

Abstract
Fibroblast activation protein (FAP) is a specific serine protease expressed in tumor stroma proven to be a stimulatory factor in the progression of some cancers. The purpose of this study was to investigate the effects of FAP knockdown on tumor growth and the tumor microenvironment. Mice bearing 4T1 subcutaneous tumors were treated with liposome-shRNA complexes targeting FAP. Tumor volumes and weights were monitored, and FAP, collagen, microvessel density (MVD), and apoptosis were measured. Our studies showed that shRNA targeting of FAP in murine breast cancer reduces FAP expression, inhibits tumor growth, promotes collagen accumulation (38%), and suppresses angiogenesis (71.7%), as well as promoting apoptosis (by threefold). We suggest that FAP plays a role in tumor growth and in altering the tumor microenvironment. Targeting FAP may therefore represent a supplementary therapy for breast cancer. [BMB Reports 2013; 46(5): 252-257]
Keywords: Angiogenesis, Breast cancer, Fibroblast activation protein, RNA interference, Tumor microenvironment
INTRODUCTION
Breast cancer is the most common neoplastic disease and the second most common cause of death in females. The high incidence and mortality of breast cancer (1) highlight the need to explore alternative therapeutic strategies. In addition, triple-negative breast cancer (TNBC) is linked to poor prognosis. Patients with TNBC are limited to cytotoxic chemotherapy, as only a few clinically validated molecular treatment targets have been identified, including human epidermal growth factor receptor 2, estrogen receptor-alpha, and progesterone receptor (2). New targets should be explored to provide supplementary therapies.
Fibroblast activated protein (FAP) is overexpressed in breast cancer and other epithelial tumors but not in normal adult tissues (3). As a marker of tumor-associated fibroblasts (TAFs), FAP is abnormally elevated in tumor stroma and has an important function in tumor growth and metastasis (4). The FAP type II transmembrane cell surface protein is a dual-specificity serine protease with both dipeptidyl peptidase and endopeptidase activity, including a collagenolytic activity capable of degrading gelatin and type I collagen (Col-I) (5,6). In vivo studies have indicated that increased FAP expression by tumor cells enhances tumorigenicity and promotes rapid tumor growth and increased microvessel density (MVD) (7,8). These studies have shown that FAP is a stimulatory factor for the progression of some cancers. As reviewed by Pietras, genes playing a role in tumor-host interactions can be targets for RNA interference (RNAi) (9). Therefore, we considered FAP to be a potential new target for RNAi-based therapy.
RNAi can selectively downregulate target gene expression and has therefore become a powerful tool for functional genomics, especially in cancer research (10). Short hairpin RNA (shRNA) (10,11) and a variety of nonviral nanoparticles (50-200 nm) and other cationic lipids have been recently reported to be suitable RNAi vehicles in experimental mouse models, providing around 50% knockdown of target gene expression in tumors (12-14). We investigated the effects of shRNA-mediated FAP silencing on the tumor microenvironment (TME) using cationic lipids in a 4T1 mouse mammary carcinoma model.
RESULTS
FAP knockdown in vitro and in vivo
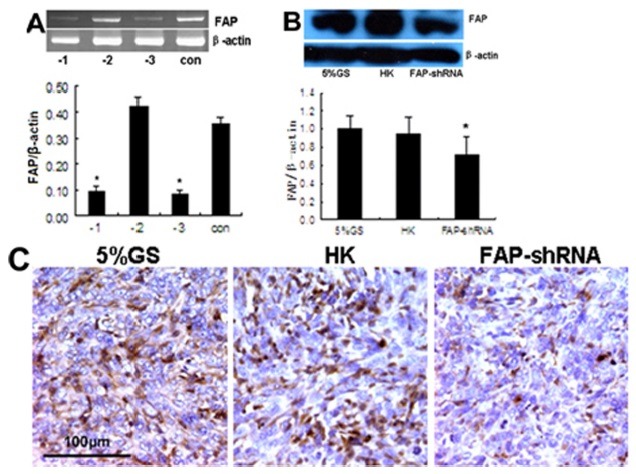
To investigate their inhibitory effect on FAP mRNA, three different mouse-specific siRNAs were transfected into pFAP-transfected 293 cells. Silencing efficiency was tested by reverse transcription-polymerase chain reaction (RT-PCR). As shown in Fig. 1A, si-m-FAP_003 caused the greatest inhibition of FAP mRNA (P<0.05). Therefore, the si-m-FAP_003 sequence was used to synthesize shRNA targeting FAP (FAP-shRNA). In the animal experiments, FAP expression was reduced in the FAP-shRNA group compared to the HK group and 5% GS group (P<0.05) (Fig. 1B and C).
Fig. 1. RNAi-mediated knockdown of FAP in vitro and in vivo. (A) RT-PCR. Representative DNA bands of FAP and β-actin, as well as normalization of FAP to β-actin. Samples from culture cells transfected Si-m-FAP_001 (-1), Si-m-FAP_002 (-2), Si-m-FAP_ 003 (-3) and blank control (con). (B) Western blotting. Representative FAP and β-actin protein bands, as well as FAP expression normalized to β-actin. (C) Immunohistochemistry staining. Sections of 4T1 tumor tissue showing randomly selected representative areas. Magnification, 40×. *P < 0.05 compared with control groups.
FAP knockdown inhibits tumor growth
Reduced tumor burden was evident upon macroscopic inspection of the FAP-shRNA group. Tumor growth was slower in the FAP-shRNA group than in the two control groups after treatment for a week (P < 0.05) (Fig. 2A). In contrast, there was no significant difference in tumor volume between the HK group and the 5% GS group (P = 0.364). In addition, a statistically significant difference was observed in tumor weight between FAP-shRNA-treated mice and controls. Tumors treated with 5% GS and HK reached 0.634 ± 0.112 g and 0.593 ± 0.102 g, respectively. However, tumor weight was reduced to 0.411 ± 0.074 g (P < 0.05) (Fig. 2B) in the FAP-shRNA group.
FAP knockdown promotes collagen accumulation and reduces angiogenesis
Col-I and MVD were measured because previous studies indicated that FAP has collagenase activity and that FAP overexpression induces angiogenesis. We found that FAP knockdown reduces tumor angiogenesis. As shown in Fig. 3A, a significant decrease in MVD was observed in tumors treated with FAP-shRNA. The average number of CD31+ cells per field was 59.8 ± 11.5 in the 5% GS group, 54.7 ± 13.2 in the HK group, and 15.4 ± 5.7 in the FAP-shRNA group. MVD in the FAP-shRNA group was reduced by 71.7% compared to control groups (P < 0.001) (Fig. 3B). We also observed an increased accumulation of disorganized collagen fibers in most tumor tissues in the FAP-shRNA group (Fig. 3A). As shown in Fig. 3C, tumors treated with FAP-shRNA contained more Col-I(an increase of 38%) (P < 0.05) than did controls.
Fig. 2. FAP-shRNA targets FAP-mediated inhibition of tumor growth. (A) Tumor sizes (mm3) (n = 7 per group) were recorded on days 8, 11, 14, 17, 19, 21, 23, and 25 after tumor inoculation. (B) Tumor weight on day 25. *P < 0.05 compared with controls.
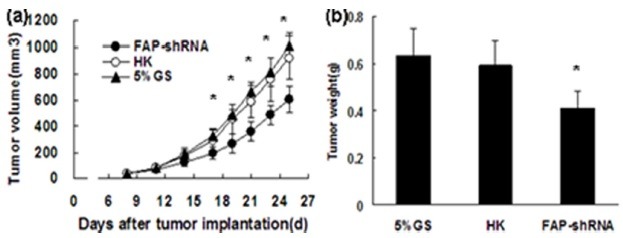
Fig. 3. FAP knockdown alters the tumor microenvironment. (A) Immunohistochemical staining for CD31 (top row) and Picric-Sirius Red staining for collagen (bottom row). Magnification, 20×. (B) Average numbers of CD31+ per high-power field (magnification, 40×). In each case, 6-10 fields were selected for counting. *P < 0.001 compared with controls. (C) Western blotting assay. Representative Col-I and β-actin protein bands, as well as Col-I expression normalized to β-actin.
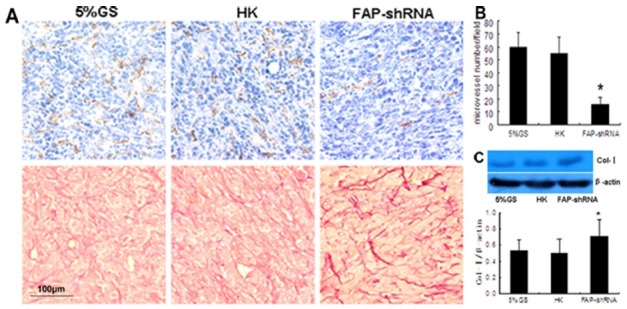
Fig. 4. FAP knockdown enhances apoptosis. TUNEL assay performed on sections from mice treated with 5% GS (A), HK (B), or FAP-shRNA (C). Magnification, 20×. (D) The average number of TUNEL-positive cells per high-power field (magnification, 20×). In each case, 6-10 fields were selected for counting. *P < 0.001 compared with controls.
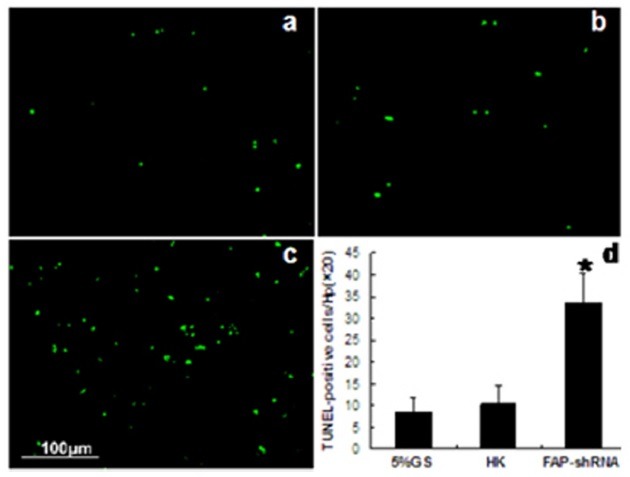
FAP knockdown enhances apoptosis
Apoptosis was evaluated by the terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay. The number of apoptotic cells was increased in tumors treated with FAP-shRNA (Fig. 4): an average of 8.2 ± 2.2 in the 5% GS group, 11.4 ± 4.5 in the HK group, and 34.8 ± 7.3 in the FAP-shRNA group. Thus, the number of apoptotic cells in the FAP-shRNA group was approximately threefold greater than in control groups (P < 0.001).
Liposome toxicity analysis
Both macroscopic and microscopic examinations were carried out to assess the toxicity of shRNA-liposome complexes. No significant adverse effects of treatment with these complexes were observed on weight, coat condition, appetite or general activity. In addition, there were no differences in mortality or in pathological changes to the heart, liver, spleen, lungs, and kidneys among the three groups (data not shown).
DISCUSSION
It is thought that the TME regulates cancer growth by providing factors required by tumor cells for survival, growth, proliferation, and metastasis (15). Stromagenesis, involving activation of TAFs, pullulation of vessels, and modulation of the extracellular matrix (ECM), occurs during tumor progression (16). FAP is a marker of TAFs that is overexpressed in > 90% of epithelial tumors but not in normal adult human tissues (3). Due to its highly specific expression and restricted tissue distribution, FAP has been identified as a potential target for antitumor therapeutics.
Previous studies aimed at blocking FAP function in tumors have included targeting FAP-expressing cells, inhibiting FAP enzyme activity, and genetic deletion of FAP. Loeffler reported that an oral DNA vaccine targeting FAP suppresses primary tumor growth and metastasis, as well as increasing intratumoral uptake of chemotherapeutic drugs in multidrug-resistant murine colon and breast carcinoma models (17). In contrast, sibrotuzumab (mAb F19), a humanized anti-FAP antibody, showed no efficacy on metastatic colorectal cancer in a Phase II trial (18). It is important to note that both DNA vaccines and antibodies work through CD8+ T cell-mediated killing of TAFs and not by FAP inhibition or through direct cytotoxic activity. However, their effects are mediated by TAF reduction. In addition, inhibitors of FAP enzyme activity, such as Val-boro-Pro (PT-100, Talabostat) and Glu-Boro-Pro (PT-630), have been used to target FAP (19-21). However, both PT-100 and PT-630 also inhibit other intracellular and extracellular dipeptidyl peptidases such as DPP IV and DPP II. Therefore, the antitumor effects of these compounds may not be directly linked to FAP inhibition.
In the current study, we used vector-based shRNA against FAP to study the effect of FAP downregulation on murine breast cancer. We report that FAP knockdown attenuates tumor growth and improves TME morphology in this system, including the accumulation of disorganized collagen and suppression of angiogenesis. The mechanism linking FAP knockdown to effects on the TME differs from that of targeting FAP-expressing cells. We chose to study the functions of FAP on tumor growth and TME IN a 4T1 breast cancer model, as breast cancers have a high stromal content and can stably express FAP at high levels.
A supporting stroma and blood vessel network is required for solid tumor growth (22,23). Thus, angiogenesis is critical for tumor volumes beyond 1-2 mm3 (24). Huang and colleagues developed FAP-expressing breast cancer cells and discovered that FAP-expressing cells have an improved ability to attract blood vessels and exhibit rapid tumor growth (7). We found that FAP knockdown strongly decreases MVD in breast cancer. Thus, targeting FAP can inhibit tumor growth at least partly through suppressing angiogenesis.
The progression of palpable breast cancers is associated with the formation of fibrillar networks, which are thought to promote tumor progression and invasion (25). Collagen is the main ECM component of tumor fibrillar networks and the presence of collagen structures radially aligned with tumor cells has been suggested to promote tumor progression and invasion (26). Our study showed that FAP knockdown causes an increased accumulation of disorganized collagen fibers. It was recently reported that FAP functions in synchrony with other proteinases, such as MMP-1, to cleave collagen into smaller peptides, a process which might regulate malignant cell growth and motility (6). FAP may therefore play a role in remodeling fibrillar networks.
In addition, we have provided evidence that FAP knockdown enhances apoptosis. Apoptosis is known to have an important function in regulating tumor growth and the tumor response to various forms of cancer therapy (27). FAP may interact with apoptosis-associated proteins through its protease activity. However, further study is required to define how FAP regulates apoptosis.
In the current study, we did not find any side effects of shRNA vector-based therapy in mice. The pVector used in this study contains the kanamycin resistance gene for selection in Escherichia coli; thus, the use of ampicillin, which may cause an allergic response, is avoided. However, exogenous shRNA expression can overload the endogenous RNAi pathway through competitive binding to exportin-5 (28). Therefore, it is possible that long-term gene silencing may trigger cytotoxicity.
In summary, the present study supports the hypothesis that targeting FAP can inhibit tumor growth and alter the structure of the TME through collagen accumulation, suppression of angiogenesis, and stimulating apoptosis. As FAP expression is cancer specific and plays a role in breast cancer growth, we suggest that FAP is a potential new target for breast cancer therapies. Thus, targeting FAP may be a supplementary therapeutic modality for breast cancer.
MATERIALS AND METHODS
Cells and animals
The 293 and 4T1 mouse mammary carcinoma cells purchased from the American Type Culture Collection (Manassas, VA, USA) were cultured in Dulbecco's modified Eagle medium supplemented with 10% heat-inactivated fetal bovine serum and 1% penicillin/streptomycin (GIBCO) at 37℃ in a 5% CO2 humidified atmosphere. Female BALB/c mice, aged 6 weeks and weighing 20-22 g, were purchased from the West China Experimental Animal Center (Chengdu, China) and housed in standard rodent cages in a light- and temperature-controlled room. All animal procedures were approved by the Institutional Animal Care and Treatment Committee of Sichuan University.
FAP mRNA inhibition screen
Three different mouse-specific siRNA molecules were designed based on the murine FAP lead siRNA sequence. The corresponding cynomolgus cDNA sequence was not available from the manufacturer. pFAP-transfected 293 cells were constructed and FAP expression was measured as described (29). Fifteen hours before transfection, pFAP-transfected 293 cells were trypsinized and seeded into 6-well culture plates at 4 × 105 cells per well. siRNA–lipoplexes (Si-m-FAP_001, Si-m-FAP_002, and Si-m-FAP_003, Si-GFP), containing 50 nM siRNA and 5 μl LipofectAMINE 2000 (Invitrogen), were prepared in 1 ml Opti-MEMI medium and incubated for 30 min at room temperature, and then added to cells. Cells transfected with Si-GFP for 24 h showed a transfection efficiency of 85% (data not shown).
RT-PCR was used to analyze FAP mRNA, using β-actin as an internal control. FAP primer sequences were: 5ʹ-CGGGATCCAAAATGAAGACATGGCTGAAAACT-3ʹ (forward) and 5ʹ-ACGCGTCGACTCAGTCTGATAAAGAAAAGCATTG-3ʹ (reverse). Primer sequences for β-actin were: 5ʹ-CGGGAAATCGTGCGTGAC-3ʹ (forward) and 5ʹ-TGGAAGGTGGACAGCGAGG-3ʹ (reverse). After 36 h, total RNA was extracted from transfected cells using TRIzol (Invitrogen, USA) as described (14). Samples (5 μl) were separated by electrophoresis in 1% agarose gels and visualized by a chemiluminescence gel imaging system (BIO-RAD). Each experiment was repeated three times. Quantitative analysis for bands was performed using Quantity One software.
Preparation of shRNA plasmid and liposome
pGenesil-3 vectors harboring 5ʹ-gatccGACAGTATCCTAGAACTAttcaagacgTAGTTCTAGGATACTGTCttttttgtcgaca-3ʹ or 5ʹ-gat ccGACTTCATAAGGCGCATGCttcaagacgGCATGCGCCTTATGAAGTCttttttgtcgaca-3ʹ inserts were prepared for expressing FAP-specific shRNAs (FAP-shRNA) or an unrelated sequence control (HK-shRNA). Colonies of E. coli containing pGenesil-3 vectors harboring FAP-shRNA or HK-shRNA were cultured in Luria Bertani broth containing 50 μg/ml kanamycin. Large-scale plasmid shRNA preparations were obtained using the EndoFree Plasmid Giga kit (Qiagen, Germany). Purified shRNA was dissolved in sterile endotoxin-free water and stored at −20℃.
A cationic liposome (DOTAP/cholesterol) was used for plasmid transfection and for animal treatment experiments. Cationic liposomes were prepared by our laboratory using a previously described procedure (29,30). Cationic liposomes (DOTAP/cholesterol) preparations comprised small multi-lamellar liposomes in the size range of 100 ± 20 nm. These were stored at 4℃ and diluted in 5% glucose solution (GS) for use.
In vivo RNAi treatment
A syngeneic transplanted 4T1 tumor model was developed. Each mouse was challenged s.c. with 1 × 106 4T1 mouse mammary carcinoma cells in the right flank on day 0. Primary tumors grew to a mean diameter of 4.5 mm by day 8. Twenty-one mice were enrolled and randomly assigned into three groups (n = 7 per group) and received the following treatments: (1) FAP-shRNA group, treated with 5 μg FAPshRNA/15 μg liposome complexes (volume = 100 μl), (2) HK group, treated with 5 μg HK-shRNA/15 μg liposome complexes (volume = 100 μl), (3) 5% GS group, treated with 100 μl 5% GS. Mice received nine intratumoral and peritumoral treatments at 48-h intervals from day 8 onwards. Tumor size was measured using Vernier calipers at 2- or 3-day intervals, and tumor volumes were calculated using the standard formula: (width)2 × length × 0.52. Mice were sacrificed on day 25, and tumors were excised and weighed. Tumor tissue samples were frozen immediately. Other samples were fixed in 4% paraformaldehyde and embedded in paraffin. Each experiment was repeated three times.
Histological and immunohistochemical examination
After embedding in paraffin, tissue sections (4-μm thick) were cut and mounted onto glass slides. Hematoxylin/eosin and Sirius Red staining were performed as previously described (29).
Rabbit anti-mouse FAP antibody (Abcam, UK) and rabbit anti-mouse CD31 antibody (Acris, Germany) at 1/100 dilution (v⁄v) were used to detect FAP expression and MVD. Standard immunohistochemistry procedures were performed for FAP (paraffin sections) and MVD (4-μm thick frozen). Sections were incubated with primary antibody at 4℃ overnight. Biotinylated goat anti-rabbit IgG was used to detect primary antibody binding and visualized using the SABC Elite kit (Boster, China) following the manufacturer’s recommendations. Reaction products were detected using the UltraVision Quanto Detection System HRP DAB kit (Thermo Scientific, USA), according to the manufacturer’s recommendations. The negative control for each double immunohistochemistry was omission of the primary antibody. Sections were counterstained with hematoxylin.
Western blotting
For protein extraction, tumor tissues were homogenized in liquid nitrogen and lysed in radioimmunoprecipitation assay buffer containing protease inhibitors. Protein extraction, quantification, and immunoblotting were carried out as described previously (20). Protein samples (80 μg) were separated by 8% SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to polyvinylidene difluoride membrane (Millipore, USA). Primary antibodies used for blotting were: rabbit anti-type I collagen (Abcam, UK), rabbit anti-mouse FAP (Abcam, UK) and mouse β-actin monoclonal antibody (Wuhan, China), all at dilutions of 1/1,000. Signal development was carried out using the Luminata Crescendo HRP substrate (Millipore, USA) and exposure to X-ray film. Quantitative analysis of bands was performed using Quantity One software.
TUNEL assay
To investigate whether FAP participates in apoptosis, apoptotic cells in paraffin sections were examined using an in situ apoptotic cell detection kit according to the manufacturer’s recommended instructions (Promega, USA). Stained sections were assessed and images were taken as described (14).
Toxicity assessment
In order to evaluate the potential side effects of treatment with shRNA plasmid-liposome complexes, mice were continuously observed for relevant indexes such as weight, coat condition, appetite, activity, and toxicity-associated death. After mice were sacrificed, their organs (heart, kidneys, liver, lung, and spleen) were fixed in 4% paraformaldehyde, embedded in paraffin, and stained with hematoxylin/eosin.
Statistical analysis
IBM SPSS 19.0 statistics software was used for all statistical analyses. All data are presented as means ± SD and analyzed by one-way ANOVA using Tukey’s multiple comparisons. Differences between means were deemed statistically significant for P values < 0.05.
Acknowledgments
This work was supported by grants from the National Natural Science Foundation of China (No. 31070818) and the National Key Basic Research Program (973 Program) of China (No. 2010CB529900). The authors thank Zhi-Yong Li for kindly providing the 293 cells transfected with pFAP or pVector.
References
- 1.Jemal A., Siegel R., Xu J., Ward E. Cancer statistics, 2010. CA Cancer J. Clin. (2010);60:277–300. doi: 10.3322/caac.20073. [DOI] [PubMed] [Google Scholar]
- 2.Verma S., Provencher L., Dent R. Emerging trends in the treatment of triple-negative breast cancer in Canada: a survey. Curr. Oncol. (2011);18:180–190. doi: 10.3747/co.v18i4.913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rettig W. J., Garin-Chesa P., Beresford H. R., Oettgen H. F., Melamed M. R., Old L. J. Cell-surface glycoproteins of human sarcomas: differential expression in normal and malignant tissues and cultured cells. Proc. Natl. Acad. Sci. U.S.A. (1988);85:3110–3114. doi: 10.1073/pnas.85.9.3110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kunz-Schughart L. A., Knuechel R. Tumor-associated fibroblasts (part I): Active stromal participants in tumor development and progression? Histol. Histopathol. (2002);17:599–621. doi: 10.14670/HH-17.599. [DOI] [PubMed] [Google Scholar]
- 5.Edosada C. Y., Quan C., Wiesmann C., Tran T., Sutherlin D., Reynolds M., Elliott J. M., Raab H., Fairbrother W., Wolf B. B. Selective inhibition of fibroblast activation protein protease based on dipeptide substrate specificity. J. Biol. Chem. (2006);281:7437–7444. doi: 10.1074/jbc.M511112200. [DOI] [PubMed] [Google Scholar]
- 6.Christiansen V. J., Jackson K. W., Lee K. N., McKee P. A. Effect of fibroblast activation protein and alpha2-antiplasmin cleaving enzyme on collagen types I, III and IV. Arch. Biochem. Biophys. (2007);457:177–186. doi: 10.1016/j.abb.2006.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Huang Y., Wang S., Kelly T. Seprase promotes rapid tumor growth and increased microvessel density in a mouse model of human breast cancer. Cancer Res. (2004);64:2712–2716. doi: 10.1158/0008-5472.CAN-03-3184. [DOI] [PubMed] [Google Scholar]
- 8.Cheng J. D., Dunbrack R. L. Jr., Valianou M., Rogatko A., Alpaugh R. K., Weiner L. M. Promotion of tumor growth by murine fibroblast activation protein, a serine protease, in an animal model. Cancer Res. (2002);62:4767–4772. [PubMed] [Google Scholar]
- 9.Pietras K., Ostman A. Hallmarks of cancer: interactions with the tumor stroma. Exp. Cell Res. (2010);316:1324–1331. doi: 10.1016/j.yexcr.2010.02.045. [DOI] [PubMed] [Google Scholar]
- 10.Sliva K., Schnierle B. S. Selective gene silencing by viral delivery of short hairpin RNA. Virol. J. (2010);7:248. doi: 10.1186/1743-422X-7-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hannon G. J., Rossi J. J. Unlocking the potential of the human genome with RNA interference. Nature. (2004);431:371–378. doi: 10.1038/nature02870. [DOI] [PubMed] [Google Scholar]
- 12.Santel A., Aleku M., Keil O., Endruschat J., Esche V., Durieux B., Loffler K., Fechtner M., Rohl T., Fisch G., Dames S., Arnold W., Giese K., Klippel A., Kaufmann J. RNA interference in the mouse vascular endothelium by systemic administration of siRNA-lipoplexes for cancer therapy. Gene Ther. (2006);13:1360–1370. doi: 10.1038/sj.gt.3302778. [DOI] [PubMed] [Google Scholar]
- 13.Pirollo K. F., Rait A., Zhou Q., Hwang S. H., Dagata J. A., Zon G., Hogrefe R. I., Palchik G., Chang E. H. Materializing the potential of small interfering RNA via a tumor-targeting nanodelivery system. Cancer Res. (2007);67:2938–2943. doi: 10.1158/0008-5472.CAN-06-4535. [DOI] [PubMed] [Google Scholar]
- 14.Wan Y., Huang A., Yang Y., Xie G., Chen X., Hu J., Yang L., Li J., Chen L., Jiang Y., Zhao X., Wei Y., Deng H. A vector-based short hairpin RNA targeting Aurora A inhibits breast cancer growth. Int. J. Oncol. (2010);36:1121–1128. doi: 10.3892/ijo_00000579. [DOI] [PubMed] [Google Scholar]
- 15.Mbeunkui F., Johann D. J. Jr. Cancer and the tumor microenvironment: a review of an essential relationship. Cancer Chemother. Pharmacol. (2009);63:571–582. doi: 10.1007/s00280-008-0881-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dvorak H. F. Tumors: wounds that do not heal. Similarities between tumor stroma generation and wound healing. N. Engl. J. Med. (1986);315:1650–1659. doi: 10.1056/NEJM198612253152606. [DOI] [PubMed] [Google Scholar]
- 17.Loeffler M., Kruger J. A., Niethammer A. G., Reisfeld R. A. Targeting tumor-associated fibroblasts improves cancer chemotherapy by increasing intratumoral drug uptake. J. Clin. Invest. (2006);116:1955–1962. doi: 10.1172/JCI26532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hofheinz R. D., al-Batran S. E., Hartmann F., Hartung G., Jager D., Renner C., Tanswell P., Kunz U., Amelsberg A., Kuthan H., Stehle G. Stromal antigen targeting by a humanised monoclonal antibody: an early phase II trial of sibrotuzumab in patients with metastatic colorectal cancer. Onkologie. (2003);26:44–48. doi: 10.1159/000069863. [DOI] [PubMed] [Google Scholar]
- 19.Adams S., Miller G. T., Jesson M. I., Watanabe T., Jones B., Wallner B. P. PT-100, a small molecule dipeptidyl peptidase inhibitor, has potent antitumor effects and augments antibody-mediated cytotoxicity via a novel immune mechanism. Cancer Res. (2004);64:5471–5480. doi: 10.1158/0008-5472.CAN-04-0447. [DOI] [PubMed] [Google Scholar]
- 20.Santos A. M., Jung J., Aziz N., Kissil J. L., Pure E. Targeting fibroblast activation protein inhibits tumor stromagenesis and growth in mice. J. Clin. Invest. (2009);119:3613–3625. doi: 10.1172/JCI38988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huang Y., Simms A. E., Mazur A., Wang S., Leon N. R., Jones B., Aziz N., Kelly T. Fibroblast activation protein-alpha promotes tumor growth and invasion of breast cancer cells through non-enzymatic functions. Clin. Exp. Metastasis. (2011);28:567–579. doi: 10.1007/s10585-011-9392-x. [DOI] [PubMed] [Google Scholar]
- 22.Barcellos-Hoff M. H. It takes a tissue to make a tumor: epigenetics, cancer and the microenvironment. J. Mammary Gland Biol. Neoplasia. (2001);6:213–221. doi: 10.1023/A:1011317009329. [DOI] [PubMed] [Google Scholar]
- 23.Roskelley C. D., Bissell M. J. The dominance of the microenvironment in breast and ovarian cancer. Semin. Cancer Biol. (2002);12:97–104. doi: 10.1006/scbi.2001.0417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Folkman J. What is the evidence that tumors are angiogenesis dependent? J. Natl. Cancer Inst. (1990);82:4–6. doi: 10.1093/jnci/82.1.4. [DOI] [PubMed] [Google Scholar]
- 25.Provenzano P. P., Eliceiri K. W., Campbell J. M., Inman D. R., White J. G., Keely P. J. Collagen reorganization at the tumor-stromal interface facilitates local invasion. BMC Med. (2006);4:38. doi: 10.1186/1741-7015-4-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Netti P. A., Berk D. A., Swartz M. A., Grodzinsky A. J., Jain R. K. Role of extracellular matrix assembly in interstitial transport in solid tumors. Cancer Res. (2000);60:2497–2503. [PubMed] [Google Scholar]
- 27.Indran I. R., Tufo G., Pervaiz S., Brenner C. Recent advances in apoptosis, mitochondria and drug resistance in cancer cells. Biochim. Biophys. Acta. (2011);1807:735–745. doi: 10.1016/j.bbabio.2011.03.010. [DOI] [PubMed] [Google Scholar]
- 28.Grimm D., Wang L., Lee J. S., Schurmann N., Gu S., Borner K., Storm T. A., Kay M. A. Argonaute proteins are key determinants of RNAi efficacy, toxicity and persistence in the adult mouse liver. J. Clin. Invest. (2010);120:3106–3119. doi: 10.1172/JCI43565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wen Y., Wang C. T., Ma T. T., Li Z. Y., Zhou L. N., Mu B., Leng F., Shi H. S., Li Y. O., Wei Y. Q. Immunotherapy targeting fibroblast activation protein inhibits tumor growth and increases survival in a murine colon cancer model. Cancer Sci. (2010);101:2325–2332. doi: 10.1111/j.1349-7006.2010.01695.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Templeton N. S., Lasic D. D., Frederik P. M., Strey H. H., Roberts D. D., Pavlakis G. N. Improved DNA: liposome complexes for increased systemic delivery and gene expression. Nat. Biotechnol. (1997);15:647–652. doi: 10.1038/nbt0797-647. [DOI] [PubMed] [Google Scholar]