

Abstract
The cell-fate specification of the anchor cell (AC) and a ventral uterine precursor cell (VU) in Caenorhabditis elegans is initiated by a stochastic interaction between LIN-12/Notch receptor and LAG-2/Delta ligand in two neighboring Z1.ppp and Z4.aaa cells. Both cells express lin-12 and lag-2 before specification, and a small difference in LIN-12 activity leads to the exclusive expressions of lin-12 in VU and lag-2 in the AC, through a feedback mechanism of unknown nature. Here we show that the expression pattern of lag-1/CSL, a transcriptional repressor itself that turns into an activator upon binding of the intracellular domain of Notch, overlaps with that of lin-12. Site-directed mutagenesis of LAG-1 binding sites in lag-1 maintains its expression in the AC, and eliminates it in the VU. Thus, AC/VU cell-fate specification appears to involve direct regulation of lag-1 expression by the LAG-1 protein, activating its transcription in VU cells, but repressing it in the AC. [BMB Reports 2013; 46(4): 219-224]
Keywords: Anchor cell/ventral uterine precursor cell, Cell-fate specification, CSL, Feedback loop, Notch signaling
INTRODUCTION
In metazoan development, cells adopt different fates, which lead to their own unique properties. There are several ways cell fates become specified, largely classified into autonomous and non-autonomous mechanisms (1-4). In autonomous specification, which usually happens early in embryogenesis, cytoplasmic determinants for cell fates are unevenly distributed into daughter cells. In non-autonomous situations, which involve several intercellular signaling pathways, such as Hedgehog, tumor growth factor-β, and Wnt, cell-fate induction signals produced in specific cells are delivered to competent, responding cells. Only the cells exposed to the signals are induced to perform specific differentiation programs, generally eliciting responses in a concentration gradient-dependent manner (5). Thus, the position of responding cells from the signal is important in this case. Unlike those paracrine signaling pathways that use secreted proteins as inductive molecules, the Notch signaling pathway mediates adjacent cell-cell interactions, through the interaction between membrane-bound ligand and receptor proteins (1).
Notch signaling is initiated by the interaction between Notch receptor and Delta/Serrate ligand in neighboring cells, which triggers the cleavage of the intracellular domain of Notch (NICD) that binds to CSL [CBF1/RBPJ-κ in vertebrates, Su(H) (Suppressor of hairless) in Drosophila, and LAG-1 in C. elegans], a sequence-specific DNA-binding protein that acts as a transcriptional repressor or activator (1,6-8). Although several biochemical studies showed that CSL itself is a transcriptional repressor, and becomes an activator upon binding of NICD, it is not clear whether both CSL repressor and activator activities are involved in the Notch-dependent cell-fate specification (1,9,10).
Currently, we do not understand the mechanism of how lateral inhibition (i.e. a competition for exclusive cell fates through membrane-bound receptor-ligand interaction between two or more neighboring cells) produces one cell to express exclusively Notch receptor and the other Delta ligand because few CSL target genes have been identified that are involved in the Notch-dependent cell-fate specification (1,7,8,11). However, the fact that CSL can repress or activate the transcription of the same targets suggests a model for how Notch signaling achieves cell-fate specification through lateral inhibition. Schematically, the cell that has more Notch receptor than its neighboring cells will produce more NICD, and thus increase the expression of CSL target genes. Neighboring cells that have less (or undetectable) Notch receptor will decrease or repress the target gene expression.
Cell-fate specification of the anchor cell (AC) and ventral uterine precursor cells (VUs) in the Caenorhabditis elegans gonad has been a good model system for understanding Notch-dependent lateral inhibition (9,10). In wild-type animals. two somatic cells in the gonad (Z1.ppa and Z4.aap) become VUs. Their sister cells (Z1.ppp and Z4.aaa) have equal potential to become the AC or a VU, and their cell-fates become specified by lateral inhibition between lin-12/Notch receptor and lag-2/Delta ligand on both cells. These sister cells express lin-12 and lag-2 before specification, and a small difference in LIN-12 activity leads to the exclusive expressions of lin-12 in VU and lag-2 in AC, through negative and positive feedback loops of unknown nature (11,12).
Here we show that the expression pattern of lag-1, C. elegans CSL, overlaps with that of lin-12 during AC/VU specification. The expression of lag-1::YFP (yellow fluorescent protein) occurs in pre-AC and pre-VU (Z1.ppp, Z1.ppa, Z4.aaa, Z4.aap) before specification. It is also expressed in VUs, but not in the AC after specification. The expression of lag-1::YFP requires clustered LAG-1 binding sites in the first intron of lag-1. The binding sites are necessary for suppressing lag-1 expression in AC, and for activating its expression in pre-AC, pre-VU, and VUs. These results suggest that the cells that become VUs more rapidly produce transcription activator complexes that contain CSL and NICD, by increasing both lag-1 and lin-12 expressions. In cells becoming AC, lin-12 expression could be more efficiently suppressed by suppressing lag-1 expression. Thus, exclusive expressions of lin-12 in VU and lag-2 in the AC during AC/VU specification could be established more efficiently by oppositely regulating the transcription of CSL, which acts as a transcriptional repressor or activator in neighboring cells.
RESULTS AND DISCUSSION
lag-1 contains a regulatory region that comprises clustered LAG-1 binding sites
Because only two cells are involved in the cell-fate specification, AC/VU specification has been a good model system for understanding the molecular mechanisms through which the Notch signaling pathway specifies different fates of neighboring cells. Extensive genetic screens have identified proteins that modify LIN-12 activity, but have not identified physiologically relevant downstream targets involved in AC/VU specification (8,13), which might be because of redundancy or pleiotropy of such targets. A bioinformatics-based approach that screens genes co-expressed from a common regulatory element, identified egl-43 as a direct downstream target of LIN-12/Notch that is involved in AC/VU specification (14,15). Two distinct regulatory elements in egl-43 are responsible for the expression during AC/VU specification. One element, similar to the element in lin-3/EGF ligand, is responsible for the egl-43 expression in the pre-AC, pre-VU, and AC. The other element, which contains clustered LAG-1/CSL binding sites, is necessary for the expression in the VUs immediately after specification (14).
In this study, we found that lag-1 also contains clustered LAG-1 binding sites, similar to those in egl-43. The expected frequency of LAG-1 binding sites (‘RTGGGAA’) in C. elegans genome sequence is approximately one occurrence per 8 kb, and 13 LAG-1 binding sites (10 ‘RTGGGAA’ and three ‘YTGGGAA’) are present in the 5’ region of the lag-1 first intron (1758 bp) (Fig. 2). Because LAG-1 itself is a transcriptional repressor, and becomes an activator upon binding NICD (16); and because lin-12 is exclusively expressed in the VUs, but not in the AC (10), we speculated that lag-1 expression is regulated through these clustered LAG-1 binding sites, during AC/VU specification.
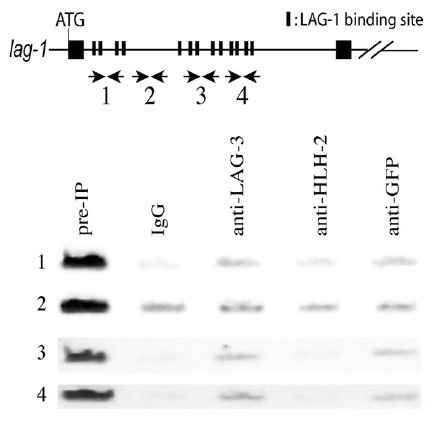
Fig. 2. In vivo LAG-1 binding to a lag-1 regulatory region. ChIP was performed with the extracts from worms expressing SEL-8::GFP. PCR analysis was performed with primers described in MATERIALS AND METHODS. LAG-1 binding sites are in the first (1sst to 4th LAG-1 binding sites in Fig. 1, as well as in the in vitro DNA binding assay section in MATERIALS AND METHODS), third (5th to 9th), and fourth (10th to 13th) sections, but not in the second section. Pre-IP represents the input extracts subjected to IP. IgG and anti-HLH-2 antibodies were used as negative controls, and anti-LAG-3 and anti-GFP antibodies were used to detect the in vivo binding of LAG-1/LAG-3 complex to LAG-1 binding sites in lag-1.
LAG-1 binds to a regulatory region in lag-1
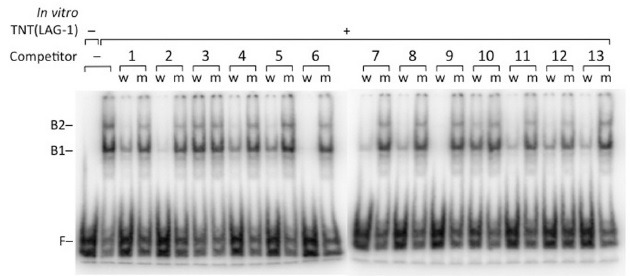
LAG-1 is the C. elegans ortholog of mammalian CBF1 and D. melanogaster Su(H), and all three of these are known to bind to ‘RTGGGAA’ in vitro (16,17). Thus, we tested whether LAG-1 binds to the 13 predicted binding sites, including the three ‘YTGGGAA’ sequences in lag-1. An electrophoretic mobility shift assay was performed with in vitro transcribed and translated LAG-1 protein, and a 32P-labeled DNA probe that contained the CSL binding site. During the mobility shift assay, double-stranded oligonucleotides that contain each of the 13 predicted LAG-1 binding sites were added as competitor. As shown in Fig. 1, binding was well-competed by adding competitor oligonucleotides that contain ‘ATGGGAA’ and ‘GTGGGAA’ sequences. Interestingly, all three competitors that contain ‘CTGGGAA’ (4 and 12 in Fig. 1) and ‘TTGGGAA’ (10 in Fig. 1) also well-competed the binding, suggesting that the presence of a purine base before ‘TGGGAA’ is not required for LAG-1 binding to its targets.
Fig. 1. In vitro LAG-1 binding to 13 LAG-1 binding sites in the first intron of lag-1. Electrophoretic mobility shift assay (EMSA) was done with in vitro transcribed and translated (TNT) LAG-1. B1 and B2 represent shifted complexes of LAG-1 and 32P-labeled DNA probe. F indicates the migration of free DNA probe. The binding was competed by adding competitor DNAs containing the wild- type (w) LAG-1 binding sites, but not by adding the mutated (m) site sequences.
To investigate whether clustered LAG-1 binding sites in the first intron of lag-1 are direct targets of LIN-12 signaling in vivo, we carried out a chromatin immunoprecipitation (ChIP) assay (Fig. 2). Activated LIN-12 signaling results in the formation of a transcriptional activator complex among LAG-1, LAG-3 (SEL-8) and NICD; therefore, we used antibodies specific for LAG-3 (SEL-8) for the assay (18). To detect the C. elegans genomic region bound to this complex in vivo, we crosslinked the LAG-3 complexes bound to genomic DNA in vivo, by treating the animals expressing sel-8::GFP (green fluorescent protein) with formaldehyde, immunoprecipitating the complexes bound to chromatin DNA fragments with anti-LAG-3 antibodies, and performing polymerase chain reaction (PCR), to detect the enrichment of specific genomic regions in the precipitate (19). This ChIP experiment showed that the regions containing LAG-1 binding sites (regions 1, 3, and 4), but not the region lacking the LAG-1 binding sites (region 2), are enriched in the LAG-3- and GFP-specific precipitates, compared with those regions precipitated with the control IgG and HLH-2-specific antibodies (Fig. 2).
Combining the results of in vitro and in vivo experiments, we conclude that clustered LAG-1 binding sites in the first intron of lag-1 appear to be involved in the transcriptional activation by the complex of LAG-1, LAG-3, and NICD.
Clustered LAG-1 binding sites are involved in the regulatory expression of lag-1 during AC/VU specification
During the L2 stage of the C. elegans gonad, all somatic cells stop dividing, and one of them moves into the mid-sagittal plane on the ventral surface of the gonad, to become the AC (20,21). The L2 stage is generally classified into three substages (early, mid, and late) (Fig. 3). In the early stage, neither the Z1.ppp nor the Z4.aaa cells, which have equal potential to become the AC, have yet determined their fates (Fig. 3A, B). In mid L2, either the Z1.ppp or the Z4.aaa cell has moved to the central position on the ventral surface of the gonad, to become the AC. In late L2 animals, cdh-3::CFP signal starts being detected in the AC (Fig. 3C, D; in red).
Fig. 3. Expression pattern of lag-1::YFP during AC/VU specification. The lag-1(wt)::YFP construct contains 13 predicted LAG-1 binding sites in the first intronic region (1,758 bp) of lag-1. All 13 LAG-1 binding sites are mutated in the lag-1(mut)::YFP construct. (A and B) lag-1 (wt)::YFP (in green) is expressed at early L2 in the pre-AC/pre-VU cells, located in two different focal planes. (C) The AC is labeled yellow, because of the co-localization of cdh-3::CFP (in red) and lag-1(wt)::YFP (in green) at late L2. (D and E) lag-1(wt)::YFP, not cdh-3::CFP, disappears in the AC at early L3, labeling the AC in red. Lateral (D) and ventral view (E). (F) The AC (cdh-3::CFP in red) and VU descendants (lag-1 (wt)::YFP in green). (G and H) lag-1(wt)::YFP is expressed in the VU descendants at late L3 stage. (I and J) The lag-1 (mut)::YFP is not expressed well in the pre-AC/pre-VU cells, and VU cells, as well as their descendants. (K and L) The AC is labeled yellow, because of the co-expression of cdh-3::CFP and lag-1 (mut)::YFP. Fluorescence images are shown in the bottom panel of each section (A to L). Overlays of fluorescence and Nomarski images are shown in the upper panels.
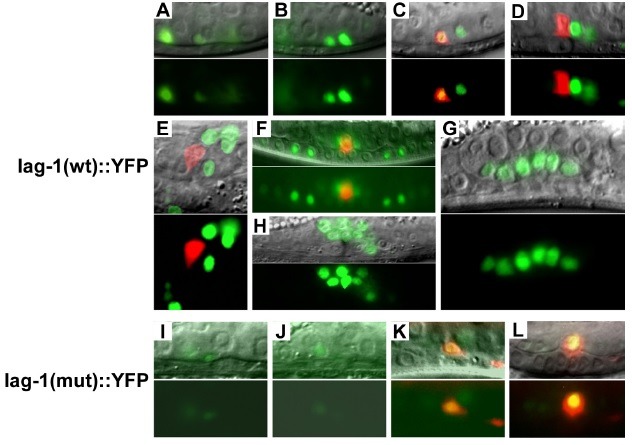
We tested whether the first intron containing clustered LAG-1 binding sites in lag-1 is involved in the lag-1 expression in the hermaphrodite somatic gonad. To do so, we examined transgenic animals containing a transcriptional reporter construct that places the lag-1 region before Δpes-10 promoter:: NLS4::YFP. As shown in Fig. 3, lag-1(wt)::YFP expression in the gonad was first detected in the pre-AC/pre-VU at early L2 (Fig. 3A, B; green), and maintained in VU, as well as in their descendants, at early L4 (Fig. 3C-G; green). The lag-1(wt)::YFP was also expressed in the AC, soon after its cell-fate becomes specified at mid L2 stage. Thus, the AC was labeled yellow by co-expressing lag-1(wt)::YFP and cdh-3::CFP in late L2 (Fig. 3C; YFP and CFP signals are merged into yellow). The YFP signal in the AC that expresses cdh-3::CFP disappeared in late L2, and in early L3 animals (Fig. 3D, E; in red). Considering the fact that nuclear-localized YFP proteins are stable for several hours, the lag-1(wt)::YFP expression appears to have decreased or disappeared in the AC from mid-L2, during which AC/VU cell-fates become specified. In some transgenic animals, the lag-1(wt)::YFP signal disappeared, or was greatly reduced in the descendants of P6.p vulval precursor cells, which have lower LIN-12 activity compared with the P5.p and P7.p descendants, which have higher LIN-12 activity (Fig. 3F).
To determine the effects of the LAG-1 binding sites on the lag-1 expression, we mutated all 13 LAG-1 binding sites in the lag-1::YFP construct, and examined the somatic gonadal expressions at different stages (Fig. 3I-L). Site-directed mutations of the LAG-1 binding sites eliminated lag-1::YFP expression in the pre-AC/pre-VU, VU, and their descendants (Fig. 3I-L). However, the mutations kept expressing lag-1(mut)::YFP in the AC, until early L4. The AC was labeled yellow by co-expressing lag-1(mut)::YFP and cdh-3::CFP (Fig. 3L). In the transgenic animals containing lag-1(wt)::YFP, the AC is labeled red by expressing cdh-3::CFP, not lag-1(wt)::YFP, from late L2, or early L3 (Fig. 3D, E). Thus, LAG-1 binding sites are necessary to activate lag-1 expression in the pre-AC/pre-VU and VU cells and their descendants. The sites also appear to be involved in turning off the lag-1 expression in the AC, after its fate becomes specified.
Significance of the lin-12/Notch and lag-1/CSL co-regulation in AC/VU specification
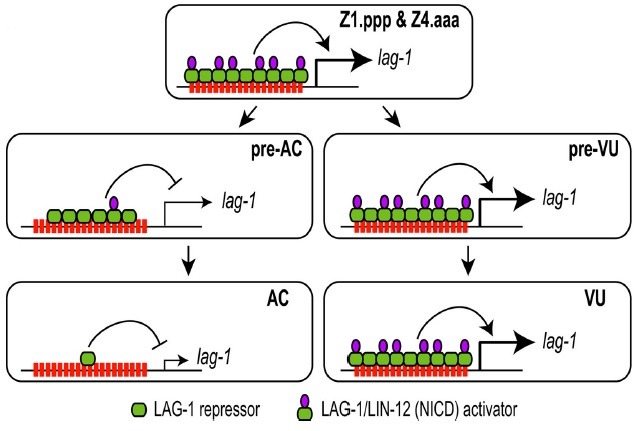
In this paper, we showed that lag-1::YFP expression overlaps with lin-12 expression in the C. elegans somatic gonad during AC/VU specification (10,12). Both genes express in the pre-AC and pre-VU (Z1.ppp, Z1.ppa, Z4.aaa, and Z4.aap) before specification. They express in VU, but not in the AC, after specification. Clustered LAG-1 binding sites in lag-1 were necessary for the transcriptional activation of lag-1 in the cells that have LIN-12 activity (pre-AC/pre-VU, VU, and their descendants). The sites were also necessary to turn off the lag-1 expression in the AC, which does not have LIN-12 activity. Because LAG-1 alone acts as a transcriptional repressor, and becomes an activator upon NICD binding, a small increase of LIN-12 in the pre-VU could rapidly amplify the LIN-12-dependent transcriptional activation activity, by increasing the expression of both LIN-12 and LAG-1. Meanwhile, the down-regulation of both lag-1 and lin-12 expressions could more rapidly lower lin-12 expression in the pre-AC (Fig. 4). Thus, AC and VU cell-fates become specified in a very short period of time, by co-regulating the expressions of both lin-12 and lag-1.
Fig. 4. Clustered LAG-1 binding sites are involved in the regulatory expression of lag-1 during AC/VU specification. The lag-1:: YFP expression patterns are summarized as a model. LAG-1/NICD transcriptional activator binds to the clustered 13 LAG-1 binding sites, to express lag-1 in the pre-AC/pre-VU and VU as well as their descendants. lag-1 expression disappears in the AC at mid L2, immediately after its fate becomes specified. When the LAG-1 binding sites are mutated, lag-1 expression in the AC is retained until L3 molt, suggesting the existence of a cryptic AC-specific enhancer in lag-1, which is suppressed by LAG-1 repressor in wild-type animals.
MATERIALS AND METHODS
General methods and strains
C. elegans strains were handled and maintained at 20℃, and were crossed according to standard protocols (22). Experiments were conducted at 20℃, unless otherwise indicated. Cell anatomy was observed with Nomarski optics, and CFP and YFP expression were noted, using a Hamamatsu ORCA-ER on a Zeiss Axioplan compound microscope. Photographs were taken with a digital camera and Improvision Openlab software, and images were overlaid using Adobe Photoshop CS. The wild-type strain used in this study is C. elegans var. Bristol strain N2. Transgenic lines were generated using a standard microinjection protocol (23). Each lag-1::YFP construct (25 or 50 μg/ml), myo-2::YFP plasmid (5 μg/ml), and pBluscript (170 μg/ml) was co-injected into unc-119(ed4); syIs57[cdh-3::CFP+pDP#MM016B] animals. After injection, animals that express myo-2::YFP in the pharynx were identified, and maintained as transgenic lines. N2 and dpy-20 (e1282); arIs50(sel-8::GFP) strains were used in the ChIP experiment.
Constructs
lag-1(wild-type)::YFP transcriptional fusion construct was prepared by cloning a part of the first intron region (1,758 bp) of lag-1, which contains 13 predicted LAG-1 binding sites, into pPD122.53(YFP), a plasmid that contains NLS4::YFP generated by replacing GFP in pPD122.53 with YFP. The 1,758 bp genomic DNA fragment (aagctttcccatcctagtttttcccacacg- - -tgggaaacccccccaactggggagaagctt) of lag-1 was PCR amplified from N2 genomic DNA, and cloned into a HindIII site in the pPD 122.53(YFP) plasmid. PCR fusion was used to generate site-directed mutations of 13 LAG-1 binding sites in the region; wild-type forms of ‘TCCCA’ and ‘TGGGA’ sites were changed to 5 ‘TCACT,’ 2 ‘AGTGA,’ 1 ‘TCGTA,’ 1 ‘TCCAT,’ 1 ‘TCCTC,’ 1 ‘TGCGT,’ 1 ‘TGGAT,’ and 1 ‘TGTGA’ sites (24). In the lag-1(mutated LAG-1 binding sites)::YFP transcriptional fusion construct, the PCR fragment containing mutations in all 13 putative LAG-1 binding sites was cloned into the pPD122.53 (YFP) plasmid.
In vitro DNA binding assay
Electrophoretic mobility shift assays (EMSA) with in vitro translated LAG-1 were performed as described previously (15,16). The sequences of DNA probe and competitors are summarized below. Sequences of the upper strand of DNA duplexes are shown here. Bold underlined characters represent wild type or mutated LAG-1 binding sites.
DNA probe: 5’- atacaagtatgaattCTCGCGACTCGTGGGAAAATGGGCGGAAGGGCACCGTGGGAAAATAGTTCCAGGaattcat acatagca-3’
Competitor DNAs:
1st competitor (wild type): 5’-ctcgaaaactttcccatcctagttttt-3’
1st competitor (mutation): 5’-ctcgaaaactttcacttcctagttttt-3’
2nd competitor (wild type): 5’-atcctagtttttcccacacgcattccc-3’
2nd competitor (mutation): 5’-ttcctagtttttcactcacgcattccc-3’
3rd competitor (wild type): 5’-ttttttagaaatgggaaggccgtcctt-3’
3rd competitor (mutation): 5’-ttttttagaaatgcgtaggccgtcctt-3’
4th competitor (wild type): 5’-ccttcaatcgttcccaggagaagaaga-3’
4th competitor (mutation): 5’-ccttcaatcgttccctggagaagaaga-3’
5th competitor (wild type): 5’-aaccgtcagtttcccatacgacaaagg-3’
5th competitor (mutation): 5’-aaccgtcagtttcgtatacgacaaagg-3’
6th competitor (wild type): 5’-cgccgacactttcccacgctcgttttt-3’
6th competitor (mutation): 5’-cgccgacactttcactcgctcgttttt-3’
7th competitor (wild type): 5’-acgctcgtttttcccaccacacacaca-3’
7th competitor (mutation): 5’-tcgctcgtttttggatccacacacaca-3’
8th competitor (wild type): 5’-caacctttttttcccactcgtcgcttt-3’
8th competitor (mutation): 5’-caacctttttttcactctcgtcgcttt-3’
9th competitor (wild type): 5’-tcgtcgctttttcccacgatattccat-3’
9th competitor (mutation): 5’-tcgtcgctttttccatcgatattccat-3’
10th competitor (wild type): 5’-ctctaaaagtttcccaatgactttggt-3’
10th competitor (mutation): 5’-ctctaaaagtttcctatagactttggt-3’
11th competitor (wild type): 5’-ctgcgtctctgtgggaatacaacaatc-3’
11th competitor (mutation): 5’-ctgcgtctctgagtgaatacaacaatc-3’
12th competitor (wild type): 5’-tgatgcgtccctgggaaacccccccaa-3’
12th competitor (mutation): 5’-tgatgcgtccctgtgaaacccccccaa-3’
13th competitor (wild type): 5’-cccccaactggtgggaagattgaaaag-3’
13th competitor (mutation): 5’-cccccaactggagtgaagattgaaaag-3’
In vivo DNA binding assay
ChIP was performed, as described previously (19). The following buffers were used in this experiment: ChIP buffer (5mMPipes,pH8.0,85mMKCl,0.5%NP-40); lysis buffer (1% sodium dodecyl sulfate [SDS], 10 mM EDTA, 50 mM Tris-HCl, pH 8.1, 1× protease inhibitor cocktail); buffer B (16.7 mM Tris-HCl, pH 8.1, 150 mM NaCl, 0.01% SDS, 1.1% Triton X-100, 1.2 mM EDTA); buffer C (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris-HCl, pH 8.1, 150 mM NaCl); buffer D (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris-HCl, pH 8.1, 500 mM NaCl); and buffer E (0.25MLiCl,1%NP-40,1%deoxycholate,1mMEDTA,10mMTris-HCl,pH8.1). Briefly, N2 and dpy-20(e1282); arIs50(sel-8::GFP) worms were fixed with 2% formaldehyde in M9 buffer at room temperature for 30 min, and excess formaldehyde was quenched with 100 mM glycine. Worms were washed sequentially with ice-cold phosphate-buffered saline and ChIP buffer, resuspended in lysis buffer, and were frozen in liquid nitrogen. After sonicating the frozen worms in ice, until generating genomic DNA fragments of average 300-500 bp long, soluble protein-DNA extracts were obtained by centrifugation for 10 min at 4℃. For each ChIP reaction, 3 mg of protein-DNA extracts was diluted in buffer B, followed by preclearing with 2 μg sheared salmon sperm DNA, and 50 μl protein A- and G-agarose mixture (50:50%) for 2 h at 4℃. ChIP reactions were performed overnight at 4℃ with 5 μg of affinity-purified LAG-3-, HLH-2-, or GFP-specific antibodies, or control IgG. After sequential washing immunoprecipitates with washing buffers C, D, and E, formaldehyde crosslinks were reversed by incubation at 65℃ overnight, in 1% SDS and 0.1 M NaHCO3. Proteins were then removed by proteinase K digestion, and DNA fragments were purified using QIAGEN PCR purification kits. The following PCR primers were used to measure the amounts of DNA fragments bound by specific antibodies in vivo:
ChIP PCR product 1 (243 bp): 5’-actttccatagaagaacgagcaacag-3’/5’-ttttgttcccgtactaacacgactac-3’
ChIP PCR product 2 (212 bp): 5’-aatcatgtttcttcaaaagaatgggg-3’/5’-ttttttcgagactcagcttggaatac-3’
ChIP PCR product 3 (232 bp): 5’-gttcggagtatcatattggaaccgtc-3’/5’-gagagaaaaaggggagggaactggtg-3’
ChIP PCR product 4 (399 bp): 5’-caccgacacagcccacaaggccaacc-3’/5’-gttactgcaatttcttagtacttttc-3’
Acknowledgments
This study was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF), funded by the Ministry of Education, Science and Technology (2010-0003785; BJH). SKP is a recipient of the Brain Korea 21 program, supported by the Ministry of Education, Science and Technology, Republic of Korea. We thank Paul W. Sternberg at the California Institute of Technology for helpful discussions.
References
- 1.Artavanis-Tsakonas S., Rand M. D., Lake R. J. Notch signaling: cell fate control and signal integration in development. Science. (1999);284:770–776. doi: 10.1126/science.284.5415.770. [DOI] [PubMed] [Google Scholar]
- 2.Rohrschneider M. R., Nance J. Polarity and cell fate specification in the control of Caenorhabditis elegans gastrulation. Dev. Dyn. (2009);238:789–796. doi: 10.1002/dvdy.21893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schuurmans C., Guillemot F. Molecular mechanisms underlying cell fate specification in the developing telencephalon. Curr. Opin. Neurobiol. (2002);12:26–34. doi: 10.1016/S0959-4388(02)00286-6. [DOI] [PubMed] [Google Scholar]
- 4.Segalen M., Bellaiche Y. Cell division orientation and planar cell polarity pathways. Semin. Cell Dev. Biol. (2009);20:972–977. doi: 10.1016/j.semcdb.2009.03.018. [DOI] [PubMed] [Google Scholar]
- 5.Guo G., Huss M., Tong G. Q., Wang C., Li Sun L., Clarke N. D., Robson P. Resolution of cell fate decisions revealed by single-cell gene expression analysis from zygote to blastocyst. Dev. Cell. (2010);18:675–685. doi: 10.1016/j.devcel.2010.02.012. [DOI] [PubMed] [Google Scholar]
- 6.Bailey A. M., Posakony J. W. Suppressor of hairless directly activates transcription of enhancer of split complex genes in response to Notch receptor activity. Genes Dev. (1995);9:2609–2622. doi: 10.1101/gad.9.21.2609. [DOI] [PubMed] [Google Scholar]
- 7.Castro B., Barolo S., Bailey A. M., Posakony J. W. Lateral inhibition in proneural clusters: cis-regulatory logic and default repression by Suppressor of Hairless. Development. (2005);132:3333–3344. doi: 10.1242/dev.01920. [DOI] [PubMed] [Google Scholar]
- 8.Kimble J., Simpson P. The LIN-12/Notch signaling pathway and its regulation. Annu. Rev. Cell Dev. Biol. (1997);13:333–361. doi: 10.1146/annurev.cellbio.13.1.333. [DOI] [PubMed] [Google Scholar]
- 9.Wilkinson H. A., Fitzgerald K., Greenwald I. Reciprocal changes in expression of the receptor lin-12 and its ligand lag-2 prior to commitment in a C. elegans cell fate decision. Cell. (1994);79:1187–1198. doi: 10.1016/0092-8674(94)90010-8. [DOI] [PubMed] [Google Scholar]
- 10.Seydoux G., Greenwald I. Cell autonomy of lin-12 function in a cell fate decision in C. elegans. Cell. (1989);57:1237–1245. doi: 10.1016/0092-8674(89)90060-3. [DOI] [PubMed] [Google Scholar]
- 11.Johnston R. J., Jr., Desplan C. Stochastic mechanisms of cell fate specification that yield random or robust outcomes. Annu. Rev. Cell Dev. Biol. (2010);26:689–719. doi: 10.1146/annurev-cellbio-100109-104113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Greenwald I. LIN-12/Notch signaling: lessons from worms and flies. Genes Dev. (1998);12:1751–1762. doi: 10.1101/gad.12.12.1751. [DOI] [PubMed] [Google Scholar]
- 13.Greenwald I. Notch and the awesome power of genetics. Genetics. (2012);191:655–669. doi: 10.1534/genetics.112.141812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hwang B. J., Meruelo A. D., Sternberg P. W. C. elegans EVI1 proto-oncogene, EGL-43, is necessary for Notch-mediated cell fate specification and regulates cell invasion. Development. (2007);134:669–679. doi: 10.1242/dev.02769. [DOI] [PubMed] [Google Scholar]
- 15.Hwang B. J., Sternberg P. W. A cell-specific enhancer that specifies lin-3 expression in the C. elegans anchor cell for vulval development. Development. (2004);131:143–151. doi: 10.1242/dev.00924. [DOI] [PubMed] [Google Scholar]
- 16.Christensen S., Kodoyianni V., Bosenberg M., Friedman L., Kimble J. lag-1, a gene required for lin-12 and glp-1 signaling in Caenorhabditis elegans, is homologous to human CBF1 and Drosophila Su(H). Development. (1996);122:1373–1383. doi: 10.1242/dev.122.5.1373. [DOI] [PubMed] [Google Scholar]
- 17.Zimber-Strobl U., Strobl L. J., Meitinger C., Hinrichs R., Sakai T., Furukawa T., Honjo T., Bornkamm G. W. Epstein-Barr virus nuclear antigen 2 exerts its transactivating function through interaction with recombination signal binding protein RBP-J kappa, the homologue of Drosophila Suppressor of Hairless. EMBO J. (1994);13:4973–4982. doi: 10.1002/j.1460-2075.1994.tb06824.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Petcherski A. G., Kimble J. LAG-3 is a putative transcriptional activator in the C. elegans Notch pathway. Nature. (2000);405:364–368. doi: 10.1038/35012645. [DOI] [PubMed] [Google Scholar]
- 19.Lee M. H., Hook B., Lamont L. B., Wickens M., Kimble J. LIP-1 phosphatase controls the extent of germline proliferation in Caenorhabditis elegans. EMBO J. (2006);25:88–96. doi: 10.1038/sj.emboj.7600901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hirsh D., Oppenheim D., Klass M. Development of the reproductive system of Caenorhabditis elegans. Dev. Biol. (1976);49:200–219. doi: 10.1016/0012-1606(76)90267-0. [DOI] [PubMed] [Google Scholar]
- 21.Kimble J., Hirsh D. The postembryonic cell lineages of the hermaphrodite and male gonads in Caenorhabditis elegans. Dev. Biol. (1979);70:396–417. doi: 10.1016/0012-1606(79)90035-6. [DOI] [PubMed] [Google Scholar]
- 22.Brenner S. The genetics of Caenorhabditis elegans. Genetics. (1974);77:71–94. doi: 10.1093/genetics/77.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mello C. C., Kramer J. M., Stinchcomb D., Ambros V. Efficient gene transfer in C. elegans: extrachromosomal maintenance and integration of transforming sequences. EMBO J. (1991);10:3959–3970. doi: 10.1002/j.1460-2075.1991.tb04966.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hobert O. PCR fusion-based approach to create reporter gene constructs for expression analysis in transgenic C. elegans. Biotechniques. (2002);32:728–730. doi: 10.2144/02324bm01. [DOI] [PubMed] [Google Scholar]