

Abstract
The translation machinery is the engine of life. Extracting the cytoplasmic milieu from a cell affords a lysate capable of producing proteins in concentrations reaching tens of micromolar. Such lysates, derivable from a variety of cells, allow the facile addition and subtraction of components that are directly or indirectly related to the translation machinery and/or the over-expressed protein. The flexible nature of such cell-free expression systems, when coupled with high throughput monitoring, can be especially suitable for protein engineering studies, allowing one to bypass multiple steps typically required using conventional in vivo protein expression.
Keywords: Cell-free protein synthesis, transcription, translation, high throughput
1. Introduction
The ability to produce a functional protein in the test tube, rather than in cells, is the essence of cell-free protein synthesis (CFPS) [1,2]. The preparation of a CFPS kit requires the separation of the cytoplasmic milieu from the cell wall, and has been applied to a variety of cell types, spanning bacteria, protozoa, plants, insects and mammals [1–8]. The cell lysate is a crowded environment of active biomolecules, capable of supporting many cellular functions. These functions include, but are not limited to, many metabolic pathways, as well as transcription and translation. The preparation of lysates for CFPS was significantly improved over the years in terms of buffer composition [3], energy recycling [9], the utilization of various mutated cell strains [10,11], the supplementation of small molecules [12,13], the addition of proteins such as T7 RNA polymerase (RNAP) for the generation of a transcription/translation coupled system [1,3] and chaperones to improve the yields of properly folded target proteins.
In E. coli CFPS the translation machinery is typically about 20-fold more dilute than in the cell, decreasing the rates of initiation, elongation and protein accumulation [14]. As well, the average distance between two adjacent ribosomes on a single mRNA strand increases and polysomes are less likely to form [15]. Despite these differences, CFPS can benefit from the relative slower synthesis rate and the distance between ribosomes by allowing nascent polypeptide chain more time and space to form desirable intra-peptide chain contacts, while decreasing the probability for undesirable, non-specific inter-peptide chain contacts, thereby increasing the probability of proper folding and decreasing the probability of aggregation.
This paper will outline progress in the field of CFPS that applies this approach in ways that would be challenging, if not impossible, to implement using the standard in vivo expression systems (Fig. 1). The use of improved fluorescent proteins, such as Emerald GFP [16], and of fluorescence detection technologies using a plate reader platform, allow real time monitoring of protein expression in a high-throughput format [17]. These advances allow the implementation of engineering and combinatorial efforts to screen the response of translation to various procedural modifications that include the introduction of exogenous chemical reagents, proteins, and nucleic acids, as well as to the substitution of mutated/modified components of the translational machinery, such as ribosomes, mRNAs and tRNAs, for their endogenous counterparts.
Figure 1.

Cell-free protein synthesis and its functionalities. Translation commences upon the addition of DNA (PCR product or plasmid) in a coupled system or by adding separately transcribed mRNA. Modified CFPS may exhibit various functionalities, some of which are depicted.
2. Methods of Cell-Free Protein Synthesis
The two basic types of CFPS are optimized cell extracts (often termed lysate-based CFPS), an approach that has been in use for more than 5 decades, and the more recently developed PURE system, which employs a mixture of a minimal set of purified components (e.g. ribosome, tRNAs, tRNA synthetases, factors, amino acids, energy sources) required for full-length protein synthesis. Below we present a brief description of each approach and discuss various factors that can influence protein yield and function, before considering some specific examples
2.1. Lysate-based CFPS, coupled transcription/translation
Some commercially available prokaryotic and eukaryotic CFPS kits produce transcribed mRNA and translated protein in a coupled fashion. Adding a DNA encoding the protein of interest along with T7 or SP6 RNA polymerase (RNAP) generally produces transcribed mRNA at a faster rate than protein synthesis, with the result that protein expression is not limited by mRNA availability. There also exists a protocol for the generation of an E. coli CFPS that utilizes endogenous RNAP [18]. In general, coupled CFPS expresses proteins in higher yields, and eliminates the separate in vitro transcription step required for mRNA-dependent CFPS (see below).
Coupled CFPS utilizes DNA in three forms: linear PCR product, linearized plasmid and circular plasmid. Circular DNA plasmid has typically been preferred to linearized plasmid or PCR products, due to the greater susceptibility of linear DNAs to nucleolytic cleavage [19]. On the other hand, use of the linear PCR product has the distinct advantage of simplicity, since it eliminates the need for time-consuming steps required when using plasmid, that include: ligation of the DNA template to a linearized plasmid, transformation of the plasmid to compatible cells, selection of colonies harboring the foreign plasmid, culture growth in culture for plasmid production, plasmid isolation, sequencing, transformation of the plasmid to compatible cells, culture growth for protein overexpression, and harvesting and lysis (Fig. 2). In contrast, addition of an amplified linear DNA fragment to a CFPS affords single step protein expression via transcription/translation coupled CFPS, either in analytic amounts amenable to a high throughput format or in preparative amounts [20,21]. Utilization of a linear PCR product on an analytical scale allows facile optimization of translation of the amplified DNA fragment, coupled with any desirable extension, as, for example, in the case of N-terminal extensions of the adiponectin hypervariable domain [21]. Moreover, the yield of expressed protein can be raised utilizing procedures that increase the stability of the linear PCR-amplified DNA via both the removal of nucleases [10,20] and the utilization of overhang extensions to cyclize PCR products, exploiting the endogenous ligase activity of lysates [22]. In the case of dengue virus NS2B/NS3 protease, the latter approach gave protein yields comparable to those obtained using plasmid-based CFPS [22].
Figure 2.

Protein over-expression using conventional in vivo techniques vs. cell-free protein synthesis. Although both methods end with a cell-lysate that contains the over-expressed protein, in vivo over-expression requires a multiple-step procedure, whereas CFPS is accomplished in a single step.
In general, these improvements make PCR product an attractive alternative to circular plasmid for applications of protein expression ranging from protein engineering to NMR structural characterization of proteins. Commercial CFPS kits, optimized for testing the coupled transcription/translation of PCR products, are available from suppliers such as: 5-prime, Promega, Jena Bioscience, New England Biolabs, Life technologies, Pierce, and Cell-Free Sciences.
2.2 PURE CFPS
The PUREexpress kit was developed by Ueda and coworkers [23] and further optimized by New England Biolabs [24,25], which also distributes it commercially. The PURExpress kit has several advantages over lysate-based kits. Because it is devoid of any biomolecules and metabolites that do not directly participate in protein synthesis it lacks both nucleases and proteases that decrease the lifetime of DNA, mRNA and proteins, and metabolic enzymes that can convert some nucleotides and amino acids necessary for transcription and translation to non-functional products not participating in these processes. In addition, it allows straightforward elimination of specific components of the translational machinery or substitution of exogenous for endogenous components. Thus, for example, by omitting the tRNA synthetase PheRS and all traces of the amino acid Phe, we could make EmGFP synthesis, measured at the single molecule level, totally dependent on the addition of fluorescently labeled Phe-tRNAPhe as the sole source of Phe incorporated into protein. In contrast, an alternative approach, based on manipulation of a lysate-based kit [17], was successful for parallel ensemble studies that could tolerate low levels of endogenous Phe and Phe-tRNAPhe, but was inadequate for single molecule experiments. The main drawback of the PURExpress kit is that the cost per mg of protein produced is considerably higher than with lysate-based kits.
3. Optimizing CFPS for specific proteins
3.1 Coupled vs. uncoupled transcription-translation
Prokaryotic and eukaryotic CFPS kits have been described that lack RNAP activity [5,7,26], such that translation is initiated by the addition of mRNA, a procedure that can also be used in transcription/translation coupled CFPS. Typical results comparing coupled and uncoupled transcription/translation in E. coli CFPS are shown in Fig. 3, from which it is clear that much greater quantities (20 – 30-fold) of mRNA than plasmid DNA are required for the generation of similar translation profile (Fig. 3). This is likely due to the instability of mRNA in the reaction medium, a problem that is alleviated when mRNA is generated in a continuous fashion.
Figure 3.
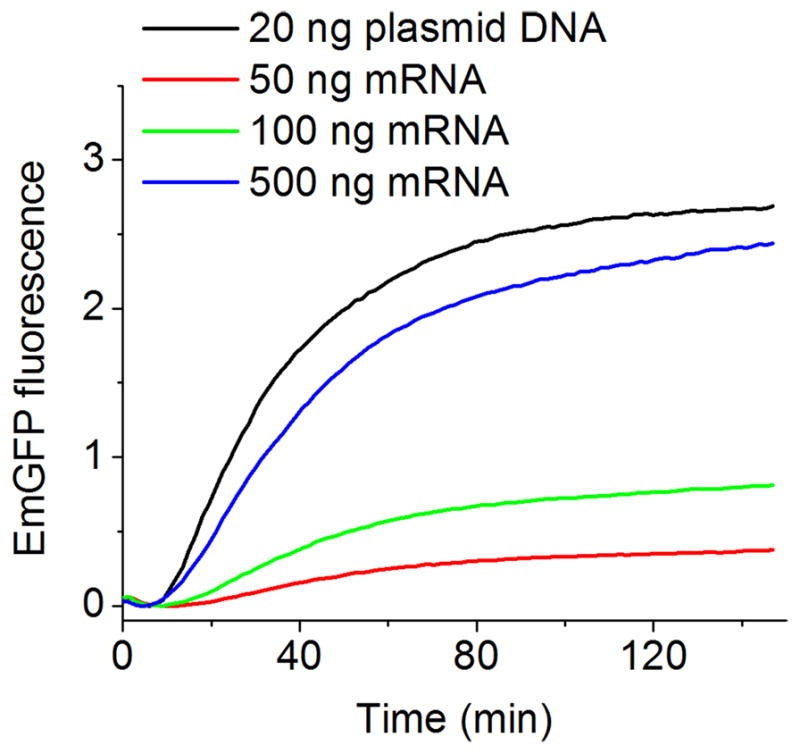
Plasmid DNA vs. mRNA. A volume of 10 μl CFPS was supplemented with the indicated amounts of plasmid DNA or mRNA encoding EmGFP. Continuous fluorescence monitoring was performed in a 384-well plate, set to 30 °C using an excitation and emission wavelength of 486 and 535 nm, respectively.
As a general rule, coupled CFPS is preferred, because it uses smaller amounts of easy-to-obtain plasmid or PCR-amplified DNA and eliminates the times and costs needed for in vitro transcription. However, there are some instances where the utilization of separately prepared mRNA is advantageous. An example is provided by a study of the effects of post-transcriptional mRNA nucleotide modifications on the yields of firefly luciferase and Renilla luciferase. [27]. In this work, synthetic mRNAs were transcribed in vitro in the presence of known amounts of modified nucleotides, which afforded mRNA preparations containing different extents of modified nucleotide. Pseudouridine modification was found to enhance translation in rabbit reticulocyte lysate, (RRL) but not in wheat germ extract or E. coli CFPS, while s2U and m6A modifications decreased translation efficiency in RRL [27]. Quantification of such effects is an important determinant in tailoring synthetic mRNAs with desirable properties such as stability, translational capacity and controlled immunogenicity.
Another example involves the effect of mRNA secondary and tertiary structures on translation. Such structures may contain stem-loops, pseudoknots and G-quadruplexes that may fully form when uncoupled transcription/translation is used, but form only partially, if at all, when the two processes are coupled. Full formation allows detailed investigation of how such structures modulate translation rate, and how modulation can be manipulated, for instance by addition of small molecules and proteins that specifically bind and stabilize these structures [28–33].
3.2 5′-UTRs
A 5′-UTR upstream from an open reading frame of the expression plasmid can be important for increasing protein yield. In bacterial 5′-UTRs such optimization can be achieved by placing the Shine-Dalgarno (SD) sequence at an optimal distance, typically 8 nucleotides, from the AUG start codon [34]. The SD sequence interacts with the 3′-end of 16S ribosomal RNA (rRNA) that is part of the small ribosomal (30S) subunit, to allow efficient initiation. Placement of an A/U-rich enhancer sequence further upstream allows the SD sequence to interact more effectively with the rRNA [35,36]. An engineered 5′-UTR was reported to induce efficient translation by CFPS kits derived from an array of cell types, including E. coli, Leishmania tarentolae, insect cells, wheat germ and rabbit reticulocyte [4]. In eukaryotes, the Internal Ribosome Entry Site (IRES), is a highly structured element found within viral mRNA that is able to induce eukaryotic initiation [37,38]. An upstream IRES was shown to be important for the expression in a HeLa (human)-derived CFPS kit of large proteins such as GCN2 (160 kD), Dicer (200 kD) and mTOR (260 kD) [8].
3.3 Optimizing temperature and kit composition
Although 37 °C is the general default temperature for CFPS, and typically affords high yields, in some cases a large fraction of the protein produced is inactive. Lowering the temperature can increase the active fraction, at the cost of low overall yield. For example, 30 °C is a reasonable compromise in the case of a GFP [39]. Other proteins may have slightly different optimal temperatures. In addition, there are cases (see below) where it is desirable to supplement the CFPS kit by addition of, e.g., ions, small molecules, proteins, and/or nucleic acids [3,12,13,20,40–43]. Here it is helpful to employ a high throughput assay to measure the effects of such additions on the synthesis of a test protein that is easy to monitor. We have recently described such an assay that monitors the synthesis of fast maturing Emerald GFP (EmGFP) [17].
3.4 Batch vs. two compartment methods
In CFPS carried out in batch mode, the yield of translated protein per ml reaction may be limiting due to depletion of reactants, in particular oxygen in systems using oxidative phosphorylation for energy generation, and accumulation of inhibitory byproducts. Both of these problems have been successfully addressed using the so-called “thin film” approach in which the batch reaction mixture is placed on a thin film [44] (Fig. 4) that provides a large gas/liquid interface allowing for continuous oxygen transfer from the air, but also a large hydrophobic surface. This latter feature facilitates protein expression and folding by binding inhibitory hydrophobic molecules such as misfolded polypeptide chains, lipids, and various small molecules. This approach achieves high concentrations of active protein (as much as 0.5 mg/mL) in a format that can be scaled up without loss of yield, while allowing the use of high throughput format for reaction mixture optimization.
Figure 4. Batch vs. two compartment methods.

A batch mode reaction can be performed in a test-tube or in a well-plate amenable to a high throughput format.. Placing the batch reaction mixture on a thin film increases the rate of oxygen transfer from the air while the hydrophobic surface facilitates protein expression and folding [44]. Two compartment approaches allow substrate and byproduct to freely diffuse between two phases and either utilize a semipermeable membrane that separates modified cell extract from a substrate feeding solution that is continuously exchanged [45] or overlay the feeding substrate solution on top of the modified extract,[47]. The latter approach is also amenable to a high throughput format
An alternative approach employs two compartments, one containing the modified extract and one containing a feeding solution that includes substrates such as amino acids, ATP and GTP, and that is renewed by continuous flow, permitting substrate replenishment and byproduct removal. In early versions, the two compartments were separated either by a semi-permeable membrane [45] or by placing the lysate in a dialysis bag immersed in the feeding solution [26,46], while maintaining a continuous flow of the feeding solution. Although both approaches were capable of maintaining translation for tens of hours while producing tens of micromolar concentration of target proteins, their mechanical complexity prevented their use in a high throughput format. In a later methodology that overcomes this limitation, a bilayer is utilized in place of the semipermeable membrane. Gently overlaying the feeding solution on top of the modified extract layer allows free diffusion of substrates and byproducts between the two phases [47], without the need for continuous flow. Such an approach can be performed in well plates using volumes as low as 10 μl [17].
Larger scale reactions utilizing semi-permeable membranes are possible (e.g. [46] reporting 10 ml scale). Indeed, a recent study reported an industrial scale 100-liter CFPS that showed linear scalability (linear relation between total protein yield and reaction volume), producing 700 mg protein/L over 10 h, in a controlled environment bioreactor [48]. Further details about scale and yields are presented in Carlson et al. [49].
4. Applications of CFPS
4.1 Hard-to-express proteins
Difficulties in obtaining a functional protein from a host cell are quite common and can, in general, be attributed to one or more of the following problems: cytotoxicity, misfolding and aggregation.
4.1.1 Toxic proteins
Proteins that are toxic to the host cell are often difficult to express in high yields. CFPS can bypass such problems. Most generally, protein toxic for a particular organism with a lysate from another organism, e.g. expressing E. coli toxin in wheat germ extract [50–53]. A special case concerns proteins that inhibit transcription, which can be expressed by adding in vitro transcribed mRNA to a CFPS kit, effectively uncoupling transcription from translation.
4.1.2 Misfolded proteins
During protein over-expression in vivo, a misfolded protein may either accumulate or be degraded, in either case leading to a potential burden on the host cell and leaving the experimenter with an inactive translation product. Some of the possible causes of misfolding can be overcome by the use of CFPS. (1) Improper disulfide bonds may be formed during expression in the host cell due to an unfavorable redox environment. E. coli CFPS supplemented with various compounds that optimized the redox potential along with addition of DsbC (prokaryotic disulfide bond isomerase) significantly improved the yield, in E. coli CFPS, of the active forms of several eukaryotic proteins, including plasminogen activator, an antibody against hemagglutinin and a urokinase protease [12,13,43]. Commercial CFPS kits that support correct disulfide bond formation are available from companies such as New England Biolabs and 5-prime. (2) During the folding pathway, a protein may be trapped in a local minimum intermediate state. Supplementing the CFPS with chaperones may assist with correct folding [20,42]. In one study using E. coli CFPS to over-express several eukaryotic proteins, the kit was supplemented with a fusion protein formed between the ribosome-binding portion of the E. coli protein trigger factor, which docked the fusion protein to the exit port of the E. coli ribosome, and the eukaryotic chaperone BiP. Such supplementation assisted the nascent eukaryotic polypeptides in acquiring their functional folds while diminishing aggregation [54].
4.1.3 Aggregated proteins
During protein over-expression, partly folded and/or misfolded protein either still bound to the ribosome or free in the cytoplasm can undergo aggregation to form so-called inclusion bodies, most likely as a result of non-covalent hydrophobic interactions [55]. The flexibility of CFPS allows the facile addition of various compounds that prevent protein aggregation in CFPS kits derived from E. coli. The compounds proven useful to alleviate aggregation in vitro include: polyethylene glycol, polysaccharide nanogel, ethanol, choline, and amino acids such as Arg, Pro, and Glu [40,41].
4.2 Ribosome replacement
Removal of endogenous ribosomes from CFPS and their replacement with exogenous ribosomes is easily performed. In lysates, an ultracentrifugation step is required [17], while in PURExpress a solution lacking ribosomes is used. Examples of such replacements include: fluorescently labeled ribosomes for single molecule experiments [17], and ribosomes containing mutations in ribosomal RNA (rRNA) [56,57] that may affect their activities. CFPS allows straightforward study of the properties of variant ribosomes that may be difficult to determine in live cells, since such variants may increase the burden on the host cell by inducing delayed growth rates, stress and even death. Examples of mutated ribosomes whose translation activities would be interesting to study via substitution into CFPS include hyperaccurate [58] and error prone [59] ribosomes that affect translation fidelity, orthogonal ribosomes (o-ribosomes) [60] recognizing specialized sets of o-mRNAs or o-tRNAs, biotinylated ribosomes [61] and ribosomes containing loop extensions within rRNA [62,63] that are used for purposes such as fluorescent labeling, surface/bead attachment or purification.
4.3 Preparation of stalled ribosome-nascent chain (RNC) complexes
RNC complexes, which also stably bind mRNA, are desirable for ribosome display [64], as substrates of chaperones for studies of co-translational protein folding [65] and for quantification of the fraction of ribosomes active in protein synthesis within a ribosome preparation [17]. Expressing RNC complexes in cells may induce stress, as they abrogate protein synthesis. CFPS permits rapid isolation of stalled RNC complexes, while minimizing proteolytic and nucleolytic cleavages of the bound nascent chain and mRNA, respectively.
4.4. Amino Acid replacement
4.4.1 Obtaining proteins with non-natural amino acids (nnAAs)
The 20 natural amino acids that serve as building blocks of proteins offer a limited repertoire of properties for the design of proteins that can be used for research and engineering purposes. This limitation can be overcome by the site-specific incorporation of nnAAs, which include a much more diverse array of functional side chains (reviewed by [66]). nnAAs are typically inserted site-specifically by exploitation of the amber stop codon, which is normally targeted by release factor 1 (RF1) for termination of protein synthesis, using suppressor tRNAs to read-through the amber codon. Orthogonal aminoacyl-tRNA synthetases (o-aaRSs) are engineered to aminoacylate suppressor tRNAs with a nnAA [67,68]. The orthogonality of the suppressor tRNA (o-tRNA) and the o-aaRS is manifested by lack of cross-talk with endogenous tRNAs and aaRSs. In order to obtain in vivo incorporation of nnAA, the culture medium is supplemented with the relevant nnAA and the bacterial strain is transfected with i) a single plasmid encoding both the o-tRNA and the o-aaRS and ii) another plasmid encoding the protein of interest that has been mutated to contain an amber stop codon located at the desired nnAA incorporation site. Upon induction, the over-expressed protein is either terminated at the amber stop codon by RF1, or is read-through by the nnAA-o-tRNA. To bias the competition so as to improve incorporation efficiency and full protein production, RF1 activity can be inhibited, using, for example, a targeted antibody [69] or RNA aptamer [70]. Incorporation of nnAA using CFPS proceeds with higher efficiency over the reported in vivo procedure. One reason is that competition between RF1 and the aa-o-tRNA in CFPS is much less than in in vivo expression systems, since the protein synthesis apparatus is much more dilute, some 20-fold in an optimized CFPS kit derived from E. coli [14]. This reduces the RF1 concentration from ~8 μM (in vivo) to 0.4 μM (in CFPS). Another reason may be ascribed to the large volume utilized for culture growth vs. relatively smaller volumes utilized for CFPS. The high cost of synthetic nnAAs favors the use of low volume CFPS as it allows relatively high nnAA concentrations to be employed, thereby increasing the rate and yields of labeled protein expression. Such high concentrations are sometimes too costly to be used in when protein is produced by culture growth.
In the case of superfolder GFP, high nnAA incorporation efficiency is achieved by supplementing the CFPS kit with high concentrations of nnAA and o-aaRS [71] This latter study well illustrates the advantages of the CFPS approach for nnAA incorporation, which yielded, per ml culture, 0.9–1.7 mg protein harboring a single p-azido-L-phenylalanine or p-propargyloxy-L-phenylalanine, with an incorporation efficiency of 50–88%, depending on the position of incorporation [71]. By comparison, in vivo expression of EGFP using a specialized RF1-knockout E. coli strain JX33 [72] that eliminates the competition between RF-1 and the amber tRNA repressor, produced, per ml culture, 3.5 μg protein containing one or two p-acetyl-L-phenylalanine (pActF) and 5.4 μg protein containing three pActFs, corresponding to a nnAA incorporation efficiency in the range 23–36%. Given the generally high costs of synthetic nnAAs, the much higher yield of nnAA-containing protein per ml of solution is an important advantage of the CFPS over the in vivo approach.
4.4.2. Obtaining proteins with isotope-labeled amino acids
Isotope-labeled amino acids allow detection of radioactive proteins, using unstable isotopes, and FT-IR and NMR characterization of proteins containing stable isotopes. For purposes of site-specific labeling, auxotrophic cells are grown in the presence of isotopically labeled amino acid. However, due to in vivo metabolic activity, some fraction of the labeled amino acids is converted to other amino acids, leading to scrambling. CFPS minimizes scrambling, since some metabolic pathways are compromised during cell extract preparation and specific metabolic inhibitors can be added to the CFPS kit [73]. Examples of expression of isotope-labeled proteins in CFPS are reviewed by [74,75].
A novel procedure was recently reported [17], making protein expression completely dependent on the addition of a specific exogenous amino acid, Phe in the example described. In this procedure all endogenous amino acids are removed from a lysate by dialysis in the presence of two inhibitors: (1) a general protease inhibitor cocktail that suppresses proteolytic cleavages that produce free amino acids and (2) a specific PheRS inhibitor that suppresses aminoacylation of tRNAPhe by free Phe. Such depleted lysates are then reconstituted with an amino acids mixture lacking Phe, such that the CFPS solution is incapable of expressing the test protein, EmGFP. Addition of increasing concentrations of Phe, resulted in EmGFP expression, with a maximum level seen at 80 μM of added Phe (Fig. 5). The total dependence of EmGFP expression on added Phe means that this procedure could be used to produce proteins that are homogeneously labeled with an isotopically labeled form of Phe.
Figure 5.

Cell-free expression of EmGFP using lysate depleted of Phe, supplemented with the amino acid Phe. Continuous fluorescence monitoring was performed as in Figure 2. EmGFP accumulation was measured as a function of added [Phe]. The figure was obtained from [17].
4.5. tRNA replacement
Substitution of endogenous aminoacyl-tRNA (aa-tRNA) by an exogenously added counterpart can be important for protein synthesis studies focused on modified amino acid (e.g. isotope-labeled or non-natural) incorporation and/or modified tRNA (e.g., mutated, chemically/enzymatically modified, fluorescently labeled) interaction with the ribosome.
Replacing the majority of the endogenous tRNA pool in a CFPS kit with exogenous tRNA was previously reported [76,77]. A specific isoacceptor tRNA can also be selectively removed from bulk tRNA and replaced with a corresponding modified aa-tRNA [78]. Alternatively, a CFPS kit can be selectively depleted for a specific amino acid (Sect. 4.3.2) such that reconstitution of the translation activity is totally dependent on the addition of the amino acid in the form of aa-tRNAaa. Figure 6 is an example of such an approach. Here, EmGFP expression reached a maximum at 8 μM of added Phe-tRNAPhe. This approach, was applied to test the activities of fluorescent-labeled tRNAs [17] that are used in single-molecule experiments examining regulatory mechanisms of protein synthesis [31]. It could also be used for testing the activities of other modified aa-tRNAaas, such as those charged with non-natural amino acids, or lacking specific posttranscriptional modifications [79].
Figure 6.
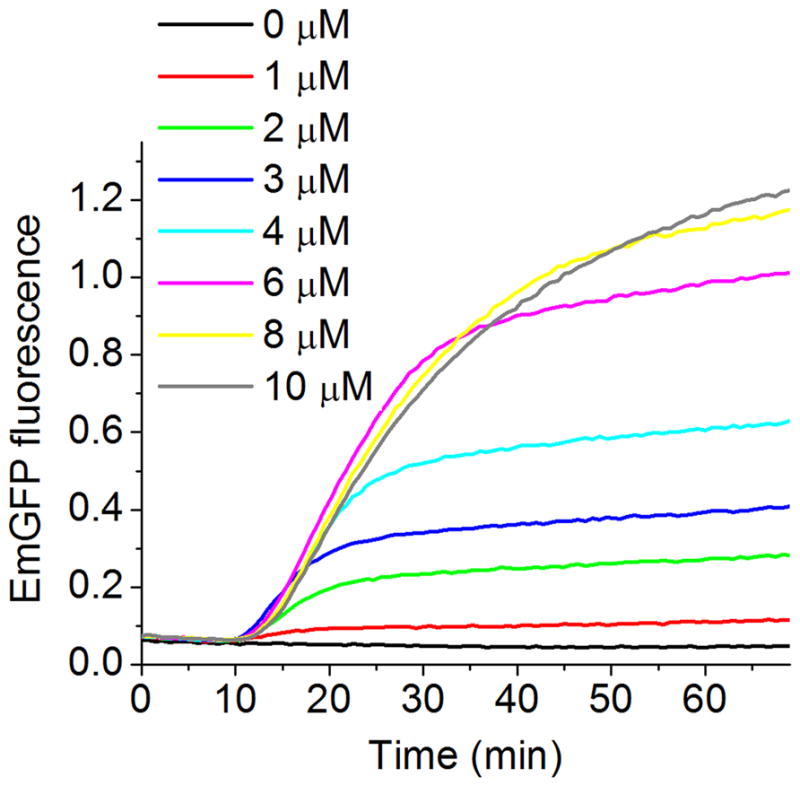
Cell-free expression of EmGFP using lysate depleted of Phe, supplemented with purified Phe-tRNAPhe. Continuous fluorescence monitoring was performed as in Fig. 2. EmGFP accumulation was measured as a function of added [Phe-tRNAPhe]. The figure was obtained from [17].
5. Conclusions and Outlook
The first CFPS system was reported in the 1950s by [80] and was later employed in the seminal work of Nirenberg and Matthaei, in the 1960s [2], as the tool that allowed deciphering the genetic code. Since then, many improvements in CFPS had been reported, directed toward both increasing expression yields and the inclusion of new functions and capabilities. The open nature of CFPS allows both changes of the concentrations of various components of the translation machinery so as to optimize functional protein synthesis and replacement of endogenous components with exogenous components (e.g., isotope-labeled and non-natural amino acids, fluorescent labeled tRNAs, mutant ribosomes) that permit many useful applications. At present, such manipulations are typically performed one at a time, but it is to be expected that future studies will increasingly involve combinations, e.g., supplementing an E. coli system that efficiently incorporates nnAAs with disulfide bond isomerases, and chaperones to insure that the modified protein is properly folded.
A particularly attractive goal for future work would be to develop a bacterial-eukaryotic CFPS hybrid to allow eukaryotic protein expression to be carried out by the more efficient bacterial translation machinery by substitution/supplementation of the bacterial machinery with eukaryotic tRNAs, aaRSs, chaperones and post-translation modification enzymes and provision of an appropriate redox environment. Ultimately, one can envision that commercial firms will offer CFPS kits tailored to meet the needs of individual scientists that will replace current protocols for protein expression that can be quite laborious. In this idealized scenario, a scientist will add a DNA coding region, amplified by PCR or ligated in a plasmid, to a customized CFPS, that will produce any protein in large quantities with high functional activity, and with amino acids that are either naturally occurring, isotopically labeled or non-natural.
Acknowledgments
This work was supported by NIH grant GM080376 to BSC and a Human Frontier Science Program postdoctoral fellowship to GR.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.DeVries JK, Zubay G. DNA-directed peptide synthesis. II. The synthesis of the alpha-fragment of the enzyme beta-galactosidase. Proc Natl Acad Sci U S A. 1967;57:1010–2. doi: 10.1073/pnas.57.4.1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nirenberg MW, Matthaei JH. The dependence of cell-free protein synthesis in E. coli upon naturally occurring or synthetic polyribonucleotides. Proc Natl Acad Sci U S A. 1961;47:1588–602. doi: 10.1073/pnas.47.10.1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zubay G. In vitro synthesis of protein in microbial systems. Annu Rev Genet. 1973;7:267–87. doi: 10.1146/annurev.ge.07.120173.001411. [DOI] [PubMed] [Google Scholar]
- 4.Mureev S, Kovtun O, Nguyen UT, Alexandrov K. Species-independent translational leaders facilitate cell-free expression. Nat Biotechnol. 2009;27:747–52. doi: 10.1038/nbt.1556. [DOI] [PubMed] [Google Scholar]
- 5.Ezure T, et al. Cell-free protein synthesis system prepared from insect cells by freeze-thawing. Biotechnol Prog. 2006;22:1570–7. doi: 10.1021/bp060110v. [DOI] [PubMed] [Google Scholar]
- 6.Sawasaki T, Morishita R, Gouda MD, Endo Y. Methods for high-throughput materialization of genetic information based on wheat germ cell-free expression system. Methods Mol Biol. 2007;375:95–106. doi: 10.1007/978-1-59745-388-2_5. [DOI] [PubMed] [Google Scholar]
- 7.Pelham HR, Jackson RJ. An efficient mRNA-dependent translation system from reticulocyte lysates. Eur J Biochem. 1976;67:247–56. doi: 10.1111/j.1432-1033.1976.tb10656.x. [DOI] [PubMed] [Google Scholar]
- 8.Mikami S, Kobayashi T, Masutani M, Yokoyama S, Imataka H. A human cell-derived in vitro coupled transcription/translation system optimized for production of recombinant proteins. Protein Expr Purif. 2008;62:190–8. doi: 10.1016/j.pep.2008.09.002. [DOI] [PubMed] [Google Scholar]
- 9.Kim DM, Swartz JR. Prolonging cell-free protein synthesis with a novel ATP regeneration system. Biotechnol Bioeng. 1999;66:180–8. [PubMed] [Google Scholar]
- 10.Michel-Reydellet N, Woodrow K, Swartz J. Increasing PCR fragment stability and protein yields in a cell-free system with genetically modified Escherichia coli extracts. J Mol Microbiol Biotechnol. 2005;9:26–34. doi: 10.1159/000088143. [DOI] [PubMed] [Google Scholar]
- 11.Jiang X, Oohira K, Iwasaki Y, Nakano H, Ichihara S, Yamane T. Reduction of protein degradation by use of protease-deficient mutants in cell-free protein synthesis system of Escherichia coli. J Biosci Bioeng. 2002;93:151–6. doi: 10.1263/jbb.93.151. [DOI] [PubMed] [Google Scholar]
- 12.Yin G, Swartz JR. Enhancing multiple disulfide bonded protein folding in a cell-free system. Biotechnol Bioeng. 2004;86:188–95. doi: 10.1002/bit.10827. [DOI] [PubMed] [Google Scholar]
- 13.Kim DM, Swartz JR. Efficient production of a bioactive, multiple disulfide-bonded protein using modified extracts of Escherichia coli. Biotechnol Bioeng. 2004;85:122–9. doi: 10.1002/bit.10865. [DOI] [PubMed] [Google Scholar]
- 14.Underwood KA, Swartz JR, Puglisi JD. Quantitative polysome analysis identifies limitations in bacterial cell-free protein synthesis. Biotechnol Bioeng. 2005;91:425–35. doi: 10.1002/bit.20529. [DOI] [PubMed] [Google Scholar]
- 15.Mikamo-Satoh E, Takagi A, Tanaka H, Matsumoto T, Nishihara T, Kawai T. Profiling of gene-dependent translational progress in cell-free protein synthesis by real-space imaging. Anal Biochem. 2009;394:275–80. doi: 10.1016/j.ab.2009.07.033. [DOI] [PubMed] [Google Scholar]
- 16.Tsien RY. The green fluorescent protein. Annu Rev Biochem. 1998;67:509–44. doi: 10.1146/annurev.biochem.67.1.509. [DOI] [PubMed] [Google Scholar]
- 17.Rosenblum G, Chen C, Kaur J, Cui X, Goldman YE, Cooperman BS. Real-time assay for testing components of protein synthesis. Nucleic Acids Res. 2012;40:e88. doi: 10.1093/nar/gks232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shin J, Noireaux V. Study of messenger RNA inactivation and protein degradation in an Escherichia coli cell-free expression system. J Biol Eng. 2010;4:9. doi: 10.1186/1754-1611-4-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang HL, Ivashkiv L, Chen HZ, Zubay G, Cashel M. Cell-free coupled transcription-translation system for investigation of linear DNA segments. Proc Natl Acad Sci U S A. 1980;77:7029–33. doi: 10.1073/pnas.77.12.7029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jun SY, Kang SH, Lee KH. Continuous-exchange cell-free protein synthesis using PCR-generated DNA and an RNase E-deficient extract. Biotechniques. 2008;44:387–91. doi: 10.2144/000112690. [DOI] [PubMed] [Google Scholar]
- 21.Kralicek AV, Radjainia M, Mohamad Ali NA, Carraher C, Newcomb RD, Mitra AK. A PCR-directed cell-free approach to optimize protein expression using diverse fusion tags. Protein Expr Purif. 2011;80:117–24. doi: 10.1016/j.pep.2011.06.006. [DOI] [PubMed] [Google Scholar]
- 22.Wu PS, Ozawa K, Lim SP, Vasudevan SG, Dixon NE, Otting G. Cell-free transcription/translation from PCR-amplified DNA for high-throughput NMR studies. Angew Chem Int Ed Engl. 2007;46:3356–8. doi: 10.1002/anie.200605237. [DOI] [PubMed] [Google Scholar]
- 23.Shimizu Y, Inoue A, Tomari Y, Suzuki T, Yokogawa T, Nishikawa K, Ueda T. Cell-free translation reconstituted with purified components. Nat Biotechnol. 2001;19:751–5. doi: 10.1038/90802. [DOI] [PubMed] [Google Scholar]
- 24.Asahara H, Chong S. In vitro genetic reconstruction of bacterial transcription initiation by coupled synthesis and detection of RNA polymerase holoenzyme. Nucleic Acids Res. 2010;38:e141. doi: 10.1093/nar/gkq377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhou Y, Asahara H, Gaucher EA, Chong S. Reconstitution of translation from Thermus thermophilus reveals a minimal set of components sufficient for protein synthesis at high temperatures and functional conservation of modern and ancient translation components. Nucleic Acids Res. 2012;40:7932–45. doi: 10.1093/nar/gks568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Madin K, Sawasaki T, Ogasawara T, Endo Y. A highly efficient and robust cell-free protein synthesis system prepared from wheat embryos: plants apparently contain a suicide system directed at ribosomes. Proc Natl Acad Sci U S A. 2000;97:559–64. doi: 10.1073/pnas.97.2.559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kariko K, Muramatsu H, Welsh FA, Ludwig J, Kato H, Akira S, Weissman D. Incorporation of pseudouridine into mRNA yields superior nonimmunogenic vector with increased translational capacity and biological stability. Mol Ther. 2008;16:1833–40. doi: 10.1038/mt.2008.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bugaut A, Balasubramanian S. 5′-UTR RNA G-quadruplexes: translation regulation and targeting. Nucleic Acids Res. 2012;40:4727–41. doi: 10.1093/nar/gks068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mandal M, Breaker RR. Adenine riboswitches and gene activation by disruption of a transcription terminator. Nat Struct Mol Biol. 2004;11:29–35. doi: 10.1038/nsmb710. [DOI] [PubMed] [Google Scholar]
- 30.Mandal M, Lee M, Barrick JE, Weinberg Z, Emilsson GM, Ruzzo WL, Breaker RR. A glycine-dependent riboswitch that uses cooperative binding to control gene expression. Science. 2004;306:275–9. doi: 10.1126/science.1100829. [DOI] [PubMed] [Google Scholar]
- 31.Rosenblum G, et al. Quantifying Elongation Rhythm during Full-Length Protein Synthesis. J Am Chem Soc. 2013;135:11322–9. doi: 10.1021/ja405205c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Winkler WC, Nahvi A, Roth A, Collins JA, Breaker RR. Control of gene expression by a natural metabolite-responsive ribozyme. Nature. 2004;428:281–6. doi: 10.1038/nature02362. [DOI] [PubMed] [Google Scholar]
- 33.Yanofsky C. Attenuation in the control of expression of bacterial operons. Nature. 1981;289:751–8. doi: 10.1038/289751a0. [DOI] [PubMed] [Google Scholar]
- 34.Shine J, Dalgarno L. The 3′-terminal sequence of Escherichia coli 16S ribosomal RNA: complementarity to nonsense triplets and ribosome binding sites. Proc Natl Acad Sci U S A. 1974;71:1342–6. doi: 10.1073/pnas.71.4.1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dreyfus M. What constitutes the signal for the initiation of protein synthesis on Escherichia coli mRNAs? J Mol Biol. 1988;204:79–94. doi: 10.1016/0022-2836(88)90601-8. [DOI] [PubMed] [Google Scholar]
- 36.Vimberg V, Tats A, Remm M, Tenson T. Translation initiation region sequence preferences in Escherichia coli. BMC Mol Biol. 2007;8:100. doi: 10.1186/1471-2199-8-100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jang SK, Krausslich HG, Nicklin MJ, Duke GM, Palmenberg AC, Wimmer E. A segment of the 5′ nontranslated region of encephalomyocarditis virus RNA directs internal entry of ribosomes during in vitro translation. J Virol. 1988;62:2636–43. doi: 10.1128/jvi.62.8.2636-2643.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pelletier J, Sonenberg N. Internal initiation of translation of eukaryotic mRNA directed by a sequence derived from poliovirus RNA. Nature. 1988;334:320–5. doi: 10.1038/334320a0. [DOI] [PubMed] [Google Scholar]
- 39.Iskakova MB, Szaflarski W, Dreyfus M, Remme J, Nierhaus KH. Troubleshooting coupled in vitro transcription-translation system derived from Escherichia coli cells: synthesis of high-yield fully active proteins. Nucleic Acids Res. 2006;34:e135. doi: 10.1093/nar/gkl462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kai L, Dotsch V, Kaldenhoff R, Bernhard F. Artificial environments for the co-translational stabilization of cell-free expressed proteins. PLoS One. 2013;8:e56637. doi: 10.1371/journal.pone.0056637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sasaki Y, Asayama W, Niwa T, Sawada S, Ueda T, Taguchi H, Akiyoshi K. Amphiphilic polysaccharide nanogels as artificial chaperones in cell-free protein synthesis. Macromol Biosci. 2011;11:814–20. doi: 10.1002/mabi.201000457. [DOI] [PubMed] [Google Scholar]
- 42.Niwa T, Kanamori T, Ueda T, Taguchi H. Global analysis of chaperone effects using a reconstituted cell-free translation system. Proc Natl Acad Sci U S A. 2012;109:8937–42. doi: 10.1073/pnas.1201380109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ryabova LA, Desplancq D, Spirin AS, Pluckthun A. Functional antibody production using cell-free translation: effects of protein disulfide isomerase and chaperones. Nat Biotechnol. 1997;15:79–84. doi: 10.1038/nbt0197-79. [DOI] [PubMed] [Google Scholar]
- 44.Voloshin AM, Swartz JR. Efficient and scalable method for scaling up cell free protein synthesis in batch mode. Biotechnol Bioeng. 2005;91:516–21. doi: 10.1002/bit.20528. [DOI] [PubMed] [Google Scholar]
- 45.Spirin AS, Baranov VI, Ryabova LA, Ovodov SY, Alakhov YB. A continuous cell-free translation system capable of producing polypeptides in high yield. Science. 1988;242:1162–4. doi: 10.1126/science.3055301. [DOI] [PubMed] [Google Scholar]
- 46.Kigawa T, Yokoyama S. A continuous cell-free protein synthesis system for coupled transcription-translation. J Biochem. 1991;110:166–8. doi: 10.1093/oxfordjournals.jbchem.a123551. [DOI] [PubMed] [Google Scholar]
- 47.Sawasaki T, Hasegawa Y, Tsuchimochi M, Kamura N, Ogasawara T, Kuroita T, Endo Y. A bilayer cell-free protein synthesis system for high-throughput screening of gene products. FEBS Lett. 2002;514:102–5. doi: 10.1016/s0014-5793(02)02329-3. [DOI] [PubMed] [Google Scholar]
- 48.Zawada JF, et al. Microscale to manufacturing scale-up of cell-free cytokine production--a new approach for shortening protein production development timelines. Biotechnol Bioeng. 2011;108:1570–8. doi: 10.1002/bit.23103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Carlson ED, Gan R, Hodgman CE, Jewett MC. Cell-free protein synthesis: applications come of age. Biotechnol Adv. 2011;30:1185–94. doi: 10.1016/j.biotechadv.2011.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Watanabe M, Miyazono K, Tanokura M, Sawasaki T, Endo Y, Kobayashi I. Cell-free protein synthesis for structure determination by X-ray crystallography. Methods Mol Biol. 2010;607:149–60. doi: 10.1007/978-1-60327-331-2_13. [DOI] [PubMed] [Google Scholar]
- 51.Xu Z, Chen H, Yin X, Xu N, Cen P. High-level expression of soluble human beta-defensin-2 fused with green fluorescent protein in Escherichia coli cell-free system. Appl Biochem Biotechnol. 2005;127:53–62. doi: 10.1385/abab:127:1:053. [DOI] [PubMed] [Google Scholar]
- 52.Renesto P, Raoult D. From genes to proteins: in vitro expression of rickettsial proteins. Ann N Y Acad Sci. 2003;990:642–52. doi: 10.1111/j.1749-6632.2003.tb07439.x. [DOI] [PubMed] [Google Scholar]
- 53.Martemyanov KA, Shirokov VA, Kurnasov OV, Gudkov AT, Spirin AS. Cell-free production of biologically active polypeptides: application to the synthesis of antibacterial peptide cecropin. Protein Expr Purif. 2001;21:456–61. doi: 10.1006/prep.2001.1400. [DOI] [PubMed] [Google Scholar]
- 54.Welsh JP, Bonomo J, Swartz JR. Localization of BiP to translating ribosomes increases soluble accumulation of secreted eukaryotic proteins in an Escherichia coli cell-free system. Biotechnol Bioeng. 2011;108:1739–48. doi: 10.1002/bit.23111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fink AL. Protein aggregation: folding aggregates, inclusion bodies and amyloid. Fold Des. 1998;3:R9–23. doi: 10.1016/S1359-0278(98)00002-9. [DOI] [PubMed] [Google Scholar]
- 56.Jewett MC, Fritz BR, Timmerman LE, Church GM. In vitro integration of ribosomal RNA synthesis, ribosome assembly, and translation. Mol Syst Biol. 2013;9:678. doi: 10.1038/msb.2013.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Vazquez-Laslop N, Ramu H, Klepacki D, Kannan K, Mankin AS. The key function of a conserved and modified rRNA residue in the ribosomal response to the nascent peptide. Embo J. 2010;29:3108–17. doi: 10.1038/emboj.2010.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gorini L, Kataja E. Phenotypic Repair by Streptomycin of Defective Genotypes in E. Coli. Proc Natl Acad Sci U S A. 1964;51:487–93. doi: 10.1073/pnas.51.3.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rosset R, Gorini L. A ribosomal ambiguity mutation. J Mol Biol. 1969;39:95–112. doi: 10.1016/0022-2836(69)90336-2. [DOI] [PubMed] [Google Scholar]
- 60.Rackham O, Chin JW. A network of orthogonal ribosome x mRNA pairs. Nat Chem Biol. 2005;1:159–66. doi: 10.1038/nchembio719. [DOI] [PubMed] [Google Scholar]
- 61.Katranidis A, et al. Fast biosynthesis of GFP molecules: a single-molecule fluorescence study. Angew Chem Int Ed Engl. 2009;48:1758–61. doi: 10.1002/anie.200806070. [DOI] [PubMed] [Google Scholar]
- 62.Dorywalska M, Blanchard SC, Gonzalez RL, Kim HD, Chu S, Puglisi JD. Site-specific labeling of the ribosome for single-molecule spectroscopy. Nucleic Acids Res. 2005;33:182–9. doi: 10.1093/nar/gki151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Petrov A, Puglisi JD. Site-specific labeling of Saccharomyces cerevisiae ribosomes for single-molecule manipulations. Nucleic Acids Res. 2010;38:e143. doi: 10.1093/nar/gkq390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hanes J, Pluckthun A. In vitro selection and evolution of functional proteins by using ribosome display. Proc Natl Acad Sci U S A. 1997;94:4937–42. doi: 10.1073/pnas.94.10.4937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Evans MS, Ugrinov KG, Frese MA, Clark PL. Homogeneous stalled ribosome nascent chain complexes produced in vivo or in vitro. Nat Methods. 2005;2:757–62. doi: 10.1038/nmeth790. [DOI] [PubMed] [Google Scholar]
- 66.Liu CC, Schultz PG. Adding new chemistries to the genetic code. Annu Rev Biochem. 2010;79:413–44. doi: 10.1146/annurev.biochem.052308.105824. [DOI] [PubMed] [Google Scholar]
- 67.Wang L, Brock A, Herberich B, Schultz PG. Expanding the genetic code of Escherichia coli. Science. 2001;292:498–500. doi: 10.1126/science.1060077. [DOI] [PubMed] [Google Scholar]
- 68.Wang L, Schultz PG. Expanding the genetic code. Angew Chem Int Ed Engl. 2004;44:34–66. doi: 10.1002/anie.200460627. [DOI] [PubMed] [Google Scholar]
- 69.Agafonov DE, Huang Y, Grote M, Sprinzl M. Efficient suppression of the amber codon in E. coli in vitro translation system. FEBS Lett. 2005;579:2156–60. doi: 10.1016/j.febslet.2005.03.004. [DOI] [PubMed] [Google Scholar]
- 70.Sando S, Ogawa A, Nishi T, Hayami M, Aoyama Y. In vitro selection of RNA aptamer against Escherichia coli release factor 1. Bioorg Med Chem Lett. 2007;17:1216–20. doi: 10.1016/j.bmcl.2006.12.013. [DOI] [PubMed] [Google Scholar]
- 71.Albayrak C, Swartz JR. Cell-free co-production of an orthogonal transfer RNA activates efficient site-specific non-natural amino acid incorporation. Nucleic Acids Res. 2013;41:5949–63. doi: 10.1093/nar/gkt226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Johnson DB, et al. RF1 knockout allows ribosomal incorporation of unnatural amino acids at multiple sites. Nat Chem Biol. 2011;7:779–86. doi: 10.1038/nchembio.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Su XC, Loh CT, Qi R, Otting G. Suppression of isotope scrambling in cell-free protein synthesis by broadband inhibition of PLP enymes for selective 15N-labelling and production of perdeuterated proteins in H2O. J Biomol NMR. 2011;50:35–42. doi: 10.1007/s10858-011-9477-5. [DOI] [PubMed] [Google Scholar]
- 74.Ozawa K, Wu PS, Dixon NE, Otting G. N-Labelled proteins by cell-free protein synthesis. Strategies for high-throughput NMR studies of proteins and protein-ligand complexes. Febs J. 2006;273:4154–9. doi: 10.1111/j.1742-4658.2006.05433.x. [DOI] [PubMed] [Google Scholar]
- 75.Takeda M, Kainosho M. Cell-free protein synthesis using E. coli cell extract for NMR studies. Adv Exp Med Biol. 2012;992:167–77. doi: 10.1007/978-94-007-4954-2_9. [DOI] [PubMed] [Google Scholar]
- 76.Ahn JH, Hwang MY, Oh IS, Park KM, Hahn GH, Cho CY, Kim DM. Preparation method for Escherichia coliS30 extracts completely dependent upon tRNA addition to catalyze cell-free protein synthesis. Biotechnology and Bioprocess Engineering. 2006;11:420–424. [Google Scholar]
- 77.Jackson RJ, Napthine S, Brierley I. Development of a tRNA-dependent in vitro translation system. RNA. 2001;7:765–73. doi: 10.1017/s1355838201002539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yokogawa T, Kitamura Y, Nakamura D, Ohno S, Nishikawa K. Optimization of the hybridization-based method for purification of thermostable tRNAs in the presence of tetraalkylammonium salts. Nucleic Acids Res. 2010;38:e89. doi: 10.1093/nar/gkp1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Phizicky EM, Alfonzo JD. Do all modifications benefit all tRNAs? FEBS Lett. 2010;584:265–71. doi: 10.1016/j.febslet.2009.11.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hoagland MB, Stephenson ML, Scott JF, Hecht LI, Zamecnik PC. A soluble ribonucleic acid intermediate in protein synthesis. J Biol Chem. 1958;231:241–57. [PubMed] [Google Scholar]