

Abstract
CY-007 and CY-049 pteridine glycosyltransferases (PGTs) that differ in sugar donor specificity to catalyze either glucose or xylose transfer to tetrahydrobiopterin were studied here to uncover the structural determinants necessary for the specificity. The importance of the C-terminal domain and its residues 218 and 258 that are different between the two PGTs was assessed via structure-guided domain swapping or single and dual amino acid substitutions. Catalytic activity and selectivity were altered in all the mutants (2 chimeric and 6 substitution) to accept both UDP-glucose and UDP-xylose. In addition, the wild type activities were improved 1.6-4.2 fold in 4 substitution mutants and activity was observed towards another substrate UDP-Nacetylglucosamine in all the substitution mutants from CY-007 PGT. The results strongly support essential role of the C-terminal domain and the two residues for catalysis as well as sugar donor specificity, bringing insight into the structural features of the PGTs. [BMB Reports 2013; 46(1): 37-40]
Keywords: Domain swapping, Pteridine glycosyltransferase, Site-directed mutagenesis, Substrate specificity, Tetrahydrobiopterin
INTRODUCTION
Pteridine glycosyltransferase (PGT) is a group of enzymes catalyzing transfer of a variety of glycosyl group to the reduced form of pteridines such as L-erythro-tetrahydrobiopterin (BH4) (1). BH4 is a well-known cofactor for aromatic amino acid hydroxylases and nitric oxide synthases in animals and is medicated for hyperphenylalaninemia (2). PGTs belong to a novel group of glycosyltransferases (GTs), which constitute a large class of proteins that are divided into 94 families according to the CAZY classification system (http://www.cazy.org/). GTs crystallized so far adopt one of three folds, termed GT-A, GT-B, and GT-A like folds (3). The GT-B fold, to which PGTs belong, comprises two domains formed by the N- and C-terminal parts, where the C-terminal domain is responsible for the majority of interactions with the sugar donor, whereas the acceptor mainly interacts with residues harbored within the N-terminal domain (4). GTs are being studied intensively for glycodiversification of therapeutically important natural products via domain swapping and amino acid substitution (5,6). PGTs are also useful for quantitative determination of oxidized and reduced forms of BH4 and possibly for production of BH4-glycosides (7). However, PGTs have not been studied for structural features. We recently discovered two PGTs from Arthrospira platensis CY-007 and Arthrospira maxima CY-049 that are 93% identical in primary structure but catalyze transfer of different sugars to BH4 (8): CY-007 PGT catalyzes glucose transfer, while CY-049 PGT mediates xylose transfer. As it was speculated that the amino acid residues different between them in the C-terminal domain would be responsible for the sugar donor specificity, we prepared here 2 chimeric enzymes via domain swapping of the C-terminal and 6 single and dual amino acid substitution mutants via site-directed mutagenesis and determined the activity towards various UDP-sugars.
RESULTS AND DISCUSSION
Structural analysis for domain swapping and amino acid substitution
The primary structures of CY-007 and CY-049 PGTs differ from each other in 25 residues among 359 amino acid residues (Fig. 1). In order to find structural information for domain swapping and amino acid substitution the putative protein structures of CY-007 and CY-049 were investigated through multiple alignment with GTs (2JJMand2IV3), for which protein structure is available (9,10), and subsequent homology modeling at Swiss-Model (http://swissmodel.expasy.org/) (11) against 2IV3. The structure-guided analysis suggested two segments having structural roles in the PGTs (Fig. 1). A linker segment spanning the residues 160-185 connects the N- and C-terminal domains. A crossover segment of over 30 residues at the C-terminal end crosses to the N-terminal domain, thereby stabilizing the bi-lobal structure. The analysis provided the assembly point for domain swapping and the amino acid residues for substitution (Fig. 1). Among 9 amino acid residues that differ between CY-007 and CY-049 PGTs in the C-terminal domain, the residues at 218 and 258 were selected for substitution, because the model structures revealed their position in the interior pocket of the C-terminal domain (Supplementary data Fig.S1), which is known to harbor the main part of interactions with donor substrate in GTs (3,4). The residues in CY-007 PGT were substituted for the corresponding ones in CY-049 PGT and vice versa, thus generating the mutants of CY-007 PGT (E218A, Q258E, and E218A/Q258E) and CY-049 PGT (A218E, E258Q, and A218E/E258Q).
Fig. 1. Amino acid sequence alignment of CY-007 and CY-049 PGTs. The secondary structure displayed on top for CY-007 was determined by DSSP (12) from the PDB file, which was obtained by homology modeling of CY-007 against 2IV3. The alignment was performed using ClustalX and ESPript (13).
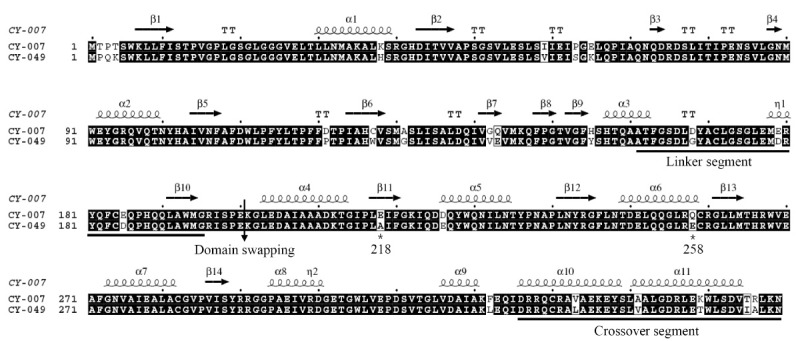
Catalytic activities of the chimeric PGTs
The chimeras (N007/C049 and N049/C007 PGTs) and 6 mutant proteins were purified as soluble forms (data not shown). N007/C049 PGT were active towards both UDP-glucose and UDP-xylose, although much less than the parental ones (Fig. 2). N049/C007 PGT also exhibited activity towards UDP-glucose but still retained 70% of the parental activity towards UDP-xylose. The results do not fully satisfy our expectation that the switched C-terminal domain would primarily determine the sugar donor specificity in the chimeras. Sugar donor may interact with both domains, as suggested for sugar acceptor specificity in plant GTs (14). In addition, domain swapping may need further structural adjustment for precise match at nanoscale level between the hybrid domains.
Fig. 2. Specific activities of chimeric PGTs compared with those of parental PGTs.
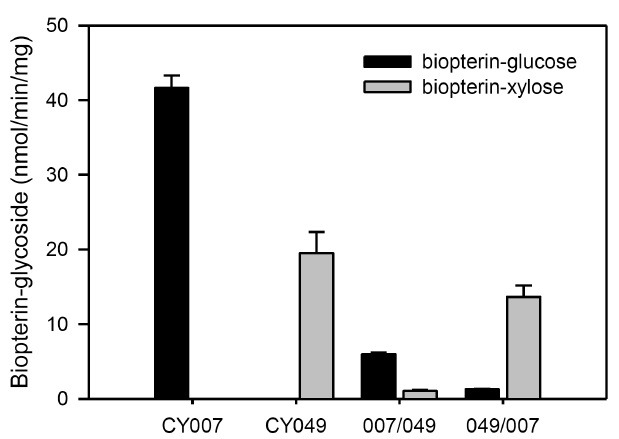
Catalytic activities of the substitution mutants
The activity and substrate specificity of both PGTs were affected profoundly by amino acid substitution (Fig. 3A, 4). The specific activity of CY-007 PGT towards UDP-glucose was increased 1.9-and 1.6-fold by Q258E and E218A/Q258E substitutions, respectively, whereas diminished to 5% by E218A substitution (Fig. 3A). All the mutants of CY-007 PGT gained catalytic activity towards UDP-xylose (Fig. 3A). Remarkably, the specific activity was 2.7-fold higher in E218A mutant than CY-049 PGT, but not significant in the others. Unexpectedly, all the mutants of CY-007 PGT gained another catalytic activity to transfer the sugar moiety of UDP-N-acetylglucosamine (UDP-NAG) to BH4 (Fig. 3B). CY-049 PGT also gained catalytic activity towards UDP-glucose by amino acid substitution, although not remarkable (Fig. 4). Interestingly, similar to the substitution in CY-007 PGT the specific activity of CY-049 PGT towards UDP-xylose was improved 1.8- and 4.2-fold by A218E and A218E/E258Q substitutions, respectively. The results strongly support that the residues 218 and 258 are essential for catalysis as well as sugar donor specificity.
Fig. 3. Specific activities of the single and dual substitution mutants of CY-007 PGT (A) and HPLC analysis of the enzymatic products (B).
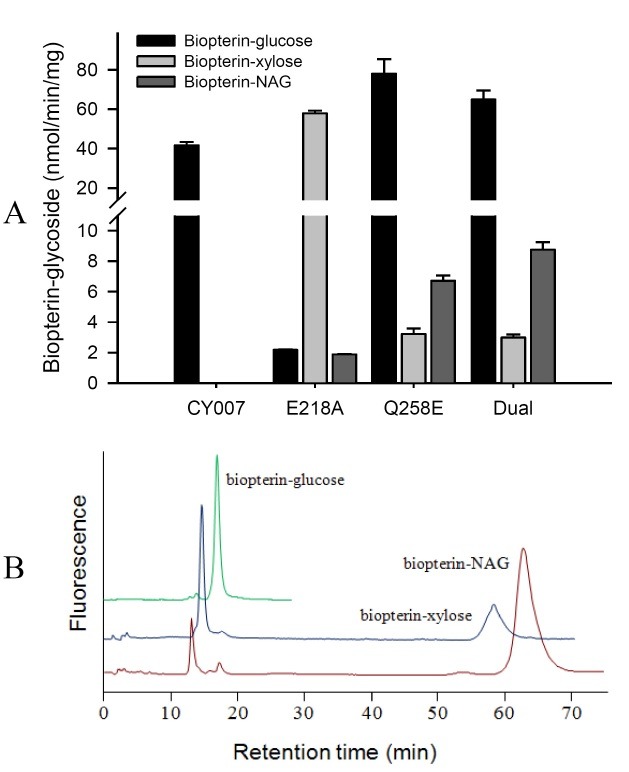
Fig. 4. Specific activities of the single and dual substitution mutants of CY-049 PGT.
Conclusion
We were able to change not only sugar donor specificity but also catalytic activity of CY-007 and CY-049 PGTs via structure-guided domain swapping or site-directed mutagenesis, thus proving the functional role of the C-terminal domain and its crucial amino acid residues 218 Glu/Ala and 258 Gln/Glu for catalysis in the PGTs. The results provide experimental insight into the structural features of the PGTs, for which protein structure has not been determined yet, and also a possibility of creating variant PGTs having different substrate specificity and enhanced catalytic activity.
MATERIALS AND METHODS
Domain swapping
Two chimeric proteins were constructed by individually replacing the C-terminal domains of CY-007 and CY-049 PGTs with the corresponding ones. The N-terminal (1-200 residues) and C-terminal (201-359 residues) halves of both PGT genes, which were obtained previously (8), were amplified by PCR. The primer sequences were 5’-CATATGACTCCAACAAGCTGGAAATTA CTATTT-3’ (N007f), 5’-GCGCTCCATTTCTAAGCCACTTC-3’ (N007r), TATCAGTTTTGTGAGCAACCCCATC (C007f), 5’-GGATCCTCAATTTTTAAGCCGGGTAACATC-3’ (C007r), 5’-CATATGCCTCAAAAAAGCTGGAAATTACTATTT-3’ (N049f), 5’-GCGATCCATTTCTAAGCCACTTCC-3’ (N049r), 5’-TATCAGTTTTGTGATCAACCCCATCAA-3’ (C049f), and 5’-GGATCCTCAATTTTTA AGCGCGATCACAT-3’ (C049r). The N- and C-terminal halves were blunt end ligated and then amplified for the full-length chimeric DNAs (N007/C049 and N049/C007) using the primer pairs N007f/C049r and N049f/C007r, respectively.
Site-directed mutagenesis
Single and dual substitutions of the amino acid residues at 218 and 258 in both CY-007 and CY-049 PGTs were performed using the overlapping-extension PCR method (15). The primer pairs for E218A substitution in CY-007 PGT were N007f/5’- GAATTTTGCCAAAAATTGCCAGAGGGATAC-3’ (E218Ar) and 5’-GTATCCCTCTGGCAATTTTTGGCAAAATTC-3’ (E218Af)/C007r. Two more primers were prepared for Q258E substitution: 5’-CCGACATTCACGCAGACCCTGTTG-3’ (Q258Er) and 5’-CAACAGGGTCTGCGTGAATGTCGG-3’ (Q258Ef). The incorporated nucleotide change is marked by underline. PCR was performed with the primer pairs using CY-007 PGT gene as a template. For dual substitution (E218A/Q258E) the E218A DNA was used as a template for Q258E substitution. The amplified DNAs were purified by agarose gel electrophoresis and then mixed in equal proportions in a PCR reaction to amplify the full-length sequence using a primer pair of N007f and C007r. The same procedure was employed to mutate CY-049 PGT gene using the following primers: N049f, C049r, 5’-GAATTTTGCCAAAAATTTCCAGAGGGATACCTG-3’ (A218Er), 5’-CAGGTATCCCTCTGGAAATTTTTGGCAAAATTC-3’ (A218Ef), 5’-CCTCGACATTGACGCAGACCCTGTTG-3’ (E258-Qr), and 5’-CAACAGGGTCTGCGTCAATGTCGAGG-3’ (E258Qf).
PCR amplifications were performed with Pfu polymerase in 1× reaction buffer, 1.5 mM MgCl2, 200 μM dNTPs, 0.2 μM each of primer pairs, and templates, under the following conditions: 4 min at 95℃, followed by 30 cycles of 94℃ for 1 min, 61℃ for 1 min, and 72℃ for 1 min, and a final polymerization at 72℃ for 10 min. The amplified DNAs were cloned into the T-Easy vector and sequenced to confirm the nucleotide change at the correct position.
Heterologous expression and purification
The chimeric and mutant DNAs were cloned as NdeI/BamHI restriction fragments into pET-28a and subsequently transformed into Escherichia coli BL21(DE3)pLysS competent cells. The cells were induced with 0.1 mM isopropyl-β-D-thio-galactopyranoside and cultured for 8 h at 22℃. The recombinant proteins containing 6xHis tags at the N-terminal were purified by chromatography on Ni-nitrilotriacetic acid gel (QIAGEN) following the manufacturer’s instructions. The proteins were eluted with 250 mM imidazole, dialyzed against 20 mM Tris-HCl (pH 7.5) and 10% (v/v) glycerol, and stored at −70℃ in aliquots until use. Purification of the proteins was confirmed by electrophoresis on a SDS-polyacrylamide gel.
PGT activity assay
PGT activity was determined following the previous method (8). Aliquots of PGT (1-5 μg) were assayed at 37℃ for 10 min in the reaction mixture of 50 μl containing 50 mM sodium phosphate, pH 7.5, 10 mM MnCl2, 0.2% ascorbic acid, 1 μM BH4, and 100 μM UDP-sugars (UDP-glucose, -xylose, -N-acetylglucosamine, -N-acetylgalactose, -N-acetylgalactosamine, -N-acetylglucuronic acid, and -N-acetylgalacturonic acid), which were available commercially. The reaction mixture was mixed with an equal volume of acidic iodine solution (2% KI and 1% I2 in 1 N HCl) and left for 1 h in the dark. After centrifugation, the supernatant was mixed in a 10 : 1 volume ratio with 5% ascorbic acid and subjected to high-performance liquid chromatography (HPLC), which was performed with a Gilson 321 pump equipped with an Inertsil ODS-3 column (5 μm, 150 × 2.3 mm, GL Science, Japan) and a fluorescence detector (Shimadzu RF-10AXL). Pteridines eluted with 10 mM potassium phosphate buffer (pH 6.0) at a flow rate of 1.2 ml/min were monitored at 350/450 nm (excitation/emission).
Statistical analysis
All of the experiments described above were performed in triplicate, and the results were expressed as the mean ± S.D.
Acknowledgments
This work was supported by the 2010 Inje University research grant.
References
- 1.Chung H. J., Kim Y.-A., Kim Y. J., Choi Y. K., Hwang Y. K., Park Y. S. Purification and characterization of UDP-glucose: tetrahydrobiopterin glucosyltransferase from Synechococcus sp. PCC 7942. Biochim. Biophys. Acta. (2000);1524:183–188. doi: 10.1016/S0304-4165(00)00156-2. [DOI] [PubMed] [Google Scholar]
- 2.Werner E. R., Blau N., Thöny B. Tetrahydrobiopterin: biochemistry and pathophysiology. Biochem. J. (2011);438:397–414. doi: 10.1042/BJ20110293. [DOI] [PubMed] [Google Scholar]
- 3.Lairson L. L., Henrissat B., Davies G. J., Withers S. G. Glycosyltransferases: structures, functions, and mechanisms. Annu. Rev. Biochem. (2008);77:521–555. doi: 10.1146/annurev.biochem.76.061005.092322. [DOI] [PubMed] [Google Scholar]
- 4.Truman A. W., Dias M. V., Wu S., Blundell T. L., Huang F., Spencer J. B. Chimeric glycosyltransferases for the generation of hybrid glycopeptides. Chem. Biol. (2009);16:676–685. doi: 10.1016/j.chembiol.2009.04.013. [DOI] [PubMed] [Google Scholar]
- 5.Park S. H., Park H. Y., Sohng J. K., Lee H. C., Liou K., Yoon Y. J., Kim B. G. Expanding substrate specificity of GT-B fold glycosyltransferase via domain swapping and high-throughput screening. Biotechnol. Bioeng. (2009);102:988–994. doi: 10.1002/bit.22150. [DOI] [PubMed] [Google Scholar]
- 6.Palcic M. M. Glycosyltransferases as biocatalysts. Curr. Opin. Chem. Biol. (2011);15:226–233. doi: 10.1016/j.cbpa.2010.11.022. [DOI] [PubMed] [Google Scholar]
- 7.Kim H.-L., Kim D. H., Lee Y. K., Park S. O., Lee Y.-W., Kwon O.-S., Park Y. S. An enzymatic method to distinguish tetrahydrobiopterin from oxidized biopterins using UDP-glucose:tetrahydrobiopterin glucosyltransferase. Anal. Biochem. (2010);397:79–83. doi: 10.1016/j.ab.2009.10.007. [DOI] [PubMed] [Google Scholar]
- 8.Lee Y. G., Kim A. H., Park M. B., Kim H. L., Lee K. H., Park Y. S. Molecular cloning of cyanobacterial pteridine glycosyltransferases that catalyze the transfer of either glucose or xylose to tetrahydrobiopterin. Appl. Environ. Microbiol. (2010);76:7658–7661. doi: 10.1128/AEM.01083-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ruane K. M., Davies G. J., Martinez-Fleites C. Crystal structure of a family GT4 glycosyltransferase from Bacillus anthracis ORF BA1558. Proteins. (2008);73:784–787. doi: 10.1002/prot.22171. [DOI] [PubMed] [Google Scholar]
- 10.Martinez-Fleites C., Proctor M., Roberts S., Bolam D. N., Gilbert H. J., Davies G. J. Insights into the synthesis of lipopolysaccharide and antibiotics through the structures of two retaining glycosyltransferases from family GT4. Chem. Biol. (2006);13:1143–1152. doi: 10.1016/j.chembiol.2006.09.005. [DOI] [PubMed] [Google Scholar]
- 11.Arnold K., Bordoli L., Kopp J., Schwede T. The SWISS-MODEL Workspace: A web-based environment for protein structure homology modelling. Bioinformatics. (2006);22:195–201. doi: 10.1093/bioinformatics/bti770. [DOI] [PubMed] [Google Scholar]
- 12.Kabsch W., Sander C. Dictionary of protein secondary structure: pattern recognition of hydrogenbonded and geometrical features. Biopolymers. (1983);22:2577–2637. doi: 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
- 13.Gouet P., Courcelle E., Stuart D. I., Metoz F. ESPript: multiple sequence alignments in PostScript. Bioinformatics. (1999);15:305–308. doi: 10.1093/bioinformatics/15.4.305. [DOI] [PubMed] [Google Scholar]
- 14.Hansen E. H., Osmani S. A., Kristensen C., Møller B. L., Hansen J. Substrate specificities of family 1 UGTs gained by domain swapping. Phytochem. (2009);70:473–482. doi: 10.1016/j.phytochem.2009.01.013. [DOI] [PubMed] [Google Scholar]
- 15.Ho S. N., Hunt H. D., Horton R. M., Pullen J. K., Pease L. R. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene. (1989);77:51–59. doi: 10.1016/0378-1119(89)90358-2. [DOI] [PubMed] [Google Scholar]