncogenic Ras activation of Raf/mitogen-activated protein kinase-independent pathways is sufficient to cause tumorigenic transformation. Mol Cell Biol. 1996;16:3923–3933. doi: 10.1128/mcb.16.7.3923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin L, Festing M, Devereux T, Crist K, Christiansen S, Wang Y, et al. Additional evidence that the K-ras protooncogene is a candidate for the major mouse pulmonary adenoma susceptibility (Pas-1) gene. Exp Lung Res. 1998;24:481–497. doi: 10.3109/01902149809087382. [DOI] [PubMed] [Google Scholar]
- Macdonald JS, McCoy S, Whitehead RP, Iqbal S, Wade JL, III, Giguere JK, et al. A phase II study of farnesyl transferase inhibitor R115777 in pancreatic cancer: a Southwest oncology group (SWOG 9924) study. Invest New Drugs. 2005;23:485–487. doi: 10.1007/s10637-005-2908-y. [DOI] [PubMed] [Google Scholar]
- Madhunapantula SV, Robertson GP. The PTEN-AKT3 signaling cascade as a therapeutic target in melanoma. Pigment Cell Melanoma Res. 2009;22:400–419. doi: 10.1111/j.1755-148X.2009.00585.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malanga D, Scrima M, De Marco C, Fabiani F, De Rosa N, De Gisi S, et al. Activating E17K mutation in the gene encoding the protein kinase AKT1 in a subset of squamous cell carcinoma of the lung. Cell Cycle. 2008;7:665–669. doi: 10.4161/cc.7.5.5485. [DOI] [PubMed] [Google Scholar]
- Manning B, Cantley L. AKT/PKB signaling: navigating downstream. Cell. 2007;129:1261–1274. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maroulakou I, Oemler W, Naber S, Tsichlis P. Akt1 ablation inhibits, whereas Akt2 ablation accelerates, the development of mammary adenocarcinomas in mouse mammary tumor virus (MMTV)-ErbB2/neu and MMTV-polyoma middle T transgenic mice. Cancer Res. 2007;67:167–177. doi: 10.1158/0008-5472.CAN-06-3782. [DOI] [PubMed] [Google Scholar]
- Ricarte-Filho JC, Ryder M, Chitale DA, Rivera M, Heguy A, Ladanyi M, et al. Mutational profile of advanced primary and metastatic radioactive iodine-refractory thyroid cancers reveals distinct pathogenetic roles for BRAF, PIK3CA, and AKT1. Cancer Res. 2009;69:4885–4893. doi: 10.1158/0008-5472.CAN-09-0727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson GP. Functional and therapeutic significance of Akt deregulation in malignant melanoma. Cancer Metastasis Rev. 2005;24:273–285. doi: 10.1007/s10555-005-1577-9. [DOI] [PubMed] [Google Scholar]
- Shoji K, Oda K, Nakagawa S, Hosokawa S, Nagae G, Uehara Y, et al. The oncogenic mutation in the pleckstrin homology domain of AKT1 in endometrial carcinomas. Br J Cancer. 2009;101:145–148. doi: 10.1038/sj.bjc.6605109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skeen JE, Bhaskar PT, Chen CC, Chen WS, Peng XD, Nogueira V, et al. Akt deficiency impairs normal cell proliferation and suppresses oncogenesis in a p53-independent and mTORC1-dependent manner. Cancer Cell. 2006;10:269–280. doi: 10.1016/j.ccr.2006.08.022. [DOI] [PubMed] [Google Scholar]
- Steelman L, Stadelman K, Chappell W, Horn S, Basecke J, Cervello M, et al. Akt as a therapeutic target in cancer. Expert Opin Ther Targets. 2008;12:1139–1165. doi: 10.1517/14728222.12.9.1139. [DOI] [PubMed] [Google Scholar]
- Swift S, Lorens J, Achacoso P, Nolan G. Rapid production of retroviruses for efficient gene delivery to mammalian cells using 293T cell-based systems. Curr Protoc Immunol. 2001 doi: 10.1002/0471142735.im1017cs31. Chapter 10, Unit.10.17C. [DOI] [PubMed] [Google Scholar]
- Trotman L, Alimonti A, Scaglioni P, Koutcher J, Cordon-Cardo C, Pandolfi P. Identification of a tumour suppressor network opposing nuclear Akt function. Nature. 2006;441:523–527. doi: 10.1038/nature04809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsurutani J, Fukuoka J, Tsurutani H, Shih J, Hewitt S, Travis W, et al. Evaluation of two phosphorylation sites improves the prognostic significance of Akt activation in non-small-cell lung cancer tumors. J Clin Oncol. 2006;24:306–314. doi: 10.1200/JCO.2005.02.4133. [DOI] [PubMed] [Google Scholar]
- West KA, Linnoila IR, Belinsky SA, Harris CC, Dennis PA. Tobacco carcinogen-induced cellular transformation increases activation of the phosphatidylinositol 3′-kinase/Akt pathway in vitro and in vivo. Cancer Res. 2004;64:446–451. doi: 10.1158/0008-5472.can-03-3241. [DOI] [PubMed] [Google Scholar]
- Witschi HP. Enhancement of tumor formation in mouse lung by dietary butylated hydroxytoluene. Toxicology. 1981;21:95–104. doi: 10.1016/0300-483x(81)90120-7. [DOI] [PubMed] [Google Scholar]
- Xu CX, Jere D, Jin H, Chang SH, Chung YS, Shin JY, et al. Poly(ester amine)-mediated, aerosol-delivered Akt1 small interfering RNA suppresses lung tumorigenesis. Am J Respir Crit Care Med. 2008;178:60–73. doi: 10.1164/rccm.200707-1022OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zilberman D, Cohen Y, Amariglio N, Fridman E, Ramon J, Rechavi G. AKT1 E17K pleckstrin homology domain mutation in urothelial carcinoma. Cancer Genet Cytogenet. 2009;191:34–37. doi: 10.1016/j.cancergencyto.2009.01.009. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.