

Abstract
Methyl-6,7-dihydroxy-1,2,3,4-tetrahydroisoquinoline (salsolinol), an endogenous neurotoxin, is known to perform a role in the pathogenesis of Parkinson’s disease (PD). In this study, we evaluated oxidative modification of cytochrome c occurring after incubation with salsolinol. When cytochrome c was incubated with salsolinol, protein aggregation increased in a dosedependent manner. The formation of carbonyl compounds and the release of iron were obtained in salsolinol- treated cytochrome c. Salsolinol also led to the release of iron from cytochrome c. Reactive oxygen species (ROS) scavengers and iron specific chelator inhibited the salsolinol-mediated cytochrome c modification and carbonyl compound formation. It is suggested that oxidative damage of cytochrome c by salsolinol might induce the increase of iron content in cells, subsequently leading to the deleterious condition which was observed. This mechanism may, in part, provide an explanation for the deterioration of organs under neurodegenerative disorders such as PD. [BMB Reports 2013; 46(2): 119-123]
Keywords: Cytochrome c, Reactive oxygen species, Salsolinol
INTRODUCTION
The catechol isoquinoline derivatives are endogenous compounds that are present in the mammalian brain (1), and are also detected in the urine of patients suffering from Parkinson’s disease (PD) who have been treated with L-DOPA (2). The representative catechol isoquinoline compound is referred to as salsolinol (1-methyl-6,7-dihydroxy-1,2,3,4-tetrahydro-isoquinoline). Increased salsolinol levels are detected in the brain of PD patients (3, 4). Previous studies have shown that salsolinol is toxic to dopaminergic neurons under both in vitro and in vivo conditions. It has also been reported that salsolinol in conjugation with iron undergoes redox cycling to generate reactive oxygen species (ROS), most notably hydroxyl radicals, which induce DNA strand scission and cell death (5).
Iron is the most abundant heavy metal in the human body, and is particularly abundant in the brain. Although iron required for oxidative metabolism, it can also be toxic, owing its ability to catalyze ROS generation (6). Iron accumulates progressively with aging in some regions of the brain, often to the high levels which are associated with oxidative stress; this process has been previously shown to promote neurodegeneration (7,8). The iron released from cytochrome c, and its associated toxicity, has also been implicated as a risk factor for age of onset for certain neurodegenerative diseases, namely PD and Alzheimer’s disease (AD) (9,10).
Cytochrome c is well known as the penultimate electron transport protein of the eukaryotic respiratory chain (11). The covalent, posttranslational attachment of a heme c moiety to the nuclear-encoded apoprotein confers the native structure (12). The heme is necessary for the protein to attain its proper three-dimensional folding pattern (13). In the mitochondria, cytochrome c transfers an electron from cytochrome c reductase to cytochrome c oxidase. These electron transfer reactions are mediated by a conserved cytochrome c-binding epitope (14,15).
Recent discoveries implicate this protein in two other biological processes, apoptosis and oxidative stress (16,17). Apoptosis, also referred to as programmed cell death, is a key to development and is linked to human diseases, such as cancer and neurodegeneration (18,19). The cellular damage induced by oxidative stress has also been associated with several diseases, including Parkinson’s disease (PD). More specifically, it has been shown that cytochrome c is co-localized with α-synuclein aggregates in Lewy bodies, the pathological hallmarks of PD (15). It has also been shown that cytochrome c catalyzes the H2O2-induced aggregation of α-synuclein (17). Although all of these observations are archetypical results inducible by free radicals, it is not yet clearly understood how free radicals are generated by the effect of cytochrome c, or which initial and cascading radical species are responsible for these damages in vivo.
In the current study, the effects of salsolinol on the oxidative modification of cytochrome c were assessed. The results demonstrated that salsolinol induced cytochrome c aggregation via free radical generation. Salsolinol-mediated cytochrome c modification, in turn, resulted in the release of iron from the protein.
RESULTS AND DISCUSSION
All cellular components are vulnerable to oxidative stress. The damage that oxidative stress causes to proteins includes side chain modification and main chain fragmentation. One of the biological markers for oxidative damage in proteins is the accumulation of carbonyl compounds (20). In the present study, salsolinol led to the oligomerization of cytochrome c (Fig. 1A), and the formation of carbonyl formation (Fig. 1B). These phenomena were increased in a salsolinol concentration-dependent manner. Metal catalyzed oxidation may cause covalent modification of proteins by introducing carbonyl groups into the amino acid residues of proteins (21). The autoxidation of salsolinol induced ROS formation that oxidizes amino acid residues at or near that cation-binding site, which then introduces carbonyl groups. Such an oxidative modification is an indicator of oxidative stress and may be significant in several physiological and pathological processes (22,23). The participation of ROS in the oligomerization of cytochrome c by salsolinol was studied by examining the protective effects of ROS scavengers. When cytochrome c was incubated with salsolinol in the presence of ROS scavengers, azide, formate, glutathione and N-acetyl-cysteine prevented the oligomerization of cytochrome c (Fig. 2A). These scavengers also prevented the formation of carbonyl compounds (Fig. 2B). The results suggest that ROS might play a critical role in the mechanism of salsolinol-mediated cytochrome c modification.
Fig. 1. Oxidative modification of cytochrome c after incubation with salsolinol. 20 μM cytochrome c was incubated with various concentrations of salsolinol in 10 mM phosphate buffer (pH 7.4) at 37℃ for 24 h. Reactions were stopped at the indicated time by freezing at −80℃. (A) The pattern of protein bands was analyzed via SDS-PAGE. The positions of the molecular weight markers (kDa) are indicated on the left. (B) To determined the formation of carbonyl compounds, the reaction mixtures were reacted with 2,4-dinitrophenyl hydrazine and then the derivatives were measured using spectrophotometry.
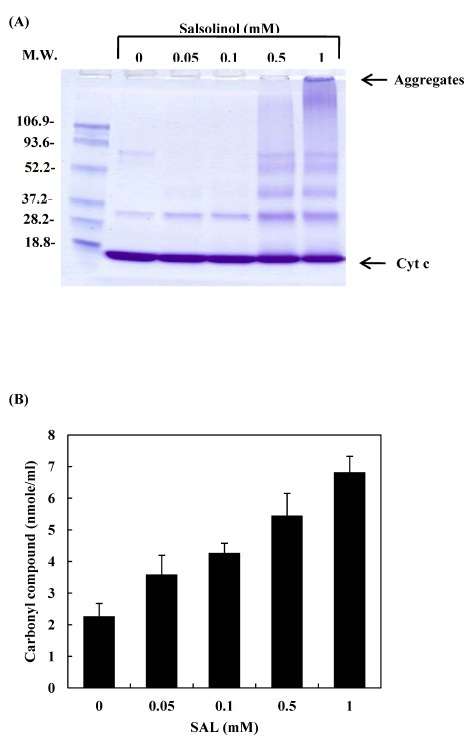
Fig. 2. The effect of ROS scavengers on salsolinol-mediated cytochrome c aggregation and carbonyl compound formation. 20 μM cytochrome c was incubated with 0.5 mM salsolinol in 10 mM phosphate buffer (pH 7.4) at 37℃ for 24 h in the presence of ROS scavengers. (A) The pattern of protein bands was analyzed via SDS-PAGE. Lane 1, cytochrome c control; lane 2, oxidized cytochrome c (without ROS scavenger); lane 3, 100 mM azide; lane 4, 100 mM formate; lane 5, 20 mM glutathione (GSH); lane 6, 20 mM N-acetyl cysteine (NAC). (B) The formation of carbonyl compounds was determined by spectrophotometry.

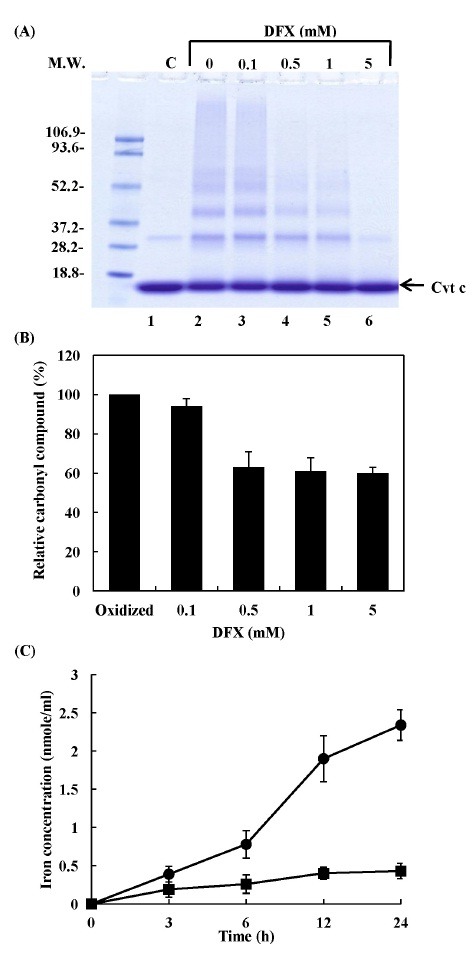
Trace metals such as iron and copper, which are present in a variety of biological systems, may interact with active oxygen species, ionizing radiation or microwave radiation, to damage macromolecules (24-26). In some biological cells, the cleavage of metalloproteins by oxidative damage may lead to increases in the levels of metal ions (27). The participation of iron in the salsolinol-mediated cytochrome c modification was investigated by an examination of the protective effects of the iron chelator, deferoxamine (DFX). When cytochrome c was incubated with salsolinol in the presence of DFX, the oligomerization of cytochrome c (Fig. 3A) and carbonyl compound formation (Fig. 3B) were inhibited in a DFX concentration-dependent manner. The reaction of cytochrome c with 0.5 mM salsolinol resulted in a time-dependent increase in the release of free iron ions (Fig. 3C). Through Fenton-like reactions, hydroxyl radicals can be formed from H2O2 in the presence of a transition metal ion, such as iron or copper. Therefore, iron ions released from salsolinol-damaged cytochrome c may lead to a pro-oxidant condition in cells.
Fig. 3. Effects of iron chelator on salsolionl-mediated cytochrome c aggregation and carbonyl compound formation and salsolinol-mediated iron release from cytochrome c. 20 μM cytochrome c was incubated with 0.5 mM salsolinol in 10 mM phosphate buffer (pH 7.4) at 37℃ for 24 h in the presence of DFX. (A) The pattern of protein bands was analyzed via SDS-PAGE. Lane 1, cytochrome c control; lane 2, oxidized cytochrome c (without DFX); lane 3, 0.1 mM DFX; lane 4, 0.5 mM DFX; lane 5, 1 mM DFX; lane 6, 5 mM DFX. (B) The formation of carbonyl compounds was determined by spectrophotometry. (C) The reaction mixture contained 20 μM cytochrome c and 0.5 mM salsolinol in 10 mM phosphate buffer at pH 7.4, and was incubated for various periods of time as: cytochrome c alone (■); cytochrome c plus salsolinol (●). Free iron ions were determined with a colorimetric reagent using bathophenanthroline sulfonate.
In order to determine the target site against the salsolinol on the protein, cytochrome c which had been reacted with salsolinol was evaluated by amino acid analysis following acid and alkaline hydrolysis of the modified proteins. Cytochrome c exposure to salsolinol produced particularly losses of histidine, isoleucine, lysine, methionine, phenylalanine and tyrosine residues (Fig. 4). Presumably, the primary, secondary and tertiary structure of a protein greatly influences the reactivity of each amino acid. The amino acid losses presented in Fig. 4 would be expected to have a dramatic effect on cellular structure and function. Cytochrome c is a small globular protein that contains a covalently-bound heme, located in an internal pocket formed by highly-conserved amino acid residues (28). Porphyrins are covalently bound to Cys-14 and Cys-17, with the fifth and sixth coordination positions of the heme-Fe interacting with His-18 and Met-80, respectively. Thus, it is suggested that iron binding sites were modified during the reaction of cytochrome c with salsolinol. Consequently, iron became almost free form the ligand, and was released from the oxidatively-damaged protein. Taken together the present findings suggest that salsolinol-mediated cytochrome c oligomerization might be due to oxidative damage resulting from ROS generated by a combination of the autoxidation of salsolinol and the Fenton-like reaction of free iron ions released from oxidatively-damaged cytochrome c.
Fig. 4. Modification of amino acid residues in cytochrome c by salsolinol. Cytochrome c was incubated in 10 mM potassium phosphate buffer (pH 7.4) at 37℃ with the following conditions: 20 μM cytochrome c alone (dark bar); 20 μM cytochrome c plus 0.5 mM salsolinol (gray bar). After incubation for 24 h, the amino acid composition of acid and alkaline hydrolysates was determined, as described in Materials and Methods.

Alterations in iron homeostasis/metabolism may be involved in the pathogenesis of neurodegenerative disease. Iron released from cytochrome c may trigger oxidative stress, which can result in the progressive neurodegeneration of the substantia nigra and may ultimately result in PD (17). The toxicity of salsolinol may be augmented by its ROS-generating activity in neurodegenerative disorders. It has been noted in previous studies that salsolinol induces DNA strand breaks in PC12 cells and neurons in the presence of copper or iron (5,29). A recent report has also shown that salsolinol increases ROS generation and effects a significant reduction in glutathione levels in SH-SY5Y cells (30).
In conclusion, the findings of the current study demonstrate that salsolinol induces the oxidative modification of cytochrome c and subsequent iron release, and that this phenomenon involves free radical generation. Salsolinol-mediated cytochrome c modification may be associated with the pathogenesis of PD, as well as related disorders.
MATERIALS AND METHODS
Materials
Horse cytochrome c, salsolinol, azide, formate, N-acetyl-L-cysteine, glutathione, trichloroacetic acid (TCA) and deferoxamine (DFX) were obtained from Sigma (St. Louis, MO, USA). Chelex 100 resin (sodium form) was obtained from Bio-Rad (Hercules, CA, USA). All solutions were treated with Chelex 100 resin in order to remove any traces of transition metal ions.
Protein oxidative modification
Protein concentrations were determined using the BCA method (31). 20 μM cytochrome c modification was induced via incubation with salsolinol in phosphate buffer (pH 7.4) at 37℃. After the incubation of the reaction mixtures, the mixtures were placed onto a Vivaspin ultrafiltration spin column (Sartorius Stedim Biotech, Goettingen, Gerrman) and were centrifuged for 1 h at 13,000 rpm to remove the salsolinol. The mixture was then washed with Chelex 100-treated water and centrifuged for 1 h at the same speed to remove any remaining salsolinol. This procedure was repeated four times, after which the filtrates were dried in a freeze-dryer and dissolved with 10 mM potassium phosphate buffer (pH 7.4). The free radical scavenger-induced protection against salsolinol-mediated cytochrome c modification was induced by the 5 min of preincubation of the enzyme with free radical scavengers at room temperature, after which the mixture was reacted with salsolinol for 24 h at 37℃. The unreacted reagent was then washed through a Microcon filter (Amicon).
Analysis of cytochrome c modification
The samples were treated with 7 μl of concentrated sample buffer (0.25 M Tris, 8% SDS, 40% glycerol, 20% β-mercaptoethanol, 0.01% bromophenol blue) and were then boiled for 10 min at 100℃ prior to electrophoresis. Each sample was then subjected to SDS-PAGE via the Laemmli method (32), using an 18% acrylamide slab gel. The gels were stained with 0.15% Coomassie Brilliant Blue R-250.
Detection of carbonyl compounds
The carbonyl contents of the proteins were determined via spectrophotometric assays, as described elsewhere (33). Both native and oxidized proteins were incubated with 10 mM 2,4-DNPH in 2.5 M HCl for 1 h at room temperature. After incubation, 20% TCA was added to the samples, and the tubes were left in an ice bucket for 10 min and then centrifuged for 5 min with a tabletop centrifuge to collect the protein precipitates, after which the supernatants were discarded. Another wash was performed using 10% TCA, and the protein pellets were mechanically broken using a pipette tip. Finally, the pellets were washed 3 times with ethanol-ethyl acetate (1:1) (v/v) to remove any free DNPH. The final precipitates were dissolved in 2 ml of 6 M guanidine hydrochloride solution and left for 10 min at 37℃ with general vortex mixing. Carbonyl contents were calculated from the absorbance (370 nm) with an absorption coefficient ε of 22,000 M-1 cm-1.
Determination of free iron ions concentration
The concentration of iron ions released from oxidatively damaged cytochrome c was determined by using bathophenanthroline sulfonate in the method described previously (34). The reaction mixture contained 20 μM cytochrome c, 0.5 mM salsolinol in a total volume of 0.5 ml. The reaction was initiated by the addition of salsolinol and was incubated for various periods of time at 37℃. After incubation, the mixtures were placed into a Vivaspin ultrafiltration spin column (Sartorius Stedim Biotech, Goettingen, Gerrman) and were then centrifuged at 13,000 rpm for 1 h. The colorimetric reagent was added to the filtrate prior to analysis by UV/VIS spectrophotometry (Shimadzu, UV-1601) at 535 nm. The final concentrations of the color reagent were 1% ascorbate, 0.02% bathophenanthroline sulfonate and 1% acetic acid-acetate buffer (pH 4.5).
Amino acid analysis
Aliquots of modified and native cytochrome c were hydrolyzed at 110℃ for 24 h after the addition of 6 N HCl. Since acid hydrolysis destroys tryptophan, the tryptophan content of oxidized and native cytochrome c preparations was determined by alkaline hydrolysis, as described previously (35). The amino acid content of acid and alkaline hydrolysates was determined by HPLC separation of their phenylisothiocyanatederivatives by using a Pico-tag free amino acid analysis column and a 996 photodiode array detector (Waters, USA).
Statistical analysis
Values are expressed as the means ± S.D of three to five separate experiments. The statistical differences between the means were determined by the Student t-test.
References
- 1.Götz M. E., Künig G, Riderer P., Youdim M. B. Oxidative stress: free radical production in neural degeneration. Pharmacol. Ther. (1994);63:37–122. doi: 10.1016/0163-7258(94)90055-8. [DOI] [PubMed] [Google Scholar]
- 2.Sandler M., Carter S. B., Hunter K. R., Stern G. M. Tetrahydroisoquinoline alkaloids: in vivo metabolites of L-dopa in man. Nature. (1973);241:439–443. doi: 10.1038/241439a0. [DOI] [PubMed] [Google Scholar]
- 3.Epp L. M., Marvec B. Chronic polysystemic candidiasis as a possible contributor to onset of idiopathic Parkinson's disease. Bratisl. Lek. Listy. (2006);107:227–230. [PubMed] [Google Scholar]
- 4.DeCuypere M., Lu Y., Miller D. D., LeDoux M. S. Regional distribution of tetrahydroisoquinoline derivatives in rodent, human, and Parkinson's disease brain. J. Neurochem. (2008);107:1398–1413. doi: 10.1111/j.1471-4159.2008.05709.x. [DOI] [PubMed] [Google Scholar]
- 5.Surh Y. J., Jung Y. J., Jung J. H., Lee J. S., Yoon H. R. Iron enhancement of oxidative DNA damage and neuronal cell death induced by salsolinol. J. Toxicol. Environ. Health Part A. (2002);65:473–488. doi: 10.1080/15287390252808127. [DOI] [PubMed] [Google Scholar]
- 6.Galaris D., Pantopoulos K. Oxidative stress and iron homeostasis: mechanistic and health aspects. Crit. Rev. Clin. Lab. Sci. (2008);45:1–23. doi: 10.1080/10408360701713104. [DOI] [PubMed] [Google Scholar]
- 7.Thomas M., Jankovic J. Neurodegenerative disease and iron storage in the brain. Curr. Opin. Neurol. (2004);17:437–442. doi: 10.1097/01.wco.0000137534.61244.d1. [DOI] [PubMed] [Google Scholar]
- 8.Zecca L., Youdim M. B., Riederer P., Connor J. R., Crichton R. R. Iron, brain ageing and neurodegenerative disorders. Nat. Rev. Neurosci. (2004);5:863–873. doi: 10.1038/nrn1537. [DOI] [PubMed] [Google Scholar]
- 9.Viña J., Lloret A., Vallés S. L., Borrás C., Badía M. C., Pallardó F. V., Sastre J., Alonso M. D. Effect of gender on mitochondrial toxicity of Alzheimer’s Abeta peptide. Antioxid. Redox. Signal. (2007);10:1677–1690. doi: 10.1089/ars.2007.1773. [DOI] [PubMed] [Google Scholar]
- 10.Jomova K., Vondrakova D., Lawson M., Valko M. Metals, oxidative stress and neurodegenerative disorders. Mol. Cell Biochem. (2010);345:91–104. doi: 10.1007/s11010-010-0563-x. [DOI] [PubMed] [Google Scholar]
- 11.Moore G. R., Pettigrew G. W. Cytochrome c: Evolution, Structure, and Physicochemical Aspects. Springer-Verlag; Berlin, Germany: (1990). [Google Scholar]
- 12.Dumont M. E., Cardillo T. S., Hayes M. K., Sherman F. Role of cytochrome c heme lyase in mitochondrial import and accumulation of cytochrome c in Saccharomyces cerevisiae. Mol. Cell Biol. (1991);11:5487–5496. doi: 10.1128/mcb.11.11.5487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dumont M. E., Corin A. F., Campbell G. A. Noncovalent binding of heme induces a compact apocytochrome c structure. Biochemistry. (1994);33:7368–7378. doi: 10.1021/bi00189a043. [DOI] [PubMed] [Google Scholar]
- 14.Pelletier H., Kraut J. Crystal structure of a complex between electron transfer partners, cytochrome c peroxidase and cytochrome c. Science. (1992);258:1748–1755. doi: 10.1126/science.1334573. [DOI] [PubMed] [Google Scholar]
- 15.Wang X., Pielak G. J. Equilibrium thermodynamics of a physiologically-relevant heme-protein complex. Biochemistry. (1999);38:16876–16881. doi: 10.1021/bi992005i. [DOI] [PubMed] [Google Scholar]
- 16.Li P., Nijhawan D., Budihardjo I., Srinivasula S. M., Ahmad M., Alnemri E. S., Alnemri X. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell. (1997);91:479–489. doi: 10.1016/S0092-8674(00)80434-1. [DOI] [PubMed] [Google Scholar]
- 17.Hashimoto M., Takeda A., Hsu L. J., Takenouchi T., Masliah E. Role of cytochrome c as a stimulator of alpha-synuclein aggregation in Lewy body disease. J. Biol. Chem. (1999);274:28849–28852. doi: 10.1074/jbc.274.41.28849. [DOI] [PubMed] [Google Scholar]
- 18.Green D. R., Evan G. I. A matter of life and death. Cancer Cell. (2002);1:19–30. doi: 10.1016/S1535-6108(02)00024-7. [DOI] [PubMed] [Google Scholar]
- 19.Friedlander R. M. Apoptosis and caspases in neurodegenerative diseases. N. Engl. J. Med. (2003);348:1365–1375. doi: 10.1056/NEJMra022366. [DOI] [PubMed] [Google Scholar]
- 20.Stadtman E. R., Berlett B. S. Reactive oxygen- mediated protein oxidation in aging and disease. Drug. Metab. Rev. (1998);30:225–243. doi: 10.3109/03602539808996310. [DOI] [PubMed] [Google Scholar]
- 21.Stadtman E. R. Oxidation of free amino acids and amino acid residues in proteins by radiolysis and by metal-catalyzed reactions. Annu. Rev. Biochem. (1993);62:797–821. doi: 10.1146/annurev.bi.62.070193.004053. [DOI] [PubMed] [Google Scholar]
- 22.Davies K. J. Intracellular proteolytic systems may function as secondary antioxidant defenses: an hypothesis. J. Free Radic. Biol. Med. (1986);2:155–173. doi: 10.1016/S0748-5514(86)80066-6. [DOI] [PubMed] [Google Scholar]
- 23.Oliver C. N., Levine R. L., Stadtman E. R. A role of mixed-function oxidation reactions in the accumulation of altered enzyme forms during aging. J. Am. Geriatr. Soc. (1987);35:947–956. doi: 10.1111/j.1532-5415.1987.tb02297.x. [DOI] [PubMed] [Google Scholar]
- 24.Goldstein S., Czapski G. Mechanism of reduction of bleomycin-Cu(II) by CO2- and oxidation of bleomycin-Cu(I) by H2O2 in the absence and presence of DNA. Free Radic. Res. Commun. (1987);51:693–706. doi: 10.1080/09553008414552221. [DOI] [PubMed] [Google Scholar]
- 25.Gutteridge J. M., Halliwell B. The role of the superoxide and hydroxyl radicals in the degradation of DNA and deoxyribose induced by a copper-phenanthroline complex. Biochem. Pharmacol. (1982);31:2801–2805. doi: 10.1016/0006-2952(82)90136-8. [DOI] [PubMed] [Google Scholar]
- 26.Imlay J. A., Chin S. M., Linn S. Toxic DNA damage by hydrogen peroxide through the Fenton reaction in vivo and in vitro. Science. (1988);240:640–642. doi: 10.1126/science.2834821. [DOI] [PubMed] [Google Scholar]
- 27.Prutz W. A. Inhibition of DNA-ethidium bromide intercalation due to free radical attack upon DNA. II. Copper(II)-catalysed DNA damage by O2-ㆍ. Radiat. Environ. Biophys. (1984);23:7–18. doi: 10.1007/BF01326732. [DOI] [PubMed] [Google Scholar]
- 28.Millett F., Scott R. A. Structual studies; in Cytochrome c, A Multidisciplinary Approach, Mauk, A. G. and Scott, R. A. (eds.) University Science Books; Sausalito, CA: (1996). pp. 475–487. [Google Scholar]
- 29.Jung Y. J., Surh Y. J. Oxidative DNA damage and cytotoxicity induced by copper-stimulated redox cycling of salsolinol, a neurotoxic tetrahydroisoquinolin alkaloid. Free Radic. Biol. Med. (2001);30:1407–1417. doi: 10.1016/S0891-5849(01)00548-2. [DOI] [PubMed] [Google Scholar]
- 30.Wanpen S., Govitrapong P., Shavali S., Sangchot P. M., Ebadi M. Salsolinol, a dopamine-derived tetrahydroisoquinoline, induces cell death by causing oxidative stress in dopaminergic SH-SY5Y cells, and the said effect is attenuated by metallothionein. Brain Res. (2004);1005:67–76. doi: 10.1016/j.brainres.2004.01.054. [DOI] [PubMed] [Google Scholar]
- 31.Smith P. K., Krohn R. I., Hermanson G. T., Mallia A. K., Gartner F. H., Provenzano M. D., Fujimoto E. K., Goeke N. M., Olson B. J., Klenk D. C. Measurement of protein using bicinchoninic acid. Anal. Biochem. (1985);150:76–85. doi: 10.1016/0003-2697(85)90442-7. [DOI] [PubMed] [Google Scholar]
- 32.Laemmli U. K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. (1970);227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 33.Reznick A. Z., Packer L. Oxidative damage to proteins: spectrophotometric method for carbonyl assay. Methods Enzymol. (1994);233:357–363. doi: 10.1016/s0076-6879(94)33041-7. [DOI] [PubMed] [Google Scholar]
- 34.An S. H., Lee M. S., Kang J. H. Oxidative modification of ferritin induced by methylglyoxal. BMB Rep. (2012);45:147–152. doi: 10.5483/BMBRep.2012.45.3.147. [DOI] [PubMed] [Google Scholar]
- 35.Kang J. H. Salsolinol, a tetrahydroisoquinoline-derived neurotoxin, induced oxidative modification of neurofilament- L: protection by histidyl dipeptides. BMB Rep. (2012);45:114–119. doi: 10.5483/BMBRep.2012.45.2.114. [DOI] [PubMed] [Google Scholar]