

Abstract
Heart failure is one of the leading causes of sudden death in developed countries. While current therapies are mostly aimed at mitigating associated symptoms, novel therapies targeting the subcellular mechanisms underlying heart failure are emerging. Failing hearts are characterized by reduced contractile properties caused by impaired Ca2+ cycling between the sarcoplasm and sarcoplasmic reticulum (SR). Sarcoplasmic/endoplasmic reticulum Ca2+ATPase 2a (SERCA2a) mediates Ca2+ reuptake into the SR in cardiomyocytes. Of note, the expression level and/or activity of SERCA2a, translating to the quantity of SR Ca2+ uptake, are significantly reduced in failing hearts. Normalization of the SERCA2a expression level by gene delivery has been shown to restore hampered cardiac functions and ameliorate associated symptoms in pre-clinical as well as clinical studies. SERCA2a activity can be regulated at multiple levels of a signaling cascade comprised of phospholamban, protein phosphatase 1, inhibitor-1, and PKCα. SERCA2 activity is also regulated by post-translational modifications including SUMOylation and acetylation. In this review, we will highlight the molecular mechanisms underlying the regulation of SERCA2a activity and the potential therapeutic modalities for the treatment of heart failure. [BMB Reports 2013; 46(5): 237-243]
Keywords: Inhibitor-1, Phospholamban, PKCα, Protein phosphatase 1, SERCA2a
INTRODUCTION
Heart failure is one of the leading causes of sudden death in developed countries (1-3). It is a complex and multi-factorial disease and its pathogenesis is poorly understood. Among the many pathological features of heart failure, the most prominent and widespread in patients and animal models of heart failure is reduced myocardial contractility caused by aberrant Ca2+ cycling (4,5). Intracellular Ca2+ concentration, the primary determinant of myocardial contractility, is regulated by a group of ion channels, transporters, and pumps in a highly coordinated manner. Upon arrival of electric signals at the sarcolemma, small amounts of Ca2+ from the extracellular space enter cardiomyocytes through voltage-gated L-type Ca2+ channels. This process, referred to as Ca2+-induced Ca2+-release, induces Ca2+-release from the sarcoplasmic reticulum (SR) through ryanodine receptor 2 (RyR2). The resulting transient increase in intracellular Ca2+ concentration drives myocardial contraction. To trigger myocardial relaxation, Ca2+ is then removed from the sarcoplasm through Ca2+ reuptake into the SR, which is mediated by sarcoplasmic /endoplasmic reticulum Ca2+ATPase 2a (SERCA2a) (Fig. 1).
Fig. 1. Calcium signaling during excitation-contraction coupling. Calcium signaling plays an essential role in cardiac muscle contraction and relaxation. Ca2+ entry via LTCC triggers SR Ca2+ release through RyR, a process termed calcium-induced calcium release (CICR). CICR causes activation of contraction of myofilaments. SR Ca2+ uptake via SERCA2a and extrusion via NCX allow relaxation of myofilaments. CICR, Ca2+-induced Ca2+ release; LTCC, L-type Ca2+ channel; NCX, Na+-Ca2+ exchanger; PLB, phospholamban; RyR, ryanodine receptor; SERCA2a, sarcoplasmic/endoplasmic reticulum Ca2+-ATPase 2a; SR, sarcoplasmic reticulum; T-tubule, transverse tubule.
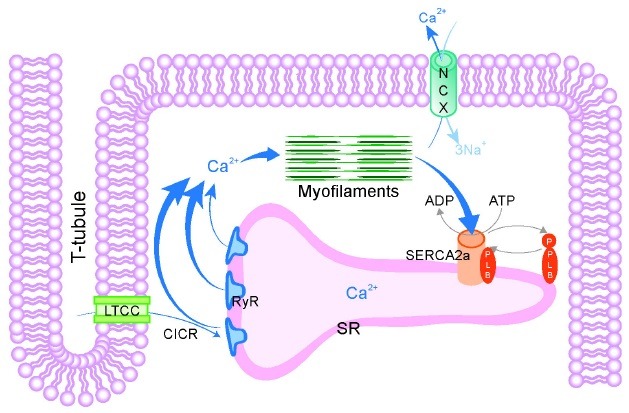
Defects in RyR2 and SERCA2a functions are partly responsible for impaired contractility in failing hearts. Under heart failing conditions, RyR2 becomes "leaky" through its oxidation, nitrosylation, phosphorylation, and altered binding to a variety of RyR2- interacting proteins including calstabins (6). The increased leakiness of RyR2 leads to a depletion of Ca2+ from the SR and abnormal depolarization of cardiomyocytes, triggering fatal arrhythmias. Therefore, reducing the leakiness of RyR2 is regarded as an effective therapeutic strategy (7). Furthermore, the expression level of SERCA2a is reduced in failing hearts. The potentiated activity of phospholamban (PLN), an endogenous inhibitor of SERCA2a, through its dephosphorylation further hampers SERCA2a activity. Restoration of SERCA2a activity is therefore a potential treatment target for heart failure.
Adeno-associated virus (AAV)-mediated delivery of SERCA2a has been shown to be safe and effective for treating heart failure in phase 1 and 2 clinical trials and is currently in phase 3 clinical studies. The activity of SERCA2a is regulated by a number of other molecules including PLN, protein phosphatase 1 (PP1), inhibitor-1 (I-1), and protein kinase Cα (PKCα), which together constitute a signaling pathway. Therefore, modulating the activities of these molecules could effectively regulate SERCA2a activity. In this mini-review, we will summarize the approaches used to restore SERCA2a levels and/or activity in animal models of heart failure and in human patients.
SERCA2a
SR Ca2+ uptake is reduced in the myocardium of patients with end-stage heart failure (8). In addition, the expression of SERCA2a, which is responsible for SR Ca2+ uptake in cardiomyocytes, is decreased at the transcript level (9) and the protein level (10) in failing hearts. Furthermore, the expression level of SERCA2a appears to play a causal role in regulating cardiac function in failing and non-failing human myocardium (11). The importance of SERCA2a in Ca2+ cycling and in the pathophysiology of heart failure has also been shown in SERCA2a knockout (KO) mice. SERCA2a heterozygous mice, in which SERCA2a protein levels are reduced by ~35%, exhibit impaired cardiac functions without showing increased heart failure phenotypes at the baseline (12). Cardiomyocytes isolated from the hearts of SERCA2a heterozygous mice show reduced SR Ca2+ load and decreased contractility (13). Under pressure overload, SERCA2a heterozygous mice show accelerated heart failure phenotypes with increased mortality and morbidity, which appears to be caused by decreased systolic and diastolic functions of the hearts (14).
Since reduced SERCA2a levels cause heart failure in patients and in model animals, restoration of reduced SERCA2a levels and/or activity is a potential treatment option for heart failure. Adenovirus-mediated gene transfer of SERCA2a improves intracellular Ca2+ handling in isolated rat cardiomyocytes in vitro (15) and in rat myocardium in vivo (16). In addition, adenoviral gene transfer of SERCA2a restores defective intracellular Ca2+ handling and contractile function in ventricular cardiomyocytes isolated from patients with end-stage heart failure (17). In a rat model of heart failure, gene transfer of SERCA2a restored reduced SERCA2a levels and concomitant defects in cardiac functions (18). Importantly, this approach significantly improved survival and the energy potential in failing hearts. Inotropic agents such as β-agonists increase contractile functions at the expense of increased mortality and decreased metabolism. The β-agonist-mediated increase in mortality is thought to be due to increased intracellular Ca2+ at diastole, which leads to activation of pro-apoptotic and pro-hypertrophic signals, which may increase the incidence of ventricular arrhythmia. Therefore, β-blockers are currently being used for the treatment of heart failure despite their negative inotropic effects. Unlike these agents, gene transfer of SERCA2a is inotropic in its effects on contractility and yet improves metabolism and survival by normalizing intracellular Ca2+ level at diastole.
The above studies suggest that gene transfer of SERCA2a could be a promising modality for the treatment of heart failure. In one study, volume overload-induced heart failure was mimicked by inducing mitral regurgitation in pigs followed by intracoronary delivery of recombinant adeno-associated virus (rAAV) carrying SERCA2a. Two months later, SERCA2a gene delivery significantly restored contractile parameters and reversed the adverse left ventricular remodeling in these pigs (19). AAV-mediated delivery of SERCA2a also improves cardiac functions in sheep with pacing-induced heart failure (20). In addition, gene delivery of SERCA2a reduces the incidence of ventricular arrhythmias after ischemia-reperfusion in pigs (21). This beneficial effect may be attributed to the modulation of post-ischemic Ca2+ overload by SERCA2a. Based on the success observed with large animal models of heart failure, gene delivery of SERCA2a was attempted in clinical trials, including a phase 1 trial of a single intracoronary infusion of AAV1 carrying SERCA2a to patients with advanced heart failure. Several of the treated patients showed significant improvements in a number of parameters, while two patients who showed no improvements had pre-existing anti-AAV1 neutralizing antibodies (22). In addition, 39 patients without neutralizing antibodies were similarly treated in a phase 2 trial, which showed that treatment with AAV1 carrying SERCA2a was beneficial and was not associated with serious safety issues (23). This approach has now moved on to phase 3 trials with a large cohort (∼400) of patients.
As described above, restoration of reduced SERCA2a levels in failing hearts using gene transfer approaches is effective in improving cardiac deficits in human patients. SERCA2a is subject to a variety of post-translational modifications, which affect its enzymatic activity and/or stability. Recently, our group showed that SERCA2a is SUMO (Small Ubiquitin-like MOdifier)-ylated at two lysine residues and that this SUMOylation is essential for preserving the activity and stability of SERCA2a (24). Notably, SUMOylation of SERCA2a is specific to SUMO1, and is not observed with SUMO2 or SUMO3. The levels of SUMO1 and the extent of SUMOylation of SERCA2a are greatly reduced in failing hearts. Gene delivery of SUMO1 rescues cardiac dysfunction, whereas shRNA-mediated down-regulation of SUMO1 decreases SERCA2a activity and accelerates cardiac dysfunction induced by pressure overload in mice. Although the exact mechanisms underlying the beneficial effects of SUMOylation are unclear, overexpression of SUMO1 via gene delivery appears to be as efficient as the overexpression of SERCA2a itself in improving cardiac function.
In conclusion, the restoration of SERCA2a levels or activity through gene delivery-mediated expression of SERCA2a or SUMO1, respectively, can be an efficient modality for the treatment of heart failure.
PHOSPHOLAMBAN
SERCA2a activity is directly modulated by a 52-amino acid endogenous inhibitor, phospholamban (PLN). The inhibitory effect of PLN on SERCA2a activity was first revealed using transgenic animal models. Cardiac-specific overexpression of PLN inhibited SR Ca2+ uptake and reduced systolic Ca2+ levels, contractile parameters, and basal systolic function in mice (25). In contrast, PLN knockout (KO) mice exhibited enhanced Ca2+ cycling and myocardial contractility with no gross developmental abnormalities. The elevated contractile parameters are associated with increased affinity of SERCA2a for Ca2+ (26). Furthermore, comparative analyses with wild type and heterozygous and homozygous PLN KO mice revealed that relative PLN levels correlate well with the affinity of SERCA2a for Ca2+ and with the rates of relaxation and contraction of isolated cardiomyocytes (27). These findings suggest that the PLN level may determine the parallel outcomes in SR function and cardiac contractility in mice.
Furthermore, studies have shown that mutations in human PLN result in dilated cardiomyopathy. A dominant Arg→Cys mutation at residue 9 (R9C) in PLN was identified in a patient with inherited dilated cardiomyopathy (Schmitt, 2003). PLNR9C indirectly inhibits SERCA2a activity by inhibiting cAMP-dependent protein kinase (PKA)-mediated phosphorylation of wild type PLN. Furthermore, PKA-mediated phosphorylation of PLN at Ser16 decreases its inhibitory activity. It appears that the chronic inhibition of SERCA2a by PLNR9C leads to dilated cardiomyopathy and premature death in families carrying this mutation. In another genetic study, a truncated PLN protein caused by the substitution of Leu39 with a termination codon (L39stop) was identified in individuals of two families with hereditary heart failure (28). Individuals homozygous for this mutation developed dilated cardiomyopathy and heart failure, whereas heterozygous individuals exhibited cardiac hypertrophy without overt heart failure. These observations were recapitulated in transgenic (Tg) mice with cardiac-specific expression of PLNR9C. PLNL39stop was neither functional nor dominant-negative when expressed in cardiomyocytes and HEK293 cells. Moreover, PLNL39stop was undetectable in the myocardium; thus, it could be viewed as a null mutation. While the PLN null mutation is beneficial in mice, it is detrimental in humans, resulting in severe heart failure. It is likely that chronic stimulation of human heart tissue resulting from the absence of functional PLN causes or accelerates the progression of heart failure. Taken together, these studies suggest that SERCA activity must be maintained within a narrow range to promote stable heart function, and that PLN may have a role in this maintenance.
The activity of PLN is critically regulated by phosphorylation at two residues; Ser16 by cAMP-dependent protein kinase (PKA), and Thr17 by Ca2+-CaM-dependent protein kinase (CaMK). Both of these phosphorylations can be reversed by protein phosphatase 1 (PP1). PLN is in dynamic equilibrium between monomeric and pentameric states. When phosphorylated, PLN appears to mainly form a pentamer, which is inactive or at least less active in inhibiting SERCA2a activity than the monomer. In contrast, the dephosphorylated form of PLN exists mainly as a monomer, which can bind to and inhibit SERCA2a activity.
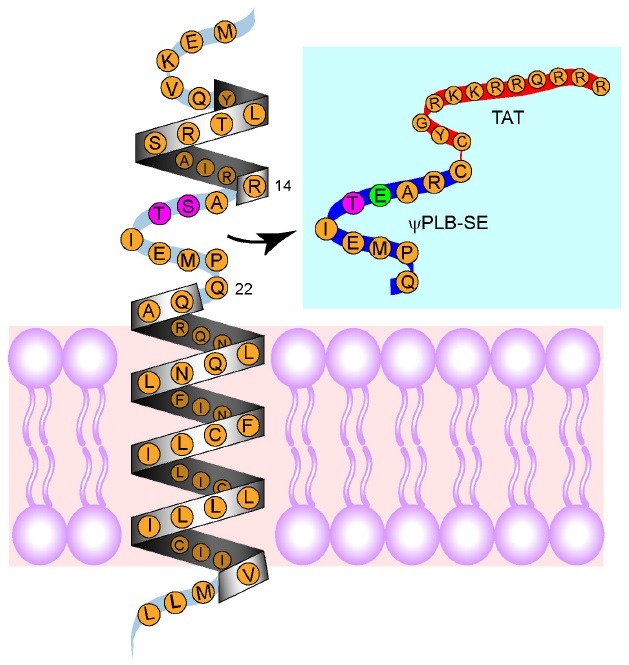
In failing hearts of patients and animals, both the level and activity of SERCA2a are lower than in normal hearts, and this has been suggested to contribute to cardiac dysfunction (29). In addition, during cardiac failure, the level of PLN is unaffected, but PLN phosphorylation is lower, which causes further loss of SERCA2a activity. Fig. 2 shows the signaling pathway involved in the decrease in SERCA2a activity in failing hearts. Decreased phosphorylation of PLN in failing hearts is mainly caused by increased PP1 activity. Therefore, inhibiting PP1-mediated dephosphorylation of PLN may be a strategy to restore SERCA2a activity during heart failure. Previously, our group tested this hypothesis using decoy peptides (30). We synthesized a 9-mer peptide that exactly matched the nine amino acids of the loop region (aa 14-22), except that the Ser residue was replaced by Glu (Fig. 3). This peptide, coupled to a cell-permeable peptide TAT, was referred to as ψPLN-SE. It mimicked the loop region of PLN phosphorylated by PKA, thereby serving as a decoy for PP1. Treatment of cardiomyocytes with phorbol 12-myristate 13-acetate (PMA) significantly reduced phosphorylation of PLN through activation of PKCα (see Fig. 2). As expected, ψPLN-SE significantly restored the phosphorylation of PLN in cardiomyocytes treated with PMA. In an ex vivo set up, ischemia-reperfusion resulted in severe loss of cardiac function and reduced phosphorylation of PLN. Addition of ψPLN-SE at the time of reperfusion significantly restored cardiac function and increased phosphorylation levels of PLN ex vivo (30) and in vivo in mice (Oh et al., unpublished data). Collectively, our data suggest that ψPLN-SE improves functional recovery after ischemia-reperfusion at least in part by increasing the phosphorylation level of PLN.
Fig. 2. SERCA2a activity and calcium handling in heart failure. The dysregulation of SERCA2a is a hallmark of heart failure. Reduction in SR Ca2+ uptake results in systolic dysfunction as well as increased cytosolic Ca2+ level and susceptibility to apoptosis. In heart failure, SERCA2a activity and expression are decreased via negative regulation by upstream modulators. I-1, protein phosphatase inhibitor 1; PKCα, the α isotype of protein kinase C; PLB, phospholamban; PP1, protein phosphatase 1; SERCA2a, sarcoplasmic/endoplasmic reticulum Ca2+-ATPase 2a; SUMO, small ubiquitin- like modifier; RyR, ryanodine receptor.
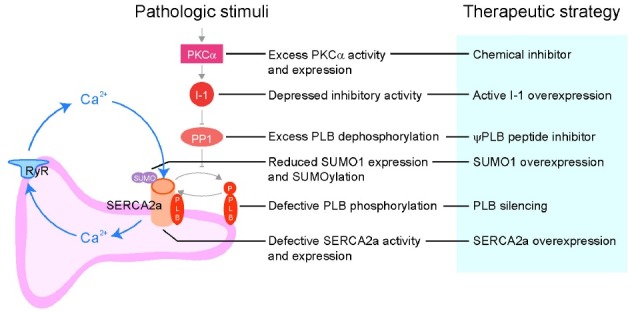
Fig. 3. Design of decoy peptides. The "L-shaped" structure of monomeric PLB is composed of a cytoplasmic helix, a connecting short loop, and a transmembrane helix. Blue box, ψPLB-SE was derived from the nine amino acids that compose the connecting loop. In the decoy peptide, the serine residue was replaced with a glutamine to mimic the peptide in its phosphorylated state (shown in green). The peptide was conjugated via a disulfide bond at the amino-termini to the cell penetrating peptide TAT (YG-RKKRRQRRR) to facilitate uptake of the peptide into cells.
PROTEIN PHOSPHATASE 1/INHIBITOR-1
PP1 was first identified as the phosphatase responsible for dephosphorylating PLN in the myocardium (31,32). PP1 activity is 2-fold higher in failing human hearts than in non-failing hearts (33). Further insights into the role of PP1 during heart failure were made through animal studies using Tg mice. Cardiac-specific overexpression of PP1 in Tg mice leads to cardiac dysfunction, dilated cardiomyopathy, and premature mortality, which are associated with decreases in the phosphorylation of PLN (34). As shown above, inhibiting PP1-mediated dephosphorylation of PLN can be an efficient strategy to activate SERCA2a. However, this strategy may have limitations as PP1 is involved in a variety of biological processes in many organs. Thus, careful arrangements for targeted inhibition of PP1 specifically in hearts are needed.
I-1 is an endogenous inhibitor of PP1. Ablation of I-1 results in increased PP1 activity and decreased phosphorylation of PLN. Moreover, basal cardiac functions are severely depressed in I-1 KO mice (34). In contrast, overexpression of I-1 in heart tissue engineered from neonatal rat cardiomyocytes or adult rat cardiomyocytes results in increased cell shortening and a concomitant increase in PLN phosphorylation (35). Both PLN phosphorylation and cardiac contractility are elevated at the cellular and organism levels in Tg mice with cardiac-specific overexpression of constitutively active truncated I-1 (I-1c) (36). Notably, adenovirus-mediated acute expression of I-1c restores cardiac function and partially reverses remodeling of the pre-established heart failure in these mice. The role of I-1 has also been evaluated by inducing the expression of I-1c in adult mouse hearts after an ischemic insult. In that study, overexpression of I-1c resulted in a significant improvement in cardiac function and associated increases in PLN phosphorylation and SERCA2a activity. Furthermore, increased I-1 activity significantly attenuated ischemia-reperfusion injury via suppression of endoplasmic reticulum stress responses (37). Collectively, these results indicate that I-1 is an endogenous inotropic molecule that inhibits PP1; thus, overexpression of I-1 may be a valuable therapy for the treatment of heart failure.
PKCα
Isoproterenol (ISO), a β-agonist, increases cardiac contractility via the activation of protein kinase A (PKA). A number of studies have shown that the inotropic effect of ISO is exerted through PKA-mediated phosphorylation of I-1 (38,39). In addition, I-1 becomes a potent inhibitor of PP1 when it is phosphorylated at Thr35 by PKA (40).
I-1 contains PKCα phosphorylation sites, Ser67 and Thr75. The role of PKCα in cardiac function was controversial until the Molkentin group showed that PKCα is associated with depressed contractility (41). Overexpression of PKCα increases PP1 activity by decreasing the interaction of I-1 with PP1 via an increase in the phosphorylation of I-1 at Ser67. Accordingly, PKCα KO hearts exhibit increased contractility and decreased phosphorylation of I-1 at Ser67. In addition, Thr75 is another primary PKCα phosphorylation site in I-1 (42). Constitutively phosphorylated forms of I-1 containing S67D and/or T75D substitutions significantly increase PP1 activity and decrease contractility by decreasing SERCA2a activity in isolated cardiomyocytes. However, the effects of these mutations are not additive. Notably, activation of the PKA signaling pathway only marginally reverses the adverse effects of the constitutively phosphorylated forms of I-1 on contractility (43). Therefore, I-1 appears to function at the intersection of PKA and PKC signaling pathways.
Inhibitors of conventional PKC isotypes such as Ro-31-8220, Ro-32-0432, and ruboxitaurin (LY333531) have been vigorously tested for their effects in different rodent models of heart failure (44). The importance of PKCα was further supported by a finding that ruboxitaurin increases cardiac contractility in wild type and PKCβγ KO mice, but not in PKCα KO mice (45). In a pig model of heart failure induced by occlusion of the left anterior descending artery, administration of ruboxitaurin resulted in a significantly better recovery of cardiac contractility and an increase in cardiac ejection fraction and cardiac output (46). These results suggest that inhibition of PKCα represents a promising approach for the treatment of heart failure.
CONCLUSION
As summarized in Fig. 2, activation of PKCα triggers a signaling pathway that leads to decreased SERCA2a activity in the failing heart. Normalization of SERCA2a activity through modulation of this signaling pathway or elevation/modification of SERCA2a itself could be a valid strategy for the treatment of heart failure. Gene delivery of SERCA2a has advanced to phase 3 clinical trials. Other approaches were also found to be effective in pre-clinical studies but will require clinical trials to elucidate their efficacy in treating heart failure. In addition to the molecules shown in Fig. 2, other molecules such as HSP20 and HAX1 are also involved in the regulation of SERCA2a activity. Taken together, the results from various laboratories and clinics show that the regulation of SERCA2a expression is complex. Further in-depth research into the SERCA2a regulatome will hopefully reveal alternative approaches for the modulation of cardiac contractility.
References
- 1.Braz J. C., Gregory K., Pathak A., Zhao W., Sahin B., Klevitsky R., Kimball T. F., Lorenz J. N., Nairn A. C., Liggett S. B., Bodi I., Wang S., Schwartz A., Lakatta E. G., DePaoli-Roach A. A., Robbins J., Hewett T. E., Bibb J. A., Westfall M. V., Kranias E. G., Molkentin J. D. PKC-alpha regulates cardiac contractility and propensity toward heart failure. Nat. Med. (2004);10:248–254. doi: 10.1038/nm1000. [DOI] [PubMed] [Google Scholar]
- 2.Byrne M. J., Power J. M., Preovolos A., Mariani J. A., Hajjar R. J., Kaye D. M. Recirculating cardiac delivery of AAV2/1SERCA2a improves myocardial function in an experimental model of heart failure in large animals. Gene Ther. (2008);15:1550–1557. doi: 10.1038/gt.2008.120. [DOI] [PubMed] [Google Scholar]
- 3.Carr A. N., Schmidt A. G., Suzuki Y., del Monte F., Sato Y., Lanner C., Breeden K., Jing S. L., Allen P. B., Greengard P., Yatani A., Hoit B. D., Grupp I. L., Hajjar R. J., DePaoli-Roach A. A., Kranias E. G. Type 1 phosphatase, a negative regulator of cardiac function. Mol. Cell. Biol. (2002);22:4124–4135. doi: 10.1128/MCB.22.12.4124-4135.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.de lMonte F., Hajjar R. J., Harding S. E. Overwhelming evidence of the beneficial effects of SERCA gene transfer in heart failure. Circ. Res. (2001);88:E66–67. doi: 10.1161/hh1101.092004. [DOI] [PubMed] [Google Scholar]
- 5.de lMonte F., Harding S. E., Schmidt U., Matsui T., Kang Z. B., Dec G. W., Gwathmey J. K., Rosenzweig A., Hajjar R. J. Restoration of contractile function in isolated cardiomyocytes from failing human hearts by gene transfer of SERCA2a. Circulation. (1999);100:2308–2311. doi: 10.1161/01.CIR.100.23.2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.El-Armouche A., Rau T., Zolk O., Ditz D., Pamminger T., Zimmermann W. H., Jäckel E., Harding S. E., Boknik P., Neumann J., Eschenhagen T. Evidence for protein phosphatase inhibitor-1 playing an amplifier role in beta-adrenergic signaling in cardiac myocytes. FASEB J. (2003);17:437–439. doi: 10.1096/fj.02-0057fje. [DOI] [PubMed] [Google Scholar]
- 7.Endo S., Zhou X., Connor J., Wang B., Shenolikar S. Multiple structural elements define the specificity of recombinant human inhibitor-1 as a protein phosphatase-1 inhibitor. Biochemistry. (1996);35:5220–5228. doi: 10.1021/bi952940f. [DOI] [PubMed] [Google Scholar]
- 8.Gupta R. C., Neumann J., Watanabe A. M., Lesch M., Sabbah H. N. Evidence for presence and hormonal regulation of protein phosphatase inhibitor-1 in ventricular cardiomyocyte. Am. J. Physiol. (1996);270:H1159–1164. doi: 10.1152/ajpheart.1996.270.4.H1159. [DOI] [PubMed] [Google Scholar]
- 9.Gwathmey J. K., Copelas L., MacKinnon R., Schoen F. J., Feldman M. D., Grossman W., Morgan J. P. Abnormal intracellular calcium handling in myocardium from patients with end-stage heart failure. Circ. Res. (1987);61:70–76. doi: 10.1161/01.RES.61.1.70. [DOI] [PubMed] [Google Scholar]
- 10.Haghighi K., Kolokathis F., Pater L., Lynch R. A., Asahi M., Gramolini A. O., Fan G. C., Tsiapras D., Hahn H. S., Adamopoulos S., Liggett S. B., Dorn G. W. 2nd, MacLennan D. H., Kremastinos D. T., Kranias E. G. Human phospholamban null results in lethal dilated cardiomyopathy revealing a critical difference between mouse and human. J. Clin. Invest. (2003);111:869–876. doi: 10.1172/JCI17892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hajjar R. J., Schmidt U., Kang J. X., Matsui T., Rosenzweig A. Adenoviral gene transfer of phospholamban in isolated rat cardiomyocytes. Rescue effects by concomitant gene transfer of sarcoplasmic reticulum Ca(2+)-ATPase. Circ. Res. (1997);81:145–153. doi: 10.1161/01.RES.81.2.145. [DOI] [PubMed] [Google Scholar]
- 12.Hajjar R. J., Schmidt U., Matsui T., Guerrero J. L., Lee K. H., Gwathmey J. K., Dec G. W., Semigran M. J., Rosenzweig A. Modulation of ventricular function through gene transfer in vivo. Proc. Natl. Acad. Sci. U.S.A. (1998);95:5251–5256. doi: 10.1073/pnas.95.9.5251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hambleton M., Hahn H., Pleger S. T., Kuhn M. C., Klevitsky R., Carr A. N., Kimball T. F., Hewett T. E., Dorn G. W. 2nd, Koch W. J., Molkentin J. D. Pharmacological- and gene therapy-based inhibition of protein kinase Calpha/beta enhances cardiac contractility and attenuates heart failure. Circulation. (2006);114:574–582. doi: 10.1161/CIRCULATIONAHA.105.592550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hasenfuss G., Reinecke H., Studer R., Meyer M., Pieske B., Holtz J., Holubarsch C., Posival H., Just H., Drexler H. Relation between myocardial function and expression of sarcoplasmic reticulum Ca(2+)- ATPase in failing and nonfailing human myocardium. Circ. Res. (1994);75:434–442. doi: 10.1161/01.RES.75.3.434. [DOI] [PubMed] [Google Scholar]
- 15.Houser S. R., Piacentino V. 3rd, Weisser J. Abnormalities of calcium cycling in the hypertrophied and failing heart. J. Mol. Cell Cardiol. (2000);32:1595–1607. doi: 10.1006/jmcc.2000.1206. [DOI] [PubMed] [Google Scholar]
- 16.Jaski B. E., Jessup M. L., Mancini D. M., Cappola T. P., Pauly D. F., Greenberg B., Borow K., Dittrich H., Zsebo K. M., Hajjar R. J. Calcium upregulation by percutaneous administration of gene therapy in cardiac disease (CUPID Trial), a first-in-human phase 1/2 clinical trial. J. Card. Fail. (2009);15:171–181. doi: 10.1016/j.cardfail.2009.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jessup M., Greenberg B., Mancini D., Cappola T., Pauly D. F., Jaski B., Yaroshinsky A., Zsebo K. M., Dittrich H., Hajjar R. J. Calcium Upregulation by Percutaneous Administration of Gene Therapy in Cardiac Disease (CUPID): a phase 2 trial of intracoronary gene therapy of sarcoplasmic reticulum Ca2+-ATPase in patients with advanced heart failure. Circulation. (2011);124:304–313. doi: 10.1161/CIRCULATIONAHA.111.022889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ji Y., Lalli M. J., Babu G. J., Xu Y., Kirkpatrick D. L., Liu L. H., Chiamvimonvat N., Walsh R. A., Shull G. E., Periasamy M. Disruption of a single copy of the SERCA2 gene results in altered Ca2+ homeostasis and cardiomyocyte function. J. Biol. Chem. (2000);275:38073–38080. doi: 10.1074/jbc.M004804200. [DOI] [PubMed] [Google Scholar]
- 19.Kadambi V. J., Ponniah S., Harrer J. M., Hoit B. D., Dorn G. W. 2nd, Walsh R. A., Kranias E. G. Cardiac-specific overexpression of phospholamban alters calcium kinetics and resultant cardiomyocyte mechanics in transgenic mice. J. Clin. Invest. (1996);97:533–539. doi: 10.1172/JCI118446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kawase Y., Ly H. Q., Prunier F., Lebeche D., Shi Y., Jin H., Hadri L., Yoneyama R., Hoshino K., Takewa Y., Sakata S., Peluso R., Zsebo K., Gwathmey J. K., Tardif J. C., Tanguay J. F., Hajjar R. J. Reversal of cardiac dysfunction after long- term expression of SERCA2a by gene transfer in a pre-clinical model of heart failure. J. Am. Coll. Cardiol. (2008);51:1112–1119. doi: 10.1016/j.jacc.2007.12.014. [DOI] [PubMed] [Google Scholar]
- 21.Kho C., Lee A., Jeong D., Oh J. G., Chaanine A. H., Kizana E., Park W. J., Hajjar R. J. SUMO1-dependent modulation of SERCA2a in heart failure. Nature. (2011);477:601–605. doi: 10.1038/nature10407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ladage D., Tilemann L., Ishikawa K., Correll R. N., Kawase Y., Houser S. R., Molkentin J. D., Hajjar R. J. Inhibition of PKCalpha/beta with ruboxistaurin antagonizes heart failure in pigs after myocardial infarction injury. Circ. Res. (2011);109:1396–1400. doi: 10.1161/CIRCRESAHA.111.255687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lehnart S. E. Novel targets for treating heart and muscle disease: stabilizing ryanodine receptors and preventing intracellular calcium leak. Curr. Opin. Pharmacol. (2007);7:225–232. doi: 10.1016/j.coph.2006.09.010. [DOI] [PubMed] [Google Scholar]
- 24.Lehnart S. E., Maier L. S., Hasenfuss G. Abnormalities of calcium metabolism and myocardial contractility depression in the failing heart. Heart. Fail. Rev. (2009);14:213–224. doi: 10.1007/s10741-009-9146-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Limas C. J., Olivari M. T., Goldenberg I. F., Levine T. B., Benditt D. G., Simon A. Calcium uptake by cardiac sarcoplasmic reticulum in human dilated cardiomyopathy. Cardiovasc Res. (1987);21:601–605. doi: 10.1093/cvr/21.8.601. [DOI] [PubMed] [Google Scholar]
- 26.Liu Q., Chen X., Macdonnell S. M., Kranias E. G., Lorenz J. N., Leitges M., Houser S. R., Molkentin J. D. Protein kinase C{alpha}, but not PKC{beta} or PKC{gamma}, regulates contractility and heart failure susceptibility: implications for ruboxistaurin as a novel therapeutic approach. Circ. Res. (2009);105:194–200. doi: 10.1161/CIRCRESAHA.109.195313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lopez A. D., Mathers C. D., Ezzati M., Jamison D. T., Murray C. J. Global and regional burden of disease and risk factors, 2001: systematic analysis of population health data. Lancet. (2006);367:1747–1757. doi: 10.1016/S0140-6736(06)68770-9. [DOI] [PubMed] [Google Scholar]
- 28.Luo W., Grupp I. L., Harrer J., Ponniah S., Grupp G., Duffy J. J., Doetschman T., Kranias E. G. Targeted ablation of the phospholamban gene is associated with markedly enhanced myocardial contractility and loss of beta-agonist stimulation. Circ. Res. (1994);75:401–409. doi: 10.1161/01.RES.75.3.401. [DOI] [PubMed] [Google Scholar]
- 29.Luo W., Wolska B. M., Grupp I. L., Harrer J. M., Haghighi K., Ferguson D. G., Slack J. P., Grupp G., Doetschman T., Solaro R. J., Kranias E. G. Phospholamban gene dosage effects in the mammalian heart. Circ. Res. (1996);78:839–847. doi: 10.1161/01.RES.78.5.839. [DOI] [PubMed] [Google Scholar]
- 30.MacDougall L. K., Jones L. R., Cohen P. Identification of the major protein phosphatases in mammalian cardiac muscle which dephosphorylate phospholamban. Eur. J. Biochem. (1991);196:725–734. doi: 10.1111/j.1432-1033.1991.tb15871.x. [DOI] [PubMed] [Google Scholar]
- 31.Mathers C. D., Boerma T., Ma Fat D. Global and regional causes of death. Br. Med. Bull. (2009);92:7–32. doi: 10.1093/bmb/ldp028. [DOI] [PubMed] [Google Scholar]
- 32.Mathers C. D., Loncar D. Projections of global mortality and burden of disease from 2002 to 2030. PLoS Med. (2006);3:e442. doi: 10.1371/journal.pmed.0030442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mercadier J. J., Lompre A. M., Duc P., Boheler K. R., Fraysse J. B., Wisnewsky C., Allen P. D., Komajda M., Schwartz K. Altered sarcoplasmic reticulum Ca2(+)-ATPase gene expression in the human ventricle during end-stage heart failure. J. Clin. Invest. (1990);85:305–309. doi: 10.1172/JCI114429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Movsesian M. A., Leveille C., Krall J., Colyer J., Wang J. H., Campbell K. P. Identification and characterization of proteins in sarcoplasmic reticulum from normal and failing human left ventricles. J. Mol. Cell Cardiol. (1990);22:1477–1485. doi: 10.1016/0022-2828(90)90990-J. [DOI] [PubMed] [Google Scholar]
- 35.Neumann J., Eschenhagen T., Jones L. R., Linck B., Schmitz W., Scholz H., Zimmermann N. Increased expression of cardiac phosphatases in patients with end-stage heart failure. J. Mol. Cell Cardiol. (1997);29:265–272. doi: 10.1006/jmcc.1996.0271. [DOI] [PubMed] [Google Scholar]
- 36.Neumann J., Gupta R. C., Schmitz W., Scholz H., Nairn A. C., Watanabe A. M. Evidence for isoproterenol-induced phosphorylation of phosphatase inhibitor-1 in the intact heart. Circ. Res. (1991);69:1450–1457. doi: 10.1161/01.RES.69.6.1450. [DOI] [PubMed] [Google Scholar]
- 37.Nicolaou P., Rodriguez P., Ren X., Zhou X., Qian J., Sadayappan S., Mitton B., Pathak A., Robbins J., Hajjar R. J., Jones K., Kranias E. G. Inducible expression of active protein phosphatase-1 inhibitor-1 enhances basal cardiac function and protects against ischemia/reperfusion injury. Circ. Res. (2009);104:1012–1020. doi: 10.1161/CIRCRESAHA.108.189811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Oh J. G., Kim J., Jang S. P., Nguen M., Yang D. K., Jeong D., Park Z. Y., Park S. G., Hajjar R. J., Park W. J. Decoy peptides targeted to protein phosphatase 1 inhibit dephosphorylation of phospholamban in cardiomyocytes. J. Mol. Cell Cardiol. (2013);56:63–71. doi: 10.1016/j.yjmcc.2012.12.005. [DOI] [PubMed] [Google Scholar]
- 39.Pathak A., de lMonte F., Zhao W., Schultz J. E., Lorenz J. N., Bodi I., Weiser D., Hahn H., Carr A. N., Syed F., Mavila N., Jha L., Qian J., Marreez Y., Chen G., McGraw D. W., Heist E. K., Guerrero J. L., DePaoli-Roach A. A., Hajjar R. J., Kranias E. G. Enhancement of cardiac function and suppression of heart failure progression by inhibition of protein phosphatase 1. Circ. Res. (2005);96:756–766. doi: 10.1161/01.RES.0000161256.85833.fa. [DOI] [PubMed] [Google Scholar]
- 40.Periasamy M., Reed T. D., Liu L. H., Ji Y., Loukianov E., Paul R. J., Nieman M. L., Riddle T., Duffy J. J., Doetschman T., Lorenz J. N., Shull G. E. Impaired cardiac performance in heterozygous mice with a null mutation in the sarco(endo)plasmic reticulum Ca2+-ATPase isoform 2 (SERCA2) gene. J. Biol. Chem. (1999);274:2556–2562. doi: 10.1074/jbc.274.4.2556. [DOI] [PubMed] [Google Scholar]
- 41.Prunier F., Kawase Y., Gianni D., Scapin C., Danik S. B., Ellinor P. T., Hajjar R. J., Del Monte F. Prevention of ventricular arrhythmias with sarcoplasmic reticulum Ca2+ ATPase pump overexpression in a porcine model of ischemia reperfusion. Circulation. (2008);118:614–624. doi: 10.1161/CIRCULATIONAHA.108.770883. [DOI] [PubMed] [Google Scholar]
- 42.Rodriguez P., Mitton B., Nicolaou P., Chen G., Kranias E. G. Phosphorylation of human inhibitor-1 at Ser67 and/or Thr75 attenuates stimulatory effects of protein kinase A signaling in cardiac myocytes. Am. J. Physiol. Heart. Circ. Physiol. (2007);293:H762–769. doi: 10.1152/ajpheart.00104.2007. [DOI] [PubMed] [Google Scholar]
- 43.Rodriguez P., Mitton B., Waggoner J. R., Kranias E. G. Identification of a novel phosphorylation site in protein phosphatase inhibitor-1 as a negative regulator of cardiac function. J. Biol. Chem. (2006);281:38599–38608. doi: 10.1074/jbc.M604139200. [DOI] [PubMed] [Google Scholar]
- 44.Schultz Jel J., Glascock B. J., Witt S. A., Nieman M. L., Nattamai K. J., Liu L. H., Lorenz J. N., Shull G. E., Kimball T. R., Periasamy M. Accelerated onset of heart failure in mice during pressure overload with chronically decreased SERCA2 calcium pump activity. Am. J. Physiol. Heart. Circ. Physiol. (2004);286:H1146–1153. doi: 10.1152/ajpheart.00720.2003. [DOI] [PubMed] [Google Scholar]
- 45.Steenaart N. A., Ganim J. R., Di Salvo J., Kranias E. G. The phospholamban phosphatase associated with cardiac sarcoplasmic reticulum is a type 1 enzyme. Arch. Biochem. Biophys. (1992);293:17–24. doi: 10.1016/0003-9861(92)90359-5. [DOI] [PubMed] [Google Scholar]
- 46.Yano M., Yamamoto T., Ikemoto N., Matsuzaki M. Abnormal ryanodine receptor function in heart failure. Pharmacol. Ther. (2005);107:377–391. doi: 10.1016/j.pharmthera.2005.04.003. [DOI] [PubMed] [Google Scholar]