

Abstract
Genome-wide association studies (GWAS) have become popular as an approach for the identification of large numbers of phenotype-associated variants. However, differences in genetic architecture and environmental factors mean that the effect of variants can vary across populations. Understanding population genetic diversity is valuable for the investigation of possible population specific and independent effects of variants. EvoSNP-DB aims to provide information regarding genetic diversity among East Asian populations, including Chinese, Japanese, and Korean. Non-redundant SNPs (1.6 million) were genotyped in 54 Korean trios (162 samples) and were compared with 4 million SNPs from HapMap phase II populations. EvoSNP-DB provides two user interfaces for data query and visualization, and integrates scores of genetic diversity (Fst and VarLD) at the level of SNPs, genes, and chromosome regions. EvoSNP-DB is a web-based application that allows users to navigate and visualize measurements of population genetic differences in an interactive manner, and is available online at [http://biomi.cdc.go.kr/EvoSNP/]. [BMB Reports 2013; 46(8): 416-421]
Keywords: Database, Genetic diversity, GWAS, Population genetics, SNP
INTRODUCTION
Recent developments in high throughput SNP chip technologies have enabled researchers to conduct large-scale genome-wide association studies (GWAS) (1-6). These have revealed an unprecedented amount of genetic variants that are associated with complex traits (7). As of August, 2012, there were 1,330 publications and 6,848 phenotype-associated SNPs in the NHGRI GWAS catalogue (http://www.genome.gov/gwastudies/). The availability of plentiful phenotype-related genomic information is expected to lead to clinically applicable personal genomics in the near future (8,9), however, a number of issues require attention before this can be widely applied. Firstly, the identification of causal variants, and a functional investigation of known loci are required (10,11). GWAS have localized associated signals to specific genomic regions, however, most identified variants are located within intergenic, intronic, and gene desert regions, and are regarded as proxies for causal variants. Further analysis, such as fine mapping and resequencing, is required to unveil causal variants of phenotypes. Only a small number of genes in close proximity to associated variants have been examined to identify possible functional relationships with phenotypes. Secondly, the majority of GWAS have been conducted on populations with European ancestry. This data of European relevence should be validated for its application to other ethnicities, such as those of Asian or African ancestry. Although some recent GWAS have been conducted on ethnic groups other than Europeans, sample sizes and numbers of target phenotypes have been relatively small compared with studies of Europeans (2,6,12). It is important to consider population specific associations for personal genomics applications, as phenotype associations regularly vary across populations (3-5).
Population specific or independent associations of variants can be identified by GWAS in a specific population, or by independent replication studies. However, these approaches require a large number of samples, compounding the high costs associated with genotyping. As an alternative, the genetic diversity of phenotype-associated regions may be examined. Large differences in genetic architecture among populations are an established cause of discrepancies in associations (3-5). The fixation index (Fst) is one of the most widely used metrics for measuring genetic differentiation between populations (13). Variation in linkage disequilibrium (VarLD) is another approach that measures population differences in LD patterns (14). Various web interfaces have been developed to provide user-friendly graphical interfaces (GUI) and browsers to access genetic variation data, including Haplotter, FstSNPHapMap3, SNP@Evolution, and Singapore Genome Variation Project (SGVP) (15-18). Three of these use only reference information, such as data from HapMap phase 2 and phase 3 (19,20). SGVP also provides information derived from three Southeast Asian populations, as compared to HapMap populations (18).
Genetic diversity among East Asian populations has not previously been provided via a web service. In particular, the Korean population is one of the most intensely studied in East Asia, but there is no web resource providing genetic diversity data which includes Koreans. Although two populations (Han Chinese in Beijing )undefined(CHB) and Japanese in Tokyo (JPT)(CHB and JPT) should not be regarded as references for Koreans (21). We therefore developed EvoSNP-DB, a web resource for genetic diversity among East Asian populations.
RESULTS AND DISCUSSION
We constructed EvoSNP-DB by integrating GBrowse and genotype data from 108 Koreans (founders) and 210 HapMap phase II release #22 samples. After quality control, 1,147,845 SNPs overlapped across Korean and other HapMap populations. The EvoSNP-DB database and web server is implemented on a 24×2.66 GHz Xeon core server running on Red Hat Enterprise Linux (version 5.2), Apache (version 2.0), Tomcat (version 5.5), and MySQL (version 5.5). It is viewable in all major web browsers and operating systems, and is available online at [http://biomi.cdc.go.kr/EvoSNP/].
Database design and organization
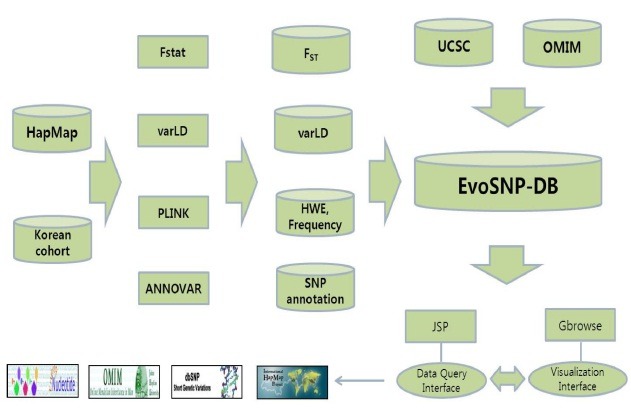
Data flow through the application is described in Fig. 1. Briefly, genotype data were analyzed to calculate Fst, VarLD, allele frequency, and Hardy-Weinberg equilibrium (HWE). Processed data are stored in the database with annotation information retrieved from UCSC and OMIM. The database is wrapped by Gbrowse and JSP for data query and visualization interfaces. Genotype datasets are derived from the International HapMap Phase II release #22 data repository (11,12), including data from 60 Utah residents with ancestry from Northern and Western Europe (CEU), 45 Han Chinese in Beijing (CHB), 45 Japanese in Tokyo (JPT), and 60 Yoruba in Ibadan, Nigeria (YRI). Considering the relatively small number of samples of CHB and JPT, we pooled the data of both as a single geographical group, and denoted it as ASN (Asian, 90 samples). The SNP information from 108 Korean founders of 54 trios was compared to those of HapMap populations. The database has been integrated with Fst and VarLD metrics to facilitate the graphical representation of the data. Fst measures polymorphism within each population and differentiation among geographical groups (13). To quantify variation in population linkage disequilibrium patterns, we used the varLD program (14). HapMap, UCSC, OMIM, and the NHGRI GWAS catalogue were major sources of annotation information.
Fig. 1. Flow diagram of EvoSNP-DB construction.
User interface and visualization
Within EvoSNP-DB there are user interfaces for data queries and visualization. Three types of query can be applied: (i) SNP identifier, (ii) mRNA ID or gene symbol, and (iii) specific chromosome region. For example, rs28218 could be used for a SNP based search, NM_002124 or ORF4F16 for a gene search, and chr1:157661000..157806000 to search for this chromosomal region.
Regardless of the query type, EvoSNP-DB returns three tables providing Region, Gene, and SNP statistics (Fig. 2). Each table contains summary variation scores. Fig. 2 illustrates the output when rs28218 was used as a search term; scores of the gene TRIO, which contains this SNP, are summarized in the gene statistics table. JSP and GMOD (http://gmod.org) were used to build the table and figure interfaces. Links to public online databases, including Entrez Nucleotide, dbSNP, OMIM (22), and HapMap (20), are provided in EvoSNP-DB, together with the results (Fig. 2). EvoSNP-DB also offers a generic genome browser, which displays overviews of chromosomes, contigs, genes, mRNAs, and SNPs (23). Figs. 3 and 4 demonstrate the output if small or large numbers of SNPs exist in the query region, respectively.
Fig. 2. A screenshot of the result table from EvoSNP-DB.
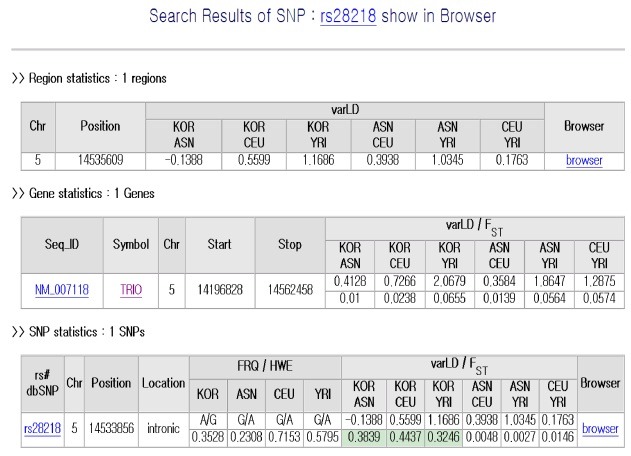
Fig. 3. A detailed screenshot showing EvoSNP-DB search results. Top track: chromosomal overview. SNP locations, diamond shapes. OMIM disease associations, rectangles. Second track: VarLD scores visualized along a 2 Mb chromosomal region. Third track: allele frequencies of SNPs, visualized as a pie chart for the Korean population or as towers for HapMap populations. Bottom track: Genes in the region.
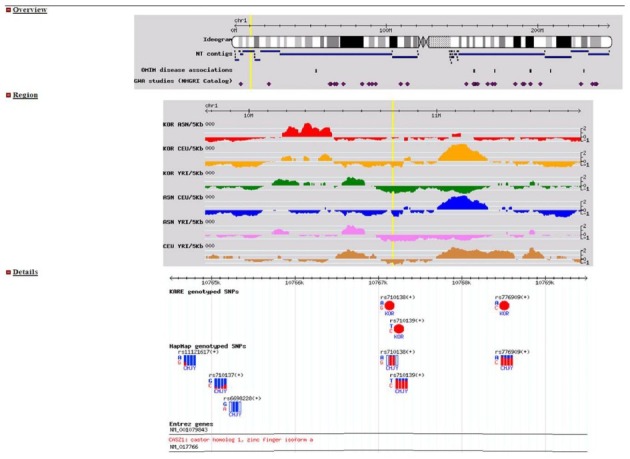
Fig. 4. A wide screenshot showing the search results with OMIM and GWAS Catalogue. Allele frequency is not displayed, but each SNP is indicated.

EvoSNP-DB provides an open-architecture website using a wiki interface for data access (a wiki is a website that allows its users to add, modify, or delete its content via a web browser), and wiki-based SNP annotation will be available in the near future. This will be particularly useful for constructing accurate and informative annotation for variants identified by the collaborative work of many researchers. MySQL, Python, JSP, and GBrowse were used in database construction, and to enhance interface utility (24).
MATERIALS AND METHODS
Korean genotype data
Previously, we conducted GWAS for two independent Korean population-based cohorts (Ansung and Ansan) as part of the Korean Genome Epidemiology Study (KoGES), and the Korean Association REsource (KARE) project, which was initiated in 2007 (2, 4). In the Ansung area, we recruited additional family members of the original participants to facilitate family based association studies. Among these, 54 trios (162 samples) were genotyped using an Affymetrix Genome-Wide Human SNP array 6.0 and an Illumina human Omni1-Quad Chip. Genotypes were called with Birdseed and BeadStudio GenCall for Affymetrix and Illumina arrays, respectively (25, 26). Initially, ∼1.9 million SNPs from the two platforms (909,622 for Affymetrix and 1,010,624 for the Illumina array) were merged. For quality control, we excluded SNPs using the following criteria: non-autosomal, mendelian errors, high missing genotype rate (> 5%), and deviation from HWE (P < 1E-6). Filtered SNPs were compared with data from HapMap SNPs, including allele, strand, and genomic position. After excluding 14 SNPs with annotation errors, 1,147,845 SNPs were overlapped with HapMap SNPs (27).
HapMap genotype data
HapMap phase II release #22 data (210 samples) were downloaded. Genotype data were converted to the PLINK binary genotype format, and genotype frequencies, allele frequencies, and P-values of HWE calculated using PLINK (28).
Analysis of genetic diversity among populations
Fst and VarLD were used as population genetic diversity metrics (13,14). Fst was calculated for each SNP by a pairwise comparison of four populations. Genome-wide VarLD analysis was performed; VarLD scores were calculated for windows of 50 SNPs, starting from the first SNP of each chromosome and ending with the last. All values from 22 chromosomes were merged and were converted to provide a standard normal distribution (mean=0, standard deviation=1). VarLD analysis procedures were performed for all pairs of populations. To access the degree of genetic difference between populations, we calculated the quartiles of Fst and VarLD score distributions.
Acknowledgments
This work was supported by a grant from the Korea Center for Disease Control and Prevention (4845-301, 4851-302, 4851-307) and intramural grant from the Korea National Institute of Health (2012-N72001-00, 2012-N73002-00).
References
- 1.come Trust Case Control Consortium. Genomewide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. (2007);447:661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cho Y. S., Go M. J., Kim Y. J., Heo J. Y., Oh J. H., Ban H. J., Yoon D., Lee M. H., Kim D. J., Park M., Cha S. H., Kim J. W., Han B. G., Min H., Ahn Y., Park M. S., Han H. R., Jang H. Y., Cho E. Y., Lee J. E., Cho N. H., Shin C., Park T., Park J. W., Lee J. K., Cardon L., Clarke G., McCarthy M. I., Lee J. Y., Oh B., Kim H. L. A large-scale genome-wide association study of Asian populations uncovers genetic factors influencing eight quantitative traits. Nat. Genet. (2009);41:527–534. doi: 10.1038/ng.357. [DOI] [PubMed] [Google Scholar]
- 3.Kato N., Takeuchi F., Tabara Y., Kelly T. N., Go M. J., Sim X., Tay W. T., Chen C. H., Zhang Y., Yamamoto K., Katsuya T., Yokota M., Kim Y. J., Ong R. T., Nabika T., Gu D., Chang L. C., Kokubo Y., Huang W., Ohnaka K., Yamori Y., Nakashima E., Jaquish C. E., Lee J. Y., Seielstad M., Isono M., Hixson J. E., Chen Y. T., Miki T., Zhou X., Sugiyama T., Jeon J. P., Liu J. J., Takayanagi R., Kim S. S., Aung T., Sung Y. J., Zhang X., Wong T. Y., Han B. G., Kobayashi S., Ogihara T., Zhu D., Iwai N., Wu J. Y., Teo Y. Y., Tai E. S., Cho Y. S., He J. Meta-analysis of genome-wide association studies identifies common variants associated with blood pressure variation in east Asians. Nat Genet. (2011);43:531–538. doi: 10.1038/ng.834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kim Y. J., Go M. J., Hu C., Hong C. B., Kim Y. K., Lee J. Y., Hwang J. Y., Oh J. H., Kim D. J., Kim N. H., Kim S., Hong E. J., Kim J. H., Min H., Kim Y., Zhang R., Jia W., Okada Y., Takahashi A., Kubo M., Tanaka T., Kamatani N., Matsuda K., Park T., Oh B., Kimm K., Kang D., Shin C., Cho N. H., Kim H. L., Han B. G., Cho Y. S. Large-scale genome-wide association studies in East Asians identify new genetic loci influencing metabolic traits. Nat. Genet. (2011);43:990–995. doi: 10.1038/ng.939. [DOI] [PubMed] [Google Scholar]
- 5.Soranzo N., Spector T. D., Mangino M., Kuhnel B., Rendon A., Teumer A., Willenborg C., Wright B., Chen L., Li M., Salo P., Voight B. F., Burns P., Laskowski R. A., Xue Y., Menzel S., Altshuler D., Bradley J. R., Bumpstead S., Burnett M. S., Devaney J., Doring A., Elosua R., Epstein S. E., Erber W., Falchi M., Garner S. F., Ghori M. J., Goodall A. H., Gwilliam R., Hakonarson H. H., Hall A. S., Hammond N., Hengstenberg C., Illig T., Konig I. R., Knouff C. W., McPherson R., Melander O., Mooser V., Nauck M., Nieminen M. S., O'Donnell C. J., Peltonen L., Potter S. C., Prokisch H., Rader D. J., Rice C. M., Roberts R., Salomaa V., Sambrook J., Schreiber S., Schunkert H., Schwartz S. M., Serbanovic-Canic J., Sinisalo J., Siscovick D. S., Stark K., Surakka I., Stephens J., Thompson J. R., Volker U., Volzke H., Watkins N. A., Wells G. A., Wichmann H. E., Van Heel D. A., Tyler-Smith C., Thein S. L., Kathiresan S., Perola M., Reilly M. P., Stewart A. F., Erdmann J., Samani N. J., Meisinger C., Greinacher A., Deloukas P., Ouwehand W. H., Gieger C. A genome-wie meta-analysis identifies 22 loci associated with eight hematological parameters in the HaemGen consortium. Nat. Genet. (2009);41:1182–1190. doi: 10.1038/ng.467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Teslovich T. M., Musunuru K., Smith A. V., Edmondson A. C., Stylianou I. M., Koseki M., Pirruccello J. P., Ripatti S., Chasman D. I., Willer C. J., Johansen C. T., Fouchier S. W., Isaacs A., Peloso G. M., Barbalic M., Ricketts S. L, Bis J. C., Aulchenko Y. S., Thorleifsson G., Feitosa M. F., Chambers J., Orho-Melander M., Melander O., Johnson T., Li X., Guo X., Li M., Shin Cho Y., Jin Go M., Jin Kim Y., Lee J. Y., Park T., Kim K., Sim X., Twee-Hee Ong R., Croteau-Chonka D. C., Lange L. A., Smith J. D., Song K., Hua Zhao J., Yuan X., Luan J., Lamina C., Ziegler A., Zhang W., Zee R. Y., Wright A. F., Witteman J. C., Wilson J. F., Willemsen G., Wichmann H. E., Whitfield J. B., Waterworth D. M., Wareham N. J., Waeber G., Vollenweider P., Voight B. F., Vitart V., Uitterlinden A. G., Uda M., Tuomilehto J., Thompson J. R., Tanaka T., Surakka I., Stringham H. M., Spector T. D., Soranzo N., Smit J. H., Sinisalo J., Silander K., Sijbrands E. J., Scuteri A., Scott J., Schlessinger D., Sanna S., Salomaa V., Saharinen J., Sabatti C., Ruokonen A., Rudan I., Rose L. M., Roberts R., Rieder M., Psaty B. M., Pramstaller P. P., Pichler I., Perola M., Penninx B. W., Pedersen N. L., Pattaro C., Parker A. N., Pare G., Oostra B. A., O C. J., Nieminen M. S., Nickerson D. A., Montgomery G. W., Meitinger T., McPherson R., McCarthy M. I., McArdle W., Masson D., Martin N. G., Marroni F., Mangino M., Magnusson P. K., Lucas G., Luben R., Loos R. J., Lokki M. L., Lettre G., Langenberg C., Launer L. J., Lakatta E. G., Laaksonen R., Kyvik K. O., Kronenberg F., Konig I. R., Khaw K. T., Kaprio J., Kaplan L. M., Johansson A., Jarvelin M. R., Janssens A. C., Ingelsson E., Igl W., Kees Hovingh G., Hottenga J. J., Hofman A., Hicks A. A., Hengstenberg C., Heid I. M., Hayward C., Havulinna A. S., Hastie N. D., Harris T. B., Haritunians T., Hall A. S., Gyllensten U., Guiducci C., Groop L. C., Gonzalez E., Gieger C., Freimer N. B., Ferrucci L., Erdmann J., Elliott P., Ejebe K. G., Doring A., Dominiczak A. F., Demissie S., Deloukas P., de Geus E. J., de Faire U., Crawford G., Collins F. S., Chen Y. D., Caulfield M. J., Campbell H., Burtt N. P., Bonnycastle L. L., Boomsma D. I., Boekholdt S. M., Bergman R. N., Barroso I., Bandinelli S., Ballantyne C. M., Assimes T. L., Quertermous T., Altshuler D., Seielstad M., Wong T. Y., Tai E. S., Feranil A. B., Kuzawa C. W., Adair L. S., Taylor H. A. Jr, Borecki I. B., Gabriel S. B., Wilson J. G., Holm H., Thorsteinsdottir U., Gudnason V., Krauss R. M., Mohlke K. L., Ordovas J. M., Munroe P. B., Kooner J. S., Tall A. R., Hegele R. A., Kastelein J. J., Schadt E. E., Rotter J. I., Boerwinkle E., Strachan D. P., Mooser V., Stefansson K., Reilly M. P., Samani N. J., Schunkert H., Cupples L. A., Sandhu M. S., Ridker P. M., Rader D. J., van Duijn C. M., Peltonen L., Abecasis G. R., Boehnke M., Kathiresan S. Biological, clinical and population relevance of 95 loci for blood lipids. Nature. (2010);466:707–713. doi: 10.1038/nature09270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hindorff L. A., Sethupathy P., Junkins H. A., Ramos E. M., Mehta J. P., Collins F. S., Manolio T. A. Potential etiologic and functional implications of genomewide association loci for human diseases and traits. Proc. Natl. Acad. Sci. U.S.A. (2009);106:9362–9367. doi: 10.1073/pnas.0903103106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ashley E. A., Butte A. J., Wheeler M. T., Chen R., Klein T. E., Dewey F. E., Dudley J. T., Ormond K. E., Pavlovic A., Morgan A. A., Pushkarev D., Neff N. F., Hudgins L., Gong L., Hodges L. M., Berlin D. S., Thorn C. F., Sangkuhl K., Hebert J. M., Woon M., Sagreiya H., Whaley R., Knowles J. W., Chou M. F., Thakuria J. V., Rosenbaum A. M., Zaranek A. W., Church G. M., Greely H. T., Quake S. R., Altman R. B. Clinical assessment incorporating a personal genome. Lancet. (2010);375:1525–1535. doi: 10.1016/S0140-6736(10)60452-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen R., Mias G. I., Li-Pook-Than J., Jiang L., Lam H. Y., Miriami E., Karczewski K. J., Hariharan M., Dewey F. E., Cheng Y., Clark M. J., Im H., Habegger L., Balasubramanian S., O M., Dudley J. T., Hillenmeyer S., Haraksingh R., Sharon D., Euskirchen G., Lacroute P., Bettinger K., Boyle A. P., Kasowski M., Grubert F., Seki S., Garcia M., Whirl-Carrillo M., Gallardo M., Blasco M. A., Greenberg P. L., Snyder P., Klein T. E., Altman R. B., Butte A. J., Ashley E. A., Gerstein M., Nadeau K. C., Tang H., Snyder M. Personal omics profiling reveals dynamic molecular and medical phenotypes. Cell. (2012);148:1293–1307. doi: 10.1016/j.cell.2012.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Park J. H., Wacholder S., Gail M. H., Peters U., Jacobs K. B., Chanock S. J., Chatterjee N. Estimation of effect size distribution from genome-wide association studies and implications for future discoveries. Nat. Genet. (2010);42:570–575. doi: 10.1038/ng.610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.On beyond GWAS. Nat. Genet. (2010);42:551. doi: 10.1038/ng0710-551. [DOI] [PubMed] [Google Scholar]
- 12.Lettre G., Palmer C. D., Young T., Ejebe K. G., Allayee H., Benjamin E. J., Bennett F., Bowden D. W., Chakravarti A., Dreisbach A., Farlow D. N., Folsom A. R., Fornage M., Forrester T., Fox E., Haiman C. A., Hartiala J., Harris T. B., Hazen S. L., Heckbert S. R., Henderson B. E., Hirschhorn J. N., Keating B. J., Kritchevsky S. B., Larkin E., Li M., Rudock M. E., McKenzie C. A., Meigs J. B., Meng Y. A., Mosley T. H., Newman A. B., Newton-Cheh C. H., Paltoo D. N., Papanicolaou G. J., Patterson N., Post W. S., Psaty B. M., Qasim A. N., Qu L., Rader D. J., Redline S., Reilly M. P., Reiner A. P., Rich S. S., Rotter J. I., Liu Y., Shrader P., Siscovick D. S., Tang W. H., Taylor H. A., Tracy R. P., Vasan R. S., Waters K. M., Wilks R., Wilson J. G., Fabsitz R. R., Gabriel S. B., Kathiresan S., Boerwinkle E. Genome-wide association study of coronary heart disease and its risk factors in 8,090 African Americans: the NHLBI CARe Project. PLoS Genet. (2011);7:e1001300. doi: 10.1371/journal.pgen.1001300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wright S. The Distribution of Gene Frequencies in Populations. Proc. Natl. Acad. Sci. U.S.A. (1937);23:307–320. doi: 10.1073/pnas.23.6.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Teo Y. Y., Fry A. E., Bhattacharya K., Small K. S., Kwiatkowski D. P., Clark T. G. Genome-wide comparisons of variation in linkage disequilibrium. Genome. Res. (2009);19:1849–1860. doi: 10.1101/gr.092189.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cheng F., Chen W., Richards E., Deng L., Zeng C. SNP@Evolution: a hierarchical database of positive selection on the human genome. BMC. Evol. Biol. (2009);9:221. doi: 10.1186/1471-2148-9-221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Voight B. F., Kudaravalli S., Wen X., Pritchard J. K. A map of recent positive selection in the human genome. PLoS. Biol. (2006);4:e72. doi: 10.1371/journal.pbio.0040072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Duan S., Zhang W., Cox N. J., Dolan M. E. FstSNP-HapMap3: a database of SNPs with high population differentiation for HapMap3. Bioinformation. (2008);3:139–141. doi: 10.6026/97320630003139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Teo Y. Y., Sim X., Ong R. T., Tan A. K., Chen J., Tantoso E., Small K. S., Ku C. S., Lee E. J., Seielstad M., Chia K. S. Singapore Genome Variation Project: a haplotype map of three Southeast Asian populations. Genome Res. (2009);19:2154–2162. doi: 10.1101/gr.095000.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.International HapMap Consortium. A haplotype map of the human genome. Nature. (2005);437:1299–1320. doi: 10.1038/nature04226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.International HapMap Consortium. Frazer K. A., Ballinger D. G., Cox D. R., Hinds D. A., Stuve L. L., Gibbs R. A., Belmont J. W., Boudreau A., Hardenbol P., Leal S. M., Pasternak S., Wheeler D. A., Willis T. D., Yu F., Yang H., Zeng C., Gao Y., Hu H., Hu W., Li C., Lin W., Liu S., Pan H., Tang X., Wang J., Wang W., Yu J., Zhang B., Zhang Q., Zhao H., Zhao H., Zhou J., Gabriel S. B., Barry R., Blumenstiel B., Camargo A., Defelice M., Faggart M., Goyette M., Gupta S., Moore J., Nguyen H., Onofrio R. C., Parkin M., Roy J., Stahl E., Winchester E., Ziaugra L., Altshuler D., Shen Y., Yao Z., Huang W., Chu X., He Y., Jin L., Liu Y., Shen Y., Sun W., Wang H., Wang Y., Wang Y., Xiong X., Xu L., Waye M. M., Tsui S. K., Xue H., Wong J. T., Galver L. M., Fan J. B., Gunderson K., Murray S. S., Oliphant A. R., Chee M. S., Montpetit A., Chagnon F., Ferretti V., Leboeuf M., Olivier J. F., Phillips M. S., Roumy S., Sallee C., Verner A., Hudson T. J., Kwok P. Y., Cai D., Koboldt D. C., Miller R. D., Pawlikowska L., Taillon-Miller P., Xiao M., Tsui L. C., Mak W., Song Y. Q., Tam P. K., Nakamura Y., Kawaguchi T., Kitamoto T., Morizono T., Nagashima A., Ohnishi Y., Sekine A., Tanaka T., Tsunoda T., Deloukas P., Bird C. P., Delgado M., Dermitzakis E. T., Gwilliam R., Hunt S., Morrison J., Powell D., Stranger B. E., Whittaker P., Bentley D. R., Daly M. J., de Bakker P. I., Barrett J., Chretien Y. R., Maller J., McCarroll S., Patterson N., Pe I., Price A., Purcell S., Richter D. J., Sabeti P., Saxena R., Schaffner S. F., Sham P. C., Varilly P., Altshuler D., Stein L. D., Krishnan L., Smith A. V., Tello-Ruiz M. K., Thorisson G. A., Chakravarti A., Chen P. E., Cutler D. J., Kashuk C. S., Lin S., Abecasis G. R., Guan W., Li Y., Munro H. M., Qin Z. S., Thomas D. J., McVean G., Auton A., Bottolo L., Cardin N., Eyheramendy S., Freeman C., Marchini J., Myers S., Spencer C., Stephens M., Donnelly P., Cardon L. R., Clarke G., Evans D. M., Morris A. P., Weir B. S., Tsunoda T., Mullikin J. C., Sherry S. T., Feolo M., Skol A., Zhang H., Zeng C., Zhao H., Matsuda I., Fukushima Y., Macer D. R., Suda E., Rotimi C. N., Adebamowo C. A., Ajayi I., Aniagwu T., Marshall P. A., Nkwodimmah C., Royal C. D., Leppert M. F., Dixon M., Peiffer A., Qiu R., Kent A., Kato K., Niikawa N., Adewole I. F., Knoppers B. M., Foster M. W., Clayton E. W., Watkin J., Gibbs R. A., Belmont J. W., Muzny D., Nazareth L., Sodergren E., Weinstock G. M., Wheeler D. A., Yakub I., Gabriel S. B., Onofrio R. C., Richter D. J., Ziaugra L., Birren B. W., Daly M. J., Altshuler D., Wilson R. K., Fulton L. L., Rogers J., Burton J., Carter N. P., Clee C. M., Griffiths M., Jones M. C., McLay K., Plumb R. W., Ross M. T., Sims S. K., Willey D. L., Chen Z., Han H., Kang L., Godbout M., Wallenburg J. C., L P., Bellemare G., Saeki K., Wang H., An D., Fu H., Li Q., Wang Z., Wang R., Holden A. L., Brooks L. D., McEwen J. E., Guyer M. S., Wang V. O., Peterson J. L., Shi M., Spiegel J., Sung L. M., Zacharia L. F., Collins F. S., Kennedy K., Jamieson R., Stewart J. A second generation human haplotype map of over 3.1 million SNPs. Nature. (2007);449:851–861. doi: 10.1038/nature06258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.He M., Gitschier J., Zerjal T., de Knijff P., Tyler-Smith C., Xue Y. Geographical affinities of the Hap Map samples. PLoS. One. (2009);4:e4684. doi: 10.1371/journal.pone.0004684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Amberger J., Bocchini C. A., Scott A. F., Hamosh A. McKusick's Online Mendelian Inheritance in Man (OMIM). Nucleic. Acids. Res. (2009);37:D793–796. doi: 10.1093/nar/gkn665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stein L. D., Mungall C., Shu S., Caudy M., Mangone M., Day A., Nickerson E., Stajich J. E., Harris T. W., Arva A., Lewis S. The generic genome browser: a building block for a model organism system database. Genome. Res. (2002);12:1599–1610. doi: 10.1101/gr.403602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Donlin M. J. Using the generic genome browser (GBrowse). Chapter 9, Unit 9 9. Curr. Protoc. Bioinformatics. (2009) doi: 10.1002/0471250953.bi0909s28. [DOI] [PubMed] [Google Scholar]
- 25.Korn J. M., Kuruvilla F. G., McCarroll S. A., Wysoker A., Nemesh J., Cawley S., Hubbell E., Veitch J., Collins P. J., Darvishi K., Lee C., Nizzari M. M., Gabriel S. B., Purcell S., Daly M. J., Altshuler D. Integrated genotype calling and association analysis of SNPs, common copy number polymorphisms and rare CNVs. Nat. Genet. (2008);40:1253–1260. doi: 10.1038/ng.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Oliphant A., Barker D. L., Stuelpnagel J. R., Chee M. S. BeadArray technology: enabling an accurate, cost-effective approach to high-throughput genotyping. Biotechniques. (2002);Suppl 56-58:60–51. [PubMed] [Google Scholar]
- 27.Hong C. B., Kim Y. J., Moon S., Shin Y. A., Cho Y. S., Lee J. Y. KAREBrowser: SNP database of Korea association resource project. BMB Rep. (2012);45:47–50. doi: 10.5483/BMBRep.2012.45.1.47. [DOI] [PubMed] [Google Scholar]
- 28.Purcell S., Neale B., Todd-Brown K., Thomas L., Ferreira M. A., Bender D., Maller J., Sklar P., de Bakker P. I., Daly M. J., Sham P. C. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. (2007);81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]