

Abstract
Neuroendocrine tumors (NET) previously called carcinoid tumors are neoplasms of enterochromaffin/neuroendocrine cell origin which display neurosecretory capacity that may result in the carcinoid syndrome. The annual incidence of patients with NET is 8.4 per 100000; yet many NET remain asymptomatic and clinically undetected. A majority of NET follows a benign course; however, some will display malignant characteristics. NET most commonly occur in the gastrointestinal tract (67%) and bronchopulmonary system (25%). Gastrointestinal NET occur within the stomach, small intestine, liver, and rectum. We report a retrospective study of 11 subjects: Eight with benign carcinoid tumors: duodenal bulb (n = 2), terminal ileum (n = 1), sigmoid colon (n = 2), and rectum (n = 3); three with malignant carcinoid: liver (n = 1) and intra-abdominal site (n = 2). The diagnosis, endoscopic images, outcome, treatment and review of the literature are presented.
Keywords: Neuroendocrine, Carcinoid, Gastrointestinal, Tumors
Core tip: Endoscopic procedures sometimes reveal submucosal lesions within the gastrointestinal tract that are resected and confirmed as neuroendocrine tumors by appropriate immunochemical stains. Most will be benign as demonstrated in our series of 11 subjects. This case series of gastrointestinal neuroendocrine tumors reminds every endoscopist to carefully examine the upper and lower gastrointestinal tract for such lesions.
INTRODUCTION
Historically described as a more indolent behaving tumor than adenocarcinoma by Oberndorfer in Germany in 1907, neuroendocrine (carcinoid) tumors (NET) are undergoing a location change within the gastrointestinal tract[1-4]. A shift in the anatomic location has occurred over the last half-century. Data from 1950 to 1971 identified the appendix as the most common site followed by rectal and ileum for NET[4]. However, a recent evaluation of carcinoid tumors identified in the Surveillance, Epidemiology and End Results Program between 1973 and 1999 found the ileum to be the most frequent site of gastrointestinal NET followed by the rectum; the appendix accounted for only 4.8% of NET[4]. Additionally, gastric NET accounted for an increasing proportion of gastrointestinal NET[4,5]. This change in location of NET has resulted from changes in diagnostic modalities used as well as reporting techniques over time[6]. The estimated incidence in the United States ranges from 2.5-5 cases per 100000[4]. A European investigation which included both surgical and autopsy specimens, reported an overall incidence of 8.4 cases per 100000[4,7,8]. Incidence estimates are limited by the clinically silent nature of many NET which remain undetected until autopsy[6].
CASE REPORT
This case series describes a wide spectrum of benign gastrointestinal NET originating in the small intestine (n = 2), terminal ileum (n = 1), colon (n = 2), rectum (n = 3), malignant NET of the liver (n = 1) and intraabdominal sites (n = 2) (Table 1).
Table 1.
Clinical data of patients with neuroendocrine tumors
| Patient (age, yr/sex) | Initial evaluation | Site | Diagnostic studies | Outcome |
| 65/F | Hematochezia, IBD epigastric pain | Duodenal bulb | 12 21 05 EGD duodenal bulb polyp; path: neuroendocrine tumor 12 30 05 repeat EGD, no residual, path: neuroendocrine tumor 11 24 08 repeat EGD no recurrence, COL mucosal prolapse syndrome | Alive and well |
| 59/M | GERD with break-through symptoms | Duodenal bulb | 11 11 08 EGD duodenal bulb polyp, path: neuroendocrine tumor 12 22 08 EGD, no residual tumor 12 30 08 PET scan negative | Alive and well |
| 50/F | 2nd opinion for liver metastatic disease | Liver | 02 09 04 EGD chronic esophagitis, HH, fundic nodularity, path: benign lymphoid aggregates 03 16 04 PET/CT innumberable larg hepatic lesions replacing R and L lobes consistent with neuroendocrine tumor | Expired 12 04 |
| 70/M | Epigastric pain and 15 lb weight loss | Intra-abdominal | 04 15 08 EGD chronic esophagitis, HH, acute and chronic gastritis; path: reactive gastropathy; COL: 1 adenomatous/2 hyperplastic polyps 04 16 08 CT Abd/Pelvis mesen-teric mass 04 24 08 CT guided bx: path: neuroendocrine tumor | 05 08 treated with sandostatin |
| 46/F | Nausea, vomiting, abdominal pain | Intra-abdominal | 01 02 10 CT Abd/Pelvis ascites small bowel and colonic obstruction 01 04 10 Gastrografin emema sigmoid Obstruction 01 04 10 exploratory laparotomy desmoplastic reaction, sigmoid colon with liver metastases and intraperitoneal implants; bx of implants positive for chromogranin and synapotophysin 01 19 10 COL 3 cm stenosis at 30 cm due to extrinsic pressure; stent placed 01 26 10 serum CGA, 27 nmole/L | Discharge To hospice |
| 40/M | Recurrent perianal abscess r/o IBD | Terminal ileum | 12 05 06 COL 10 mm sessile polyp in terminal ileum, path: neuroendocrine tumor | Lost to follow-up |
| 50/F | GERD and CRCS | Sigmoid | 04 04 08 EGD chronic esophagitis, HH, path: mild reactive gastropathy, COL 4 mm sigmoid neuroendocrine tumor resected 04 30 08 normal octreotide scan 03 30 09 COL negative bx at prior polypectomy site | Alive and well |
| 75/F | Breast cancer and CRCS | Sigmoid | 02 06 08 COL 7 mm sigmoid submucosal nodule resected; cells positive for synaptophysin, but negative for chromogranin 03 11 08 Urinary 5-HIAA negative 04 22 08 Repeat COL with resection of remaining neuroendocrine tumor 05 19 09 COL negative for recurrence | Alive and well |
| 55/M | LLQ tenderness, CRCS | Rectum | 08 22 06 COL sigmoid tubulovillous adenoma and 6 mm rectal neuro-endocrine tumor 09 01 09 COL hyperplastic polyp, no recurrence of neuroendocrine tumor | Alive and well |
| 55/F | CRCS | Rectum | 05 01 09 COL 8 mm neuroendocrine tumor COL 1 yr later no recurrence | Alive and well |
| 60/F | CRCS | Rectum | 11 29 07 COL submucosal nodule neuroendocrine tumor 01 28 08 COL no recurrence | Alive and well |
IBD: Inflammatory bowel disease; CGA: Chromogranin A; EGD: Esophagoduodenoscopy; COL: Colonoscopy; GERD: Gastroesophageal reflux disease; HH: Hiatal hernia; R: Right; L: Left; bx: Biopsy; CRCS: Colorectal cancer screening; F: Female; M: Male; 5-HIAA: 5-hydroxyindoleacetic acid; PET/CT: Positron emission tomography/computed tomography; LLQ: Left lower quadrant.
Patient 1
A 65-year-old female with a history of possible inflammatory bowel disease presented for evaluation of epigastric pain and occasional hematochezia. Colonoscopy revealed multiple polypoid lesions throughout the colon with biopsies consistent with mucosal prolapse syndrome. Esophagogastrastroduodenoscopy (EGD) revealed mild esophagitis, chronic gastritis, and a 5 mm polyp in the duodenal bulb biopsied with cold forceps (Figure 1A). Pathology demonstrated duodenal mucosa with atypical organized nests of cells with expression of low molecular cytokeratin, neuron-specific enolase (NSE), chromogranin, and synaptophysin on immunohistochemistry consistent with a neuroendocrine tumor (Figure 1B and C). Repeat EGD was performed 35 mo later and revealed no residual neuroendocrine tumor.
Figure 1.
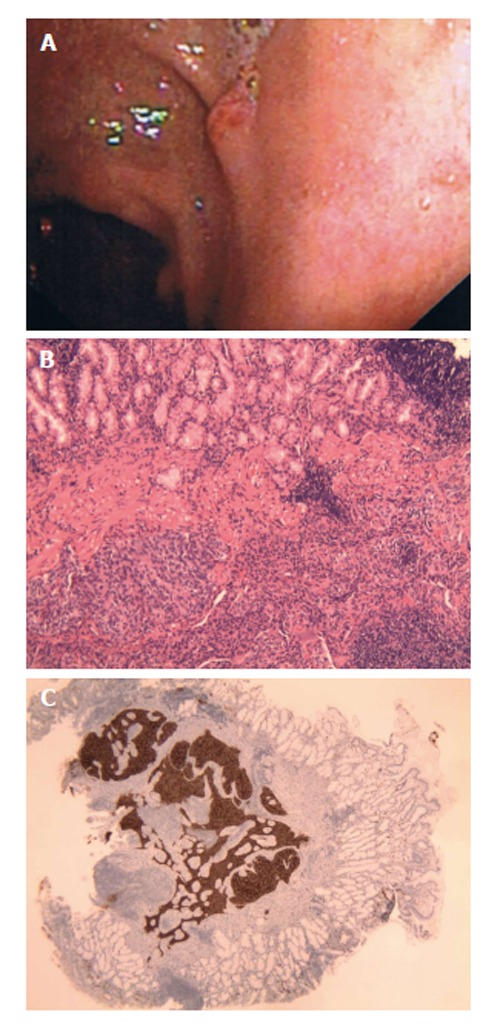
A 65-year-old female with a history of possible inflammatory bowel disease presented for evaluation of epigastric pain and occasional hematochezia. A: Patient 1, neuroendocrine (carcinoid) tumors as duodenal nodule at endoscopy; B: Solid growth pattern with organoid architecture and bland monotonous cells with lack of significant atypia and increased mitoses. H and E, × 10; C: Neoplastic neuroendocrine cells show diffuse positivity for Chromogranin. Chromogranin, × 20.
Patient 2
A 61-year-old male with a history of gastroesophageal reflux disease (GERD) underwent EGD for evaluation of chest discomfort with breakthrough reflux symptoms while taking a proton pump inhibitor daily. LA Grade C esophagitis and ulcerated mucosa were present in the distal esophagus. A 6 mm sessile polyp also observed in the duodenal bulb and resected by snare. Pathology revealed a neuroendocrine tumor of the duodenum. Positron emission tomography-computed tomography (PET-CT) was performed and demonstrated no evidence of hypermetabolic malignancy. A repeat EGD with biopsies from the previous polypectomy site six weeks later demonstrated reactive duodenopathy with foveolar metaplasia but no residual neuroendocrine tumor.
Patient 3
A 50-year-old female with chronic diarrhea was found to have metastatic liver disease of unknown primary origin on CT. The largest lesion measured 9 cm × 6 cm in the right hepatic lobe and PET-CT demonstrated only moderate metabolic activity consistent with a neuroendocrine tumor. CT-guided liver biopsy demonstrated metastatic neuroendocrine tumor with positive synaptophysin, chromogranin, NSE, and CD57 reactions on immunohistochemistry. EGD was performed that showed chronic esophagitis, hiatal hernia, and nodularity in the gastric fundus. Pathology from gastric biopsies revealed only benign lymphoid aggregates. Follow-up CT findings included a 2.4 cm partially calcified mass in the mid-abdominal mesentery suggestive of a neuroendocrine tumor of small bowel origin. The patient was started on long-acting octreotide and entered into hospice care 28 mo after initial presentation.
Patient 4
A 70-year-old male presenting with epigastric pain and 15 pound weight loss underwent upper endoscopy revealing chronic esophagitis, hiatal hernia, acute and chronic gastritis involving the antrum, and a small polypoid lesion which was found in the duodenal bulb. Biopsies were consistent with chronic duodenitis. Colonoscopy revealed one tubular adenoma < 1 cm and multiple hyperplastic polyps. A 3 cm mesenteric mass with surrounding desmoplastic reaction, small bowel thickening, and a 2 cm liver lesion were found on CT of the abdomen and pelvis (Figure 2A). CT guided biopsy of the mesenteric mass demonstrated a metastatic well-differentiated neuroendocrine tumor with immunohistochemistry positive for cytokeratin, NSE, synaptophysin, chromogranin, and CD56 (Figure 2B and C); however, biopsy of the liver lesion was negative for malignancy. PET-CT demonstrated heterogenous metabolic activity of the mesenteric mass with metabolic activity of the liver lesion similar to the surrounding hepatic parenchyma. Urinary 5-hydroxyindoleacetic acid (5-HIAA) was within normal range. The overall presentation was most consistent with a neuroendocrine tumor originating in the small bowel. The patient was started on long-acting octreotide therapy and did not undergo surgical resection of the tumor.
Figure 2.
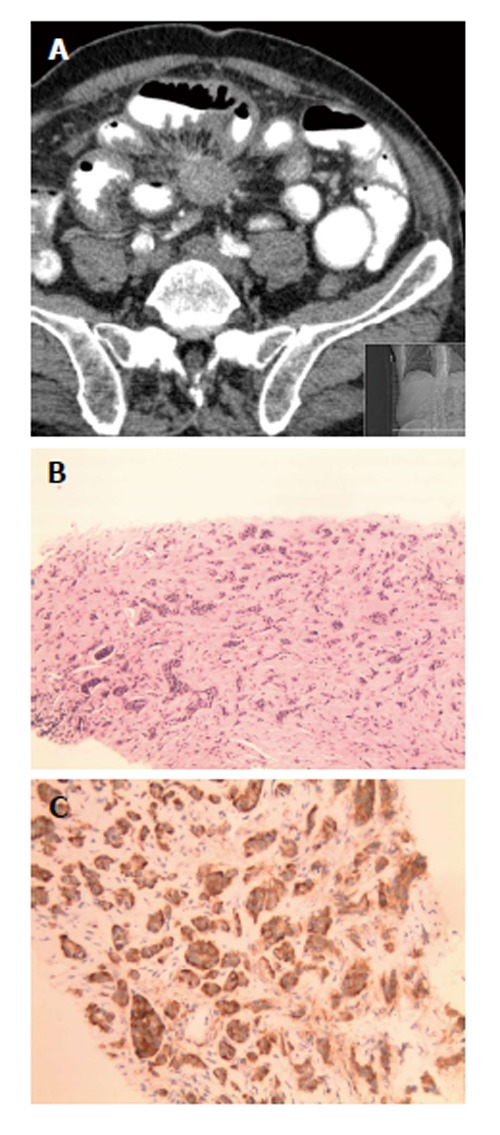
A 70-year-old male presenting with epigastric pain and 15 pound weight loss underwent upper endoscopy revealing chronic esophagitis, hiatal hernia, acute and chronic gastritis involving the antrum, and a small polypoid lesion which was found in the duodenal bulb. A: Patient 4, neuroendocrine (carcinoid) tumors as solid spiculated mesenteric mass on computed tomography of abdomen; B: Diffuse infiltration by monotonous bland cells with trabecular growth pattern. Mitoses, atypia and necrosis are not identified. H and E, × 10; C: The tumor cells are diffusely and strongly positive for CD56 immunohistochemical stain. CD56, × 20.
Patient 5
A 45-year-old female presented to an outside facility with nausea, vomiting, and abdominal pain and had dilation of the small bowel and colon and ascitic fluid on CT scan. Gastrografin enema demonstrated an obstruction in the sigmoid colon. An area of desmoplastic reaction involving the sigmoid colon was found during exploratory laparotomy along with multiple metastatic lesions to the liver and mesenteric and peritoneal implants. Surgical decompression of the small bowel and colon was performed and the patient was transferred for further care. Biopsies obtained from the peritoneal implants were consistent with a low-grade neuroendocrine tumor with immunohistochemistry positive for chromogranin and synaptophysin. Serum chromogranin A level was elevated at 27 nmol/L. Following transfer to our facility, the patient underwent colonoscopy which revealed a 3 cm area of stenosis due to extrinsic compression 30 cm from the anal verge. As no further surgical intervention was deemed appropriate, two overlapping metal colonic stents (Wallstent, 22 mm × 90 mm and 22 mm × 60 mm) were placed across the area of stenosis. The patient was later discharged for hospice care.
Patient 6
A 40-year-old male with recurrent perianal fistulous disease underwent colonoscopy to rule out inflammatory bowel disease. Colonoscopy revealed normal colonic mucosa and a 1 cm sessile polyp at the terminal ileum (Figure 3A). Snare polypectomy was performed and pathology revealed a submucosal neuroendocrine tumor with well formed nests of cells and diffuse expression of synaptophysin and chromogranin. KI-67 proliferative index was < 5% (Figure 3B and C). The patient was lost to follow-up.
Figure 3.
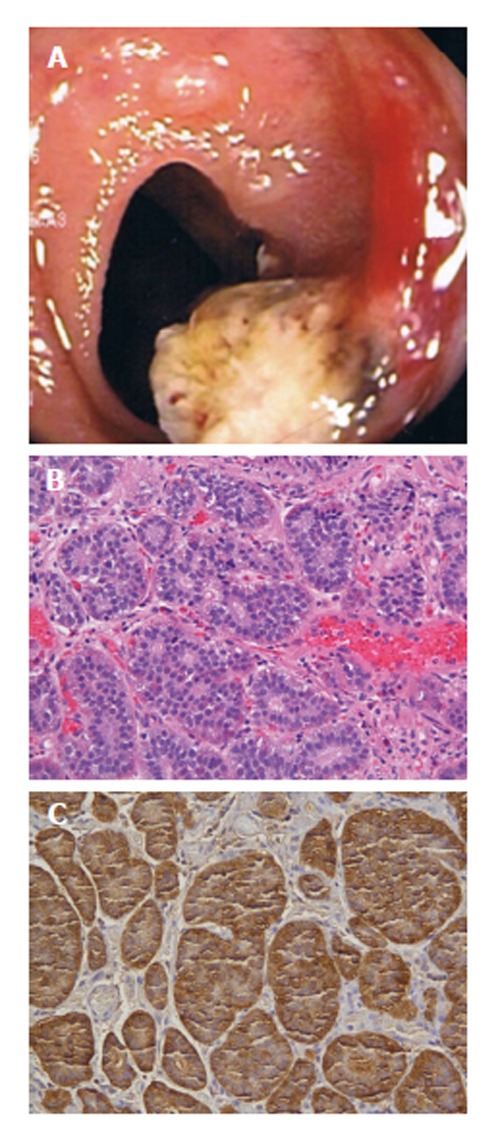
A 40-year-old male with recurrent perianal fistulous disease underwent colonoscopy to rule out inflammatory bowel disease. A: Patient 6, neuroendocrine (carcinoid) tumors as 10 mm ileocecal sessile polyp at colonoscopy; B: Nests of monotonous cells with bland nuclei arranged in organoid pattern. H and E, × 10; C: Carcinoid tumor; Chromogranin A: Marked cytoplasmic positivity.
Patient 7
A 50-year-old female presented for evaluation of GERD and colon cancer screening. EGD revealed a hiatal hernia, chronic esophagitis, and chronic gastritis. On colonoscopy, benign polyps were removed from the cecum and transverse colon. A 5 mm sessile polyp resected with hot forceps in the sigmoid colon (Figure 4A); pathology demonstrated atypical proliferation of cells and glandular-like inflammation with monotonous nuclei indicative of a neuroendocrine tumor. Immunostains were positive for chromogranin, synaptophysin, and CD56 consistent with a neuroendocrine tumor (Figure 4B and C). Somatostatin receptor scintigraphy demonstrated no evidence of other carcinoid tumors. Surveillance colonoscopy performed one year later revealed a scar at the site of the previously resected tumor without tumor recurrence.
Figure 4.
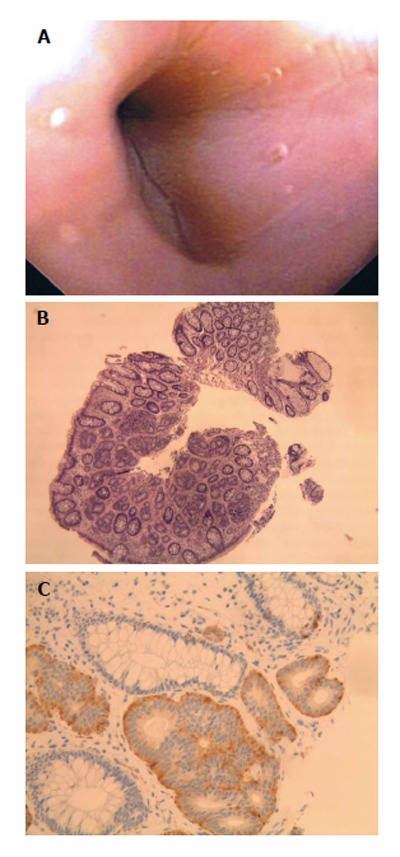
A 50-year-old female presented for evaluation of gastroesophageal reflux disease and colon cancer screening. A: Patient 7, neuroendocrine (carcinoid) tumors as sessile sigmoid polyp at colonoscopy; B: Organoid growth pattern with regular bland nuclei with indistinct cell borders. H and E, ×10; C: The neuroendocrine cells are positive for Synaptophysin and adjacent colonic glands are negative. Synaptophysin, × 20.
Patient 8
A 77-year-old female with Stage I right breast cancer presented for screening colonoscopy. A 7 mm submucosal nodule was biopsied from the sigmoid colon; pathology revealed tumor cells positive for synaptophysin and negative for chromogranin but overall consistent with a neuroendocrine tumor. Urinary 5-HIAA levels and octreotide scan were unremarkable. Endoscopic mucosal resection was subsequently performed with a snare. Excisional biopsy consisted of a 7 mm × 6 × mm × 3 mm submucosal neuroendocrine tumor. Colonoscopy one year later revealed no recurrence.
Patient 9
A 55-year-old male presented for evaluation of left lower quadrant tenderness and colon cancer screening. Colonoscopy revealed a 1.4 cm tubulovillous adenoma in the sigmoid colon. A 6 mm rectal polyp removed by snare was consistent with a neuroendocrine tumor. Surveillance colonoscopy three years later revealed a 6 mm hyperplastic polyp in the rectum and no evidence of recurrence of a neuroendocrine tumor.
Patient 10
A 54-year-old female presented for colon cancer screening. On colonoscopy, an 8 mm nodule was found in the rectum. Snare polypectomy was performed. Pathology demonstrated atypical proliferation of cells and glandular-like inflammation with monotonous nuclei suggestive of a neuroendocrine tumor. Colonoscopy one year later was negative for recurrence.
Patient 11
A 60-year-old female presented for colon cancer screening. On colonoscopy, a 5 mm submucosal nodule was found in the rectum and removed snare polypectomy. The biopsy was consistent with a neuroendocrine tumor involving the submucosa with tumor cells positive for synaptophysin and focally positive for chromogranin. Fourteen months later, colonoscopic biopsies from the polypectomy site revealed no recurrence.
DISCUSSION
Our case series describes a wide spectrum of benign gastrointestinal NET originating in the small intestine, colon, and rectum and malignant NET originating in the liver and intraabdominal sites. The following discussion will focus on the diagnosis and management of NET originating from the luminal gastrointestinal tract and will not include pancreatic NET.
Advances in our understanding of both the biologic and morphologic heterogeneity of NET have left the term “carcinoid” nearly obsolete[7]. Gastroenteropancreatic NET (GEP-NET), encompassing both traditional gastrointestinal carcinoids and pancreatic endocrine tumors, are replacing the less descriptive and often times pathologically and clinically more confusing term “carcinoid”[3,9]. In 2000, the World Health Organization (WHO) classification replaced “carcinoid” with the terms neuroendocrine tumors and neuroendocrine carcinomas to describe gastrointestinal neoplasms originating from the diffuse system of neuroendocrine cells[9]. Along with developing tumor node metastasis staging and grading systems[10-14], the WHO classification[9] provides an improved system for determining prognosis and treatment and includes three main groups subdivided by organ of tumor origin: (1) well differentiated neuroendocrine tumors (benign behavior or uncertain malignant potential-“carcinoids”); (2) well differentiated neuroendocrine carcinomas (low-grade malignancy-“malignant carcinoids”); and (3) poorly-differentiated carcinomas (high-grade malignancy). This classification replaces the previous outdated system which was based on embryologic cell of origin (foregut, midgut, hindgut) and shared little correlation between tumor behavior and tumor location especially for neoplasms originating in the foregut (tracheobronchopulmonary, gastric, and pancreatic tumors)[3,9]. Histologically, tumor proliferation capacity is measured by Ki-67 staining with Ki-67 Index < 2% seen in grade I tumors, 2%-20% in grade II tumors, and > 20% tumor cell involvement in grade III GEP-NET[11].
Cells originating from the diffuse system of neuroendocrine cells within the gastrointestinal tract share phenotypic similarities with neural cells in their expression of synaptophysin, NSE, and chromogranin A[3,10]. Useful as GEP-NET markers found on the secretory vesicles of neuroendocrine cells, these proteins usually remain independent of cellular production of hormones that are stored within the vesicles[3,10,15]. Hormone production and biologic activity generally varies by GEP-NET location (Table 2) and less than half of the known hormones originating from at least 15 different types of endocrine cells are expressed by GEP-NET[15]. Many tumors remain clinically silent and may present with intestinal obstruction as a result of tumor-induced fibrosis rather than signs or symptoms of secretory products[3]. The classic carcinoid syndrome (cutaneous flushing and secretory diarrhea) occurs in less than 10% of patients[3] and typically in the setting of hepatic metastases.
Table 2.
| Location | Hormones |
| Stomach | Histamine, Gastrin, Serotonin, Somatostatin, Gastrin Releasing Peptide |
| Duodenum/Upper Jejunum | Gastrin, Serotonin, Somatostatin, Gastrin Releasing Peptide |
| Ileum/Cecum | Enteroglucagon, Serotonin, Substance P, Tachykinins |
| Appendix | Enteroglucagon, Peptide YY, Serotonin, Somatostatin |
| Colon/Rectum | Enteroglucagon, Serotonin, Somatostatin |
| Pancreas | ACTH, Calcitonin, Cholecystokinin, Corticotropin-Releasing Hormone, Gastrin, Glucagon, Growth Hormone-Releasing Hormone, Growth Hormone-Releasing Factor, Insulin, Neurotensin, Pancreatic Polypeptide, Parathyroid Hormone-Related Peptide, Prolactin, Somatostatin, Vasoactive Intestinal Peptide |
Diagnostic evaluation
Initial evaluation of patients with a suspected GEP-NET should include a serum chromogranin A level[3,16]. Elevated in approximately 80% of patients with neuroendocrine tumors regardless of location and functional activity, chromogranin A levels also appear to correlate with overall tumor burden[17]. Twenty-four-hour urinary 5-HIAA levels as well as serum gastrin, histamine, serotonin, and substance P levels should be included as part of the initial evaluation when the presentation is consistent with carcinoid syndrome[3]. Urinary 5-HIAA elevation sensitivity is as high as 100% with a specificity of 88% for the carcinoid syndrome[18]. Care must be taken to avoid medications and foods that may affect urinary 5-HIAA excretion; large amounts of serotonin are in foods as avocados, bananas, eggplant, kiwi, pineapple, plums, tomatoes, and walnuts and may cause false positive results[16].
Patients with positive biochemical markers should be evaluated with somatostatin receptor scintigraphy (111Indium-labeled octreotide scan) for tumor localization as well as either CT or magnetic resonance imaging (MRI) to identify mass lesions, mesenteric fibrosis, and lymphadenopathy[3,16]. 111Indium-labeled octreotide scan is useful in detection of both primary and metastatic tumors with sensitivity as high as 90%[3,19]. CT and MRI play an important role in identification of primary tumors and metastatic disease; however, they may underestimate the extent of disease in up to 25% of cases[20,21] and overall sensitivities around 80% are lower than 111Indium-labeled octreotide scanning[3]. Radiolabeled metaiodobenzylguanide (123I-MIBG) scanning may be used in patients on long-acting octreotide medications which interfere with somatostatin receptor scintigraphy[3]. Radiolabeled 5-HTP positron emission tomography has demonstrated better sensitivities than CT and octreotide scanning; however, it is not widely available and is generally still considered an investigational modality[3,21,22]. Barium studies, including small-bowel-follow-through, play little if any role in tumor localization with the availability of other diagnostic modalities with increased sensitivity[23].
Following tumor localization, biopsy for tissue diagnosis should be obtained including performing upper endoscopy and colonoscopy with ileoscopy as clinically indicated[3,16]. Small bowel enteroscopy has low diagnostic sensitivities as well as a limited ability to evaluate the distal jejunum and ileum and has largely been replaced by capsule endoscopy in both diagnostic and surveillance roles[3,24,25]. Endoscopic ultrasound (EUS) plays in important role in guiding management as it is accurate in assessing tumor size and depth of invasion especially in gastric, duodenal, and rectal carcinoid tumors[26,27].
Site specific information
Gastric carcinoids are typically divided into Type I, II, and III tumors with some classifications including Type IV tumors[9]. Type I and II gastric carcinoid tumors develop in response to hypergastrinemia effects on enterochromaffin-like cells of the oxyntic mucosa found in the gastric fundus and body[28-30]. Type I are the most common gastric NET tumors usually presenting as small multifocal lesions associated with autoimmune chronic atrophic gastritis and hypergastrinemia in the setting of low gastric acid output[3,9,30]. They have an excellent prognosis with 5-year survival rates > 95%[3]. Type II gastric NET develop in patients with Multiple Endocrine Neoplasia type-1 (MEN-1) associated Zollinger-Ellison syndrome (ZES) as a result of tumor driven hypergastrinemia in the setting of an autosomal dominant mutation of the MEN-1 gene located on chromosome 11q13[9,29,30]. Type II gastric NET rarely develop in patients with sporadic ZES[29]. Prognosis is good with 5-year survival rates of 70%-90%[3].
Type I and II tumors < 1 cm in size without extension into the muscularis propria on EUS can initially be managed with endoscopic mucosal resection[3,26,31]. When more than 5 lesions are present, tumor size is > 1 cm, or recurrence occurs at a site of previous endoscopic resection, local surgical excision is recommended[3]. Type II lesions may require aggressive gastrectomy as well as surgical resection of the underlying gastrinoma[3]. Surveillance endoscopy with biopsy should be performed every six months following both endoscopic and surgical tumor removal[3].
Type III tumors are sporadic gastric carcinoids which develop in normal gastric mucosa in the setting of normal gastrin levels[3,9]. They are aggressive with deep invasion and the potential for metastatic disease characteristic of even small primary tumors[26]. Five-year survival rates are < 35%[3]. Type IV tumors are neuroendocrine carcinomas which are indistinguishable from gastric adenocarcinomas with the exception of the presence of neuroendocrine cells within the tumor matrix[3]. Both type III and IV tumors should be managed surgically with complete or partial gastrectomy[3,9].
Small intestine
Duodenal: Five types of duodenal neuroendocrine tumors have been described[32]: (1) gastrinomas which may occur sporadically or in the setting of MEN-1/ZES and are the most common duodenal NET[3,9,32]; (2) somatostatinomas which usually occur in the ampullary/periampullary region and are more likely to be associated with von Recklinghausen’s disease (neurofibromatosis type 1)[3,33]; (3) gangliocytic paraganglionomas[3,9,32]; (4) nonfunctioning NET which contain serotonin-, gastrin-, or calcitonin-positive cells[3,9]; and (5) neuroendocrine carcinomas[3,32,33]. Overall 5-year survival for duodenal carcinoid lesions is 60%[3]. Endoscopic resection may be considered for nonmetastatic duodenal (and ampullary) lesions measuring up to 2 cm if the tumor is confined to the mucosa and submucosa on EUS examination[3,33-35]. Surgical resection should be performed on tumors > 2 cm[34,35]. While distant metastases rarely occurs with duodenal NET, lymph node metastases may occur in tumors < 1 cm and surgical resection should be performed in all patients with evidence of lymph node involvement on pretreatment imaging studies[35].
Jejuno-Ileal: Terminal ileum NET are the most common GEP-NET. They are frequently found at an advanced stage with metastatic disease to the liver present in 50% and regional lymph node involvement in up to 70% of patients regardless of primary tumor size[21]. Associated mesenteric fibrosis, nodal metastases, and desmoplastic reactions involving mesenteric vessels may lead to nonspecific abdominal pain, gastrointestinal bleeding, intermittent ischemia, or bowel obstruction. These symptoms may prompt emergent surgical intervention and subsequent diagnosis of a previously unidentified jejunal or ileal NET in up to 40% of patients[3,21]. Ileal NET are associated with the carcinoid syndrome in the setting of liver metastases in approximately 20% of cases[9,21]. While the 5-year survival rate is 60% for both jejunal and ileal tumors, it is as low as 18% when hepatic metastases are present[3]. Surgical resection of the primary tumor as well as en bloc resection of regional lymph nodes is recommended and should be performed even when hepatic metastases is present in order to delay progression and local complications of disease[21,31].
Appendix: Appendiceal NET are the most common appendiceal tumor[21]. They are often found incidentally during appendectomy with the majority (90%) of tumors < 1 cm in size. Overall 5-year survival for appendiceal NETs is 98% for benign tumors and 27% for malignant tumors[3]. Metastatic disease rarely occurs with tumors < 2 cm and the occurrence of metastases increases with increasing tumor size over 2 cm[3,21,36]. Tumors > 2 cm should be managed with right hemicolectomy. Appendectomy should be performed in tumors < 2 cm in size with right hemicolectomy considered for tumors 1-2 cm based on pathologic criteria (invasion into mesoappendix, serosal or lymphovascular invasion, involvement of tumor margins, positive lymph nodes, or Ki67 index > 2% on immunohistochemistry staining)[21]. Variant mixed endocrine/exocrine goblet-cell (adenocarcinoid) tumors are more aggressive lesions associated with a poorer prognosis and higher rates of both metastatic and recurrent disease and should be managed with right hemicolectomy regardless of tumor size[21,36].
Colon: Neuroendocrine tumors rarely occur in the colon with many previously reported cecal NET representing appendiceal tumors[3,9]. Clinical presentation of colonic NET includes change in bowel habits, gastrointestinal bleeding, abdominal pain, weight loss, and asymptomatic lesions found during screening colonoscopy is generally indistinguishable from other mass lesions of the colon[3,9]. Most tumors are > 2 cm in size with invasion into the muscularis propria at the time of diagnosis and overall prognosis is poor with 5-year survival rates of only 33%-42%[3]. Wide surgical resection with lymph node dissection is recommended for management of colonic NET[3] as metastatic disease is common at the time of diagnosis[9]. Local excision may be considered for tumors < 2 cm in size[3]; however, data regarding metastatic disease in this setting are limited.
Rectum: Frequently found as small, asymptomatic submucosal tumors during endoscopic evaluation, rectal NET have an excellent overall prognosis with 5-year survival rates of 87%[3,9]. When present, symptoms may include change in bowel habits, gastrointestinal bleeding, anorectal discomfort, and pruritis ani[3]. Submucosal tumors < 1 cm in size account for 80% of rectal carcinoids[3] and can be managed endoscopically in the absence of muscularis invasion or pararectal lymph node metastases on EUS examination[31,37]. Rectal NETs 1-2 cm in size may be managed with wide surgical excision if there is no evidence of muscularis invasion or lymph node metastasis[3]. Low anterior resection or abdominoperineal resection is recommended for tumors > 2 cm as the risk of metastatic disease increases with tumors > 2 cm in size and with invasion of the muscularis propria[3,9].
Medical therapy
Following surgical resection of a GEP-NET, medical therapy may be required for symptom management related to functional tumor syndromes as well as management of progressive metastatic and residual disease[31,38,39]. Patients with symptomatic functional NET should be considered for somatostatin (SST) analog (short- or long-acting octreotide) or interferon- α therapy alone or in combination[3,31,38,39]. In addition to reducing symptoms in patients with carcinoid syndrome[40,41], VIPoma associated Verner-Morrison syndrome (watery diarrhea, hypokalemia, and achlorhydria)[40,42], and glucagonoma associated necrolytic migratory erythema[4], SST analogs may also play a role in growth inhibition of nonfunctioning NET[39,41]. Interferon-α therapy may be considered in patients who become intolerant or resistant to SST analog therapy as it has also been shown to reduce diarrhea and flushing in patients with carcinoid syndrome[39].
Systemic chemotherapy or peptide receptor radionuclide therapy with I-131 MIBG, Yttrium90, or Lutetium177 should be considered in patients with metastatic disease with transarterial embolization/chemoembolization or radiofrequency ablation considered when metastases are limited to the liver[3,31,38,39].
Patients undergoing biologic or cytotoxic therapies should have their clinical response to treatment monitored every 3 mo[11]. Biochemical markers (based on the functional status of their underlying tumor) should be followed every 3-6 mo along with CT or MRI scanning every 6 mo for 5 years following curative surgical resection[11].
In a conclusion, GEP-NET are relatively rare neoplasms of the gastrointestinal tract with variable clinical presentation, morbidity, and mortality dependent on tumor location, metastatic potential, and functional biologic status. Staging and classification systems for GEP-NET are likely to continue to evolve along with further development of tumor directed diagnostic and therapeutic modalities as our understanding of GEP-NET continues to expand over time.
COMMENTS
Case characteristics
This case series describes a wide spectrum of benign gastrointestinal neuroendocrine (carcinoid) tumors (NET).
Clinical diagnosis
The diagnosis and management of NET originating from the luminal gastrointestinal tract and will not include pancreatic NET.
Imaging diagnosis
Computed tomography and magnetic resonance imaging play an important role in identification of primary tumors and metastatic disease.
Experiences and lessons
Gastroenteropancreatic NET are relatively rare neoplasms of the gastrointestinal tract with variable clinical presentation, morbidity, and mortality dependent on tumor location, metastatic potential, and functional biologic status.
Peer review
This is a very good example of a case series combined with a good review.
Footnotes
P- Reviewer: Albuquerque A, Anadol Z, Hasanein P S- Editor: Wen LL L- Editor: A E- Editor: Liu SQ
References
- 1.Klöppel G. Oberndorfer and his successors: from carcinoid to neuroendocrine carcinoma. Endocr Pathol. 2007;18:141–144. doi: 10.1007/s12022-007-0021-9. [DOI] [PubMed] [Google Scholar]
- 2.Oberndorfer S. Karzinoide Tumoren des Dunndarms. Frank Z Pathol. 1907;1:426–429. [Google Scholar]
- 3.Modlin IM, Kidd M, Latich I, Zikusoka MN, Shapiro MD. Current status of gastrointestinal carcinoids. Gastroenterology. 2005;128:1717–1751. doi: 10.1053/j.gastro.2005.03.038. [DOI] [PubMed] [Google Scholar]
- 4.Modlin IM, Lye KD, Kidd M. A 5-decade analysis of 13,715 carcinoid tumors. Cancer. 2003;97:934–959. doi: 10.1002/cncr.11105. [DOI] [PubMed] [Google Scholar]
- 5.Modlin IM, Lye KD, Kidd M. A 50-year analysis of 562 gastric carcinoids: small tumor or larger problem? Am J Gastroenterol. 2004;99:23–32. doi: 10.1046/j.1572-0241.2003.04027.x. [DOI] [PubMed] [Google Scholar]
- 6.Kulke MH, Mayer RJ. Carcinoid tumors. N Engl J Med. 1999;340:858–868. doi: 10.1056/NEJM199903183401107. [DOI] [PubMed] [Google Scholar]
- 7.Modlin IM, Oberg K, Chung DC, Jensen RT, de Herder WW, Thakker RV, Caplin M, Delle Fave G, Kaltsas GA, Krenning EP, et al. Gastroenteropancreatic neuroendocrine tumours. Lancet Oncol. 2008;9:61–72. doi: 10.1016/S1470-2045(07)70410-2. [DOI] [PubMed] [Google Scholar]
- 8.Berge T, Linell F. Carcinoid tumours. Frequency in a defined population during a 12-year period. Acta Pathol Microbiol Scand A. 1976;84:322–330. [PubMed] [Google Scholar]
- 9.Klöppel G, Perren A, Heitz PU. The gastroenteropancreatic neuroendocrine cell system and its tumors: the WHO classification. Ann N Y Acad Sci. 2004;1014:13–27. doi: 10.1196/annals.1294.002. [DOI] [PubMed] [Google Scholar]
- 10.Klöppel G, Couvelard A, Perren A, Komminoth P, McNicol AM, Nilsson O, Scarpa A, Scoazec JY, Wiedenmann B, Papotti M, et al. ENETS Consensus Guidelines for the Standards of Care in Neuroendocrine Tumors: towards a standardized approach to the diagnosis of gastroenteropancreatic neuroendocrine tumors and their prognostic stratification. Neuroendocrinology. 2009;90:162–166. doi: 10.1159/000182196. [DOI] [PubMed] [Google Scholar]
- 11.Oberg K, Jelic S. Neuroendocrine gastroenteropancreatic tumors: ESMO clinical recommendation for diagnosis, treatment and follow-up. Ann Oncol. 2009;20 Suppl 4:150–153. doi: 10.1093/annonc/mdp158. [DOI] [PubMed] [Google Scholar]
- 12.Pape UF, Jann H, Müller-Nordhorn J, Bockelbrink A, Berndt U, Willich SN, Koch M, Röcken C, Rindi G, Wiedenmann B. Prognostic relevance of a novel TNM classification system for upper gastroenteropancreatic neuroendocrine tumors. Cancer. 2008;113:256–265. doi: 10.1002/cncr.23549. [DOI] [PubMed] [Google Scholar]
- 13.Rindi G, Klöppel G, Alhman H, Caplin M, Couvelard A, de Herder WW, Erikssson B, Falchetti A, Falconi M, Komminoth P, et al. TNM staging of foregut (neuro)endocrine tumors: a consensus proposal including a grading system. Virchows Arch. 2006;449:395–401. doi: 10.1007/s00428-006-0250-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rindi G, Klöppel G, Couvelard A, Komminoth P, Körner M, Lopes JM, McNicol AM, Nilsson O, Perren A, Scarpa A, et al. TNM staging of midgut and hindgut (neuro) endocrine tumors: a consensus proposal including a grading system. Virchows Arch. 2007;451:757–762. doi: 10.1007/s00428-007-0452-1. [DOI] [PubMed] [Google Scholar]
- 15.Ardill JE. Circulating markers for endocrine tumours of the gastroenteropancreatic tract. Ann Clin Biochem. 2008;45:539–559. doi: 10.1258/acb.2008.008039. [DOI] [PubMed] [Google Scholar]
- 16.Ghevariya V, Malieckal A, Ghevariya N, Mazumder M, Anand S. Carcinoid tumors of the gastrointestinal tract. South Med J. 2009;102:1032–1040. doi: 10.1097/SMJ.0b013e3181b67356. [DOI] [PubMed] [Google Scholar]
- 17.Stivanello M, Berruti A, Torta M, Termine A, Tampellini M, Gorzegno G, Angeli A, Dogliotti L. Circulating chromogranin A in the assessment of patients with neuroendocrine tumours. A single institution experience. Ann Oncol. 2001;12 Suppl 2:S73–S77. doi: 10.1093/annonc/12.suppl_2.s73. [DOI] [PubMed] [Google Scholar]
- 18.Tormey WP, FitzGerald RJ. The clinical and laboratory correlates of an increased urinary 5-hydroxyindoleacetic acid. Postgrad Med J. 1995;71:542–545. doi: 10.1136/pgmj.71.839.542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kwekkeboom DJ, Kam BL, van Essen M, Teunissen JJ, van Eijck CH, Valkema R, de Jong M, de Herder WW, Krenning EP. Somatostatin-receptor-based imaging and therapy of gastroenteropancreatic neuroendocrine tumors. Endocr Relat Cancer. 2010;17:R53–R73. doi: 10.1677/ERC-09-0078. [DOI] [PubMed] [Google Scholar]
- 20.Chambers AP, Pasieka JL, Dixon E, Rorstad O. The role of imaging studies in the staging of midgut neuroendocrine tumors. J Am Coll Surg. 2009;207:S18. doi: 10.1016/j.jamcollsurg.2010.07.016. [DOI] [PubMed] [Google Scholar]
- 21.Pasieka JL. Carcinoid tumors. Surg Clin North Am. 2009;89:1123–1137. doi: 10.1016/j.suc.2009.06.008. [DOI] [PubMed] [Google Scholar]
- 22.Orlefors H, Sundin A, Garske U, Juhlin C, Oberg K, Skogseid B, Langstrom B, Bergstrom M, Eriksson B. Whole-body (11)C-5-hydroxytryptophan positron emission tomography as a universal imaging technique for neuroendocrine tumors: comparison with somatostatin receptor scintigraphy and computed tomography. J Clin Endocrinol Metab. 2005;90:3392–3400. doi: 10.1210/jc.2004-1938. [DOI] [PubMed] [Google Scholar]
- 23.Sippel RS, Chen H. Carcinoid tumors. Surg Oncol Clin N Am. 2006;15:463–478. doi: 10.1016/j.soc.2006.05.002. [DOI] [PubMed] [Google Scholar]
- 24.Bellutti M, Fry LC, Schmitt J, Seemann M, Klose S, Malfertheiner P, Mönkemüller K. Detection of neuroendocrine tumors of the small bowel by double balloon enteroscopy. Dig Dis Sci. 2009;54:1050–1058. doi: 10.1007/s10620-008-0456-y. [DOI] [PubMed] [Google Scholar]
- 25.Coates SW, DeMarco DC. Metastatic carcinoid tumor discovered by capsule endoscopy and not detected by esophagogastroduodenoscopy. Dig Dis Sci. 2004;49:639–641. doi: 10.1023/b:ddas.0000026311.62364.0b. [DOI] [PubMed] [Google Scholar]
- 26.Ichikawa J, Tanabe S, Koizumi W, Kida Y, Imaizumi H, Kida M, Saigenji K, Mitomi H. Endoscopic mucosal resection in the management of gastric carcinoid tumors. Endoscopy. 2003;35:203–206. doi: 10.1055/s-2003-37256. [DOI] [PubMed] [Google Scholar]
- 27.Ishii N, Horiki N, Itoh T, Maruyama M, Matsuda M, Setoyama T, Suzuki S, Uchida S, Uemura M, Iizuka Y, et al. Endoscopic submucosal dissection and preoperative assessment with endoscopic ultrasonography for the treatment of rectal carcinoid tumors. Surg Endosc. 2010;24:1413–1419. doi: 10.1007/s00464-009-0791-x. [DOI] [PubMed] [Google Scholar]
- 28.Borch K, Ahrén B, Ahlman H, Falkmer S, Granérus G, Grimelius L. Gastric carcinoids: biologic behavior and prognosis after differentiated treatment in relation to type. Ann Surg. 2005;242:64–73. doi: 10.1097/01.sla.0000167862.52309.7d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Delle Fave G, Capurso G, Milione M, Panzuto F. Endocrine tumours of the stomach. Best Pract Res Clin Gastroenterol. 2005;19:659–673. doi: 10.1016/j.bpg.2005.05.002. [DOI] [PubMed] [Google Scholar]
- 30.von Rosenvinge EC, Wank SA, Lim RM. Gastric masses in multiple endocrine neoplasia type I-associated Zollinger-Ellison syndrome. Gastroenterology. 2009;137:1222, 537. doi: 10.1053/j.gastro.2009.03.050. [DOI] [PubMed] [Google Scholar]
- 31.Ramage JK, Davies AH, Ardill J, Bax N, Caplin M, Grossman A, Hawkins R, McNicol AM, Reed N, Sutton R, et al. Guidelines for the management of gastroenteropancreatic neuroendocrine (including carcinoid) tumours. Gut. 2005;54 Suppl 4:iv1–iv16. doi: 10.1136/gut.2004.053314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bordi C, D’Adda T, Azzoni C, Canavese G, Brandi ML. Gastrointestinal endocrine tumors: recent developments. Endocr Pathol. 1998;9:99–115. [Google Scholar]
- 33.Makhlouf HR, Burke AP, Sobin LH. Carcinoid tumors of the ampulla of Vater: a comparison with duodenal carcinoid tumors. Cancer. 1999;85:1241–1249. doi: 10.1002/(sici)1097-0142(19990315)85:6<1241::aid-cncr5>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 34.Chou JW, Huang WH, Lai HC. A case with duodenal bleeding. Gastroenterology. 2009;137:e1–e2. doi: 10.1053/j.gastro.2008.12.035. [DOI] [PubMed] [Google Scholar]
- 35.Mullen JT, Wang H, Yao JC, Lee JH, Perrier ND, Pisters PW, Lee JE, Evans DB. Carcinoid tumors of the duodenum. Surgery. 2005;138:971–977; discussion 977-978. doi: 10.1016/j.surg.2005.09.016. [DOI] [PubMed] [Google Scholar]
- 36.Bamboat ZM, Berger DL. Is right hemicolectomy for 2.0-cm appendiceal carcinoids justified? Arch Surg. 2006;141:349–352; discussion 352. doi: 10.1001/archsurg.141.4.349. [DOI] [PubMed] [Google Scholar]
- 37.Onozato Y, Kakizaki S, Iizuka H, Sohara N, Mori M, Itoh H. Endoscopic treatment of rectal carcinoid tumors. Dis Colon Rectum. 2010;53:169–176. doi: 10.1007/DCR.0b013e3181b9db7b. [DOI] [PubMed] [Google Scholar]
- 38.Oberg K, Astrup L, Eriksson B, Falkmer SE, Falkmer UG, Gustafsen J, Haglund C, Knigge U, Vatn MH, Välimäki M. Guidelines for the management of gastroenteropancreatic neuroendocrine tumours (including bronchopulmonary and thymic neoplasms). Part I-general overview. Acta Oncol. 2004;43:617–625. doi: 10.1080/02841860410018575. [DOI] [PubMed] [Google Scholar]
- 39.Srirajaskanthan R, Toumpanakis C, Meyer T, Caplin ME. Review article: future therapies for management of metastatic gastroenteropancreatic neuroendocrine tumours. Aliment Pharmacol Ther. 2009;29:1143–1154. doi: 10.1111/j.1365-2036.2009.03988.x. [DOI] [PubMed] [Google Scholar]
- 40.Arnold R, Trautmann ME, Creutzfeldt W, Benning R, Benning M, Neuhaus C, Jürgensen R, Stein K, Schäfer H, Bruns C, et al. Somatostatin analogue octreotide and inhibition of tumour growth in metastatic endocrine gastroenteropancreatic tumours. Gut. 1996;38:430–438. doi: 10.1136/gut.38.3.430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Oberg K, Kvols L, Caplin M, Delle Fave G, de Herder W, Rindi G, Ruszniewski P, Woltering EA, Wiedenmann B. Consensus report on the use of somatostatin analogs for the management of neuroendocrine tumors of the gastroenteropancreatic system. Ann Oncol. 2004;15:966–973. doi: 10.1093/annonc/mdh216. [DOI] [PubMed] [Google Scholar]
- 42.Ghaferi AA, Chojnacki KA, Long WD, Cameron JL, Yeo CJ. Pancreatic VIPomas: subject review and one institutional experience. J Gastrointest Surg. 2008;12:382–393. doi: 10.1007/s11605-007-0177-0. [DOI] [PubMed] [Google Scholar]
- 43.Massironi S, Sciola V, Peracchi M, Ciafardini C, Spampatti MP, Conte D. Neuroendocrine tumors of the gastro-entero-pancreatic system. World J Gastroenterol. 2008;14:5377–5384. doi: 10.3748/wjg.14.5377. [DOI] [PMC free article] [PubMed] [Google Scholar]