

Abstract
Chloroplast sequence contamination in 16S ribosomal RNA gene (16S) analyses can be particularly problematic when sampling microbial communities in plants and folivorous arthropods. We previously encountered high levels of plastid contamination in herbivorous insect samples when we used the predominant 454 pyrosequencing 16S methodologies described in the literature. 799F, a primer previously found to exclude chloroplast sequences, was modified to enhance its efficacy, and we describe, in detail, our methodology throughout amplicon pyrosequencing. Thirteen versions of 799F were assessed for the exclusion of chloroplast sequences from our samples. We found that a shift in the mismatch between 799F and chloroplast 16S resulted in significant reduction of chloroplast reads. Our results also indicate that amplifying sequences from environmental samples in a two-step PCR process, with the addition of the multiplex identifiers and 454 adapters in a second round of PCR, further improved primer specificity. Primers that included 3′ phosphorothioate bonds, which were designed to block primer degradation, did not amplify consistently across samples. The different forward primers do not appear to bias the bacterial communities detected. We provide a methodological framework for reducing chloroplast reads in high-throughput sequencing data sets that can be applied to a number of environmental samples and sequencing techniques.
Keywords: insect, symbiosis, 454 pyrosequencing, 16S rRNA gene, chloroplast
1. Introduction
The advent of next-generation sequencing technologies has revolutionized methods for studying microbial ecology and symbiosis. Barcoded amplicon pyrosequencing has enabled researchers to multiplex samples and generate a tremendous amount of data in a variety of systems (Hamady et al., 2008; Weinstock, 2012). Microbial communities associated with arthropods and plants have received increased attention during this time. Studies on insects such as those describing establishment of a core microbiome (Moran et al., 2012), microbial transmission (Sudakaran et al., 2012), community dynamics through development (Wong et al., 2011), phylogenies (Hulcr et al., 2012), and across populations (Jones et al., 2011) have been made possible from these technological advancements. Additionally, these advances have improved our understanding of the mechanisms and ecological consequences of bacterial community acquisition in plants (Bengtsson et al., 2012; Gottel et al., 2011; Koopman et al., 2010; Redford et al., 2010).
While these studies utilize a diversity of model insects and plants, their investigations lack information on bacterial communities of folivorous insects. There are major methodological obstacles in addressing questions pertaining to foliage-feeding herbivores as several of the published primer pairs used in bacterial 16S ribosomal RNA gene (henceforth abbreviated 16S) pyrosequencing are not feasible for projects studying these systems. The homology between bacterial 16S, chloroplast 16S, plant nuclear and mitochondrial 18S rRNA genes (18S), and arthropod 18S leads to challenges in choosing the appropriate primer pairs. For example, the commonly used universal bacterial primers 926F and 1392R are sufficiently similar to arthropod 18S, resulting in the amplification of these non-bacterial sequences (Dams et al., 1988). The universal 16S primers targeting hypervariable regions V1–V3 (27F, 338R, 519R), V3–V6 (534F, 926F, 1114R), and V6–V8 (926F, 1392R), used in other insect systems (Andreotti et al., 2011; Fagen et al., 2012; Hail et al., 2012, 2011; Hulcr et al., 2012; Jones et al., 2013, 2010; Lalzar et al., 2012; Moran et al., 2012; Osei-Poku et al., 2012; Palavesam et al., 2012; Reid et al., 2011; Wang et al., 2011; Wong et al., 2011) are homologous to chloroplast 16S (Dams et al., 1988; Lane, 1991; Rastogi et al., 2010). Many of the phytophagous insects covered in the pyrosequencing literature consume substrates other than leaves, such as phloem, wood, xylem, and seeds, that contain low abundances of chloroplasts (Fagen et al., 2012; Hail et al., 2012, 2011; Köhler et al., 2012; Reid et al., 2011; Salem et al., 2013; Sudakaran et al., 2012). However, in several pyrosequencing studies focusing on insects, authors have reported unintended chloroplast contamination of varying degrees (Hulcr et al., 2012; Kelley and Dobler, 2011; Moran et al., 2012; Reid et al., 2011; Russell et al., 2013).
For the study of maize roots, Chelius and Triplett (2001) designed 799F, a primer intended to amplify bacterial 16S sequences while avoiding chloroplast 16S sequences, by using a two base pair mismatch on the 3′ end of the primer (Chelius and Triplett, 2001). This primer, and other versions designed from it (799F2, 783R, 783Rabc), have been sufficient for some plant systems (Edwards et al., 2007; Rastogi et al., 2010; Sakai et al., 2004; Sun et al., 2008), but it has not been entirely effective in eliminating amplification of chloroplast 16S sequences in all samples (Bodenhausen et al., 2013; Bulgarelli et al., 2012; Leveau and Tech, 2011; Sagaram et al., 2009; Shade et al., 2013). Others have made no direct comments on the presence or absence of chloroplast sequences in their data sets (Jones et al., 2013; Lundberg et al., 2012; Rastogi et al., 2012; Redford and Fierer, 2009; Redford et al., 2010). To our knowledge, this primer has not been applied in arthropod systems.
Chloroplasts are evolutionarily descended from bacteria, so it is not surprising that the 16S genes are nearly homologous between the two. One of the only regions appropriate for primer design that allows broad bacterial 16S amplification while potentially blocking chloroplast 16S exists between positions 783–799 of the 16S gene (E. coli numbering system). Chloroplast 16S genes have two base pair mismatches at positions 798 and 799 that Chelius and Triplett took advantage of in designing 799F and two additional mismatches at positions 783 and 784 (Chelius and Triplett, 2001). Mismatches between the 3′ end of the primer and the targeted sequence are commonly thought to block amplification (Klindworth et al., 2013; Kumar et al., 2011; Lane, 1991; Nossa et al., 2010; Rastogi et al., 2010; Sipos et al., 2007; Wang and Qian, 2009). As noted by Chelius and Triplett, these minor differences can be exploited in an attempt to avoid chloroplast 16S sequences (Chelius and Triplett, 2001; Sakai et al., 2004). Primer design may also have a major influence on data analysis and interpretation of bacterial community data sets. There is an extensive literature debating which of the various primer combinations are best suited for 16S pyrosequencing projects, but much of this work has been done using marine ecosystems (Klindworth et al., 2013) or the Human Microbiome Project (Kumar et al., 2011; Nossa et al., 2010) as models.
Additional complications in primer design include length of the primer, and potential exonuclease activity from error correcting polymerases. Primers that are too short will anneal to non-targeted sequences while primers that are too long will be overly specific which may incorporate additional biases in pyrosequencing (Sergeant et al., 2012). Moreover, exonuclease activities observed in error correcting polymerases can degrade primers, causing unintended amplification of undesired targets (Ahn et al., 2012). Modifications on the 3′ ends of primers, such as phosphorothioate bonds, create highly stable primers that are not degraded by polymerase exonuclease activities and may be used to circumvent this polymerase issue.
We found that following the current methodology in the published insect bacterial 16S literature was inadequate for our samples, resulting in significant loss of sequencing reads to chloroplast or arthropod sequences, rendering our data useless. Here we test modifications that improve upon the previously published primer, 799F, and show how these modifications perform in 454 pyrosequencing of herbivorous insect samples (Chelius and Triplett, 2001). We tested these primers in two distantly related arthropods that consume plant substrates with high chloroplast abundances, gypsy moth (Lymantria dispar) and the herbivorous ant Azteca constructor, along with a mock community consisting of 50% chloroplast 16S and 50% Streptomyces 16S. We designed 13 modifications to 799F for use in pyrosequencing, with the objective of complete elimination of chloroplast 16S reads in our arthropod systems. Our results indicate that 799F does reduce chloroplast abundance relative to other published primers, but primer length and various 3′ modifications appear to be more effective at further minimizing plastid contamination. These primer modifications are appropriate for use in folivorous arthropod bacterial community analysis and in other samples that may contain chloroplasts, such as plants and algae.
2. Materials & Methods
2.1. Sample collection, preparation, and DNA extraction
Two herbivorous insects and a mock mixture of chloroplast and bacterial 16S were used for the analysis. Five Azteca constructor workers were collected sterilely from a Cecropia insignis tree at Carara Biological Reserve, Costa Rica, May 2011, pooled, stored in 99% ethanol, and transported to the laboratory in Madison, WI, for DNA extraction. Gypsy moth (Lymantria dispar) larvae were reared in petri dishes from egg masses obtained from a wild population in Temperance, MI. Larvae were fed white birch (Betula papyrifera) obtained from WI-DNR (Wilson Nursery, Boscobel, WI), with diet being replaced daily. At fifth instar, ten larvae were starved for 24 hours, and then anesthetized by placing at −20°C for 15 min. Larvae were surface sterilized in 70% ethanol for 30 s, air-dried for 1 min, and midguts were dissected with flame sterilized instruments. Midguts were pooled and stored at −80°C until DNA extraction.
DNA was extracted from both insect tissues with the Epicenter Master Pure Complete DNA and RNA Purification Kit (Illumina, Madison, WI) with modifications. Tissues were homogenized in 2.0 mL screw-cap vials with one 3 mm diameter steel bead in 500 μL T&C buffer, samples were centrifuged at 500×g for 3 min, supernatant was collected, and the remaining manufacturer’s directions were followed. DNA was resuspended in TE and stored at −20°C until use.
2.2. Construction of mock mixture
To test the effectiveness of the newly designed primers, a mock mixture was constructed to contain half chloroplast 16S and half Streptomyces 16S. The chloroplast 16S was cloned from an Azteca alfari larvae sample found to contain 99% chloroplast reads from a previous 454 pyrosequencing run (data not shown). A near-full length sequence of the 16S was PCR amplified in 50 μL total volume, containing 100 ng template DNA, 0.5 μL Herculase II DNA polymerase (Agilent Technologies, Santa Clara, CA), 1.0 nM dNTPs, 1.25 μL DMSO, 10 μL buffer, 250 nM each of the primers 27F (5′AGAGTTTGATCNTGGCTCAG) and 1492R (5′ TACGGYTACCTTGTTACG). Reaction conditions were as follows: 95°C for 2 min, 32 cycles of 95°C for 20 s, 55°C for 20 s, 72°C for 1:30 min, and a final elongation of 72°C for 3 min. PCR products were cleaned using Promega Wizard SV gel and PCR clean-up system as per manufacturer’s directions (Promega, Madison, WI). One μL of cleaned PCR product was used in Novagen Perfectly Blunt Cloning kit with the pT7 Blue Blunt vector, performed as per manufacturer’s directions (EMD Millipore, Billerica, MA). Colonies were selected based on blue/white screening and colony PCRs were performed, with a total volume of 20 μL containing 4 μL of boiled and centrifuged colony supernatant, 0.4 μL Herculase II DNA polymerase, 1.0 nM dNTPs, 0.5 μL DMSO, 4 μL buffer, and 100 nM of each of the primers M13F (5′GTTTTCCCAGTCACGAC) and M13R (5′CAGGAAACAGCTATGAC). Reaction conditions were as follows: 95°C for 2 min, 35 cycles at 95°C for 20 s, 55°C for 20 s, 72°C for 2 min, and a final elongation of 72°C for 3 min. Three colonies were positive and selected for sequencing using Big Dye chemistry. A 10 μL total volume PCR contained 1 μL Big Dye, 1.5 μL Big Dye buffer, 100 nM primer (27F or 1492R), 0.5 μL DMSO, and 0.5 μL PCR template. Reaction conditions were as follows: 96°C for 2 min, 36 cycles of 96°C for 10 s, 52°C for 15 s, 60°C for 3 min, and a final elongation of 72°C for 2 min. Products were cleaned with Agencourt CleanSeq beads (Beckman Coulter, Brea, CA), as per manufacturer’s directions, and sequenced at the University of Wisconsin-Madison Biotechnology Center. Sequences were edited and aligned in Sequencher v4.5 (Gene Codes Corporation, Ann Arbor, MI), and confirmed via NCBI blastN to be chloroplast (Altschul et al., 1990).
A laboratory strain of Streptomyces spp. was used as the bacterial 16S, and amplified as described above using 27F and 1492R. The PCR products from both chloroplast and Streptomyces were quantified by Invitrogen Qubit Fluorometer (Life Sciences, Grand Island, NY) and mixed in equal concentrations to create the 50:50 mock mixture.
2.3. Cross-phyla small subunit ribosomal DNA alignment
A framework by Dams et al (1988) was used for the alignment between bacterial 16S, chloroplast 16S, arthropod 18S, plant nuclear 18S, and plant mitochondrial 18S consensus sequences (Dams et al., 1988). The 16S consensus sequence for bacteria from Baker et al (2003) was used here with no modifications (Baker et al., 2003). A chloroplast 16S consensus sequence was created from the chloroplast sequenced above, the top 15 full length Blast matches to that sequence representing different genera within the rosids clade (DQ226511, JN884817, JQ041763, AP012207, HQ336405, HQ664605, JF317356, HQ244500, HQ664560, HQ664552, FJ895895, HQ664600, EF207453, HQ664619, EU431223), and 5 partial Betula chloroplast 16S sequences (GQ284846-50). Sequences were aligned in MEGA 5.2.1 using default ClustalW parameters, and the chloroplast consensus sequence was manually made (Tamura et al., 2011) (Chloroplast consensus sequence in Supplemental Text 1). Azteca ovaticeps 18S (EF012842) and L. dispar 18S (DQ186972.1) were used as representatives of arthropod 18S. A plant mitochondrial 18S consensus sequence was created, as described for the chloroplast 16S consensus, using two near full length rosid sequences, Oenothera berteriana (X61277) and Arabidopsis thaliana (Y08501). A plant nuclear 18S consensus sequence was created, as described for the chloroplast 16S consensus, using Arabidopsis thaliana (X16077), Betula pendula (GU476453), and three sequences from the same family as Cecropia (Urticaceae), Pilea cadierei (JF317373.1), Debregeasia saeneb (JF317363), and Boehmeria nivea (AF206870). All sequences were assessed for the presence of universal bacterial primers, including the forward primers 27F, 338F, 534F, 799F, 926F, and 1114F, in addition to the reverse primers 338R, 519R, 1114R, 1392R, and 1492R (Table 1).
Table 1.
Cross phyla alignment of the universal bacterial primers with 16S- and 18S-rRNA genes from bacteria and eukaryotes. Primer sequences written 5′ → 3′. Shaded bases differ from the corresponding nucleotides in the primer. Numbers ending in F are forward primers and numbers ending in R are reverse primers.
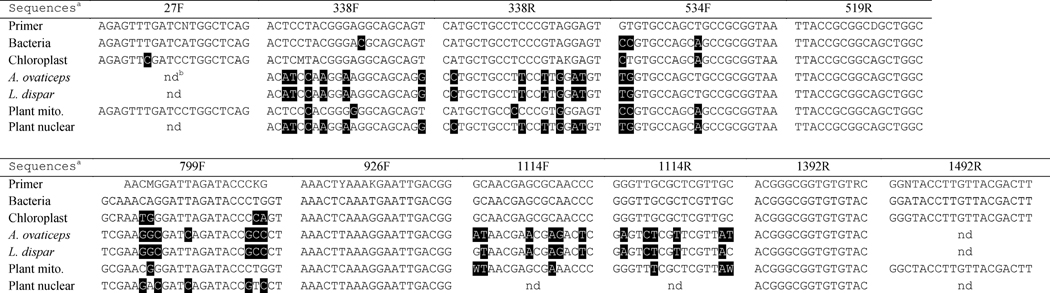
|
Primer: Universal primers, Bactera: Bacterial consensus 16S, Chloroplast: Chloroplast consensus 16S, A ovaticeps: Azteca ovaticeps 18S, L. dispar: Lymantria dispar 18S, Plant mito: Plant mitochondrial 18S, Plant nuclear: Plant nuclear 18S
nd = Sequence not detected
2.4. Primer design and assessment
The primers used in this study were developed to minimize chloroplast contamination and amplify the V5–V8 region of the 16S. Chloroplast and bacterial 16S are, as expected, highly conserved, severely limiting the number of potential primer mismatches (Table 1, Supplemental Table 1). Besides the four mismatches around 799F, no other conserved region of the bacterial 16S appropriate for primer design contains sufficient mismatches to chloroplast 16S. Consensus sequences of chloroplast and bacterial 16S were aligned along with the previously published 799F (Chelius and Triplett, 2001). 799F was modified on both the 5′ and 3′ ends, resulting in 9 different versions of the primer (Table 2). 799F-mod2, -mod4, and -mod7 were also synthesized with phosphorothioate bonds on the 3′ end, resulting in 13 total versions of 799F (Table 2). Primers were synthesized by Integrated DNA Technologies (Coralville, IA).
Table 2.
Sequence and coverage rate of forward primers.
| Primer Name | Primer Sequence (5′ → 3′) | Primer Length (bp) | % RDP predicted coverageb
|
||
|---|---|---|---|---|---|
| M=0 | M=1 | M=2 | |||
| 799F | AACMGGATTAGATACCCKG | 19 | 81.8 | 88.1 | 95.9 |
| 799F-mod1 | AACMGGATTAGATACCCKGG | 20 | 81.6 | 88.1 | 95.8 |
| 799F-mod2 | AACMGGATTAGATACCCKGGT | 21 | 81.4 | 88.0 | 95.7 |
| 799F-mod3 | CMGGATTAGATACCCKGG | 18 | 82.1 | 88.2 | 95.9 |
| 799F-mod4 | GGATTAGATACCCKGG | 16 | 82.4 | 95.5 | 99.3 |
| 799F-mod5 | ATTAGATACCCKGG | 14 | 82.7 | 95.7 | 99.5 |
| 799F-mod6 | CMGGATTAGATACCCKGGT | 19 | 81.9 | 88.1 | 95.9 |
| 799F-mod7 | GGATTAGATACCCKGGT | 17 | 82.2 | 95.4 | 99.3 |
| 799F-mod8 | ATTAGATACCCKGGT | 15 | 82.5 | 95.6 | 99.4 |
| 799F-mod9 | TAGATACCCKGGT | 13 | 85.8 | 95.8 | 99.6 |
| 799F-thio | AACMGGATTAGATACCCKG | 19 | 81.8 | 88.1 | 95.9 |
| 799F-mod2thio | AACMGGATTAGATACCCKGGT | 21 | 81.4 | 88.0 | 95.7 |
| 799F-mod4thio | GGATTAGATACCCKGG | 16 | 82.4 | 95.5 | 99.3 |
| 799F-mod7thio | GGATTAGATACCCKGGT | 17 | 82.2 | 95.4 | 99.3 |
Underlined bases are bonded with phosphorothioate bonds. The number of mismatches (M), zero, one, or two, allowed between the primer and target region in a RDP search.
All versions of 799F, including 799F-tags which includes the 454 B adapter, were assessed for their ability to amplify chloroplast 16S, bacterial 16S, the 50:50 mock mixture, and environmental samples, in addition to their predicted coverage rate using probe match in the Ribosomal Database Project (RDP) (Cole et al., 2009). 19.2 ng of chloroplast 16S, bacterial 16S and 50:50 mock mixture was used as template, while 50 ng of environmental DNA was used. A 50 μL total volume PCR contained 0.5 μL Herculase II DNA polymerase, 1.0 nM dNTPs, 1 μL DMSO, 10 μL buffer, 300 nM 1392R (5′ ACGGGCGGTGTGTRC) and 300 nM forward primer. Reaction conditions were as follows: 95°C for 3 min, 30 cycles of 95°C for 20 s, 45°C for 30 s, 72°C for 30 s, and a final elongation of 72°C for 3 min. PCR products were visualized and assessed on a 2% agarose gel. Primers not capable of amplifying the Streptomyces 16S (799F-mod5, -mod9, and -mod4thio) were excluded from further testing. Likewise, primers unable to amplify either environmental sample (799F-mod4, -mod8 and -mod2thio) were removed. In addition to 799F and 799F-tags, the modified versions 799F-mod2, -mod3, -mod6, -mod7, -thio and -mod7thio were included in further assessment.
2.5. PCR for 454 Pyrosequencing
With two exceptions, PCRs were carried out using a two-step PCR procedure, utilizing a gel extraction of the 16S band between the first and second steps (Berry et al., 2011). The first PCR was done with primers that lacked the 454 adapters or multiplex identifiers (MIDs), while the second added the required 454 A- and B-adapters along with a 5 bp MID for each respective sample. PCRs with the primer pairs 799F-tags/1392R and 27F/519R were tagged in the first round of PCR, and were not done in a two-step process. All tested versions of the forward primer 799F (799F, 799F-tags, 799F-mod2, 799F-mod3, 799F-mod6, 799F-mod7, 799F-thio, and 799F-mod7thio) were paired with the reverse primer 1392R.
PCRs were done in triplicates containing 30–50 ng template DNA, 0.5 μL Herculase II DNA polymerase, 1.0 nM dNTPs, 1.0 μL DMSO, 10 μL buffer, 300 nM of forward and reverse primers, and water amounting to a final volume of 50 μL. Reaction conditions were as follows: 95°C for 2 min, 30 cycles of 95°C for 20 s, 48–50°C for 30 s, 72°C for 30 s, and a final elongation of 72°C for 3 min. The optimal annealing temperature differed between the two environmental samples, 50°C for A. constructor, and 48°C for L. dispar. Triplicate reactions were pooled and an aliquot was used for gel extraction. The ~600 bp band expected from the 16S amplicon was extracted from a low-melt agarose gel using a Zymoclean Gel DNA Recovery Kit (Zymo Research, Irvine, CA) by visualizing on a blue light transilluminator (Clare Chemical Research, Dolores, CO). A second PCR was done on all primers pairs, except 799F-tags/1392R and 27F/519R, using 2 μL of the extraction product using primers that included the A- and B- adaptors along with the MIDs. All conditions for the second PCR step were identical except that thermocycling was done for 10 cycles instead of 30. Five μL of the second PCR product was loaded on a 2.0% agarose gel to verify the presence of a ~700 bp fragment while lacking one at ~600 bp. PCR products were cleaned with three rounds of cleanup using Agencourt AMPure XP beads (Beckman Coulter, Brea, CA) as per manufacturer’s directions. PCR products were quantified with a Qubit, and, according to Roche 454 pyrosequencing protocols, diluted and pooled at 10−6 DNA molecules/μl.
2.6. 454 pyrosequencing and long read modifications
The expected fragment size from 799F and 1392R, including 454 adapters and MIDs (~700 bp), is longer than recommended for 454 pyrosequencing (Roche, Indianapolis, IN) on a GS Junior with FLX Titanium chemistry. Therefore, emPCR was modified to accommodate the longer fragment. The modifications were based on suggestions in technical bulletin TCB-11001 “Amplicon Sequencing on GS FLX System with Various emPCR Conditions” and from Roche technical support (personal communication). Modifications included increased Amp mix to 297 μL, Amp Primer to 104 μL, and decreased water to 359 μL. Thermocycler conditions were changed to: 94°C for 4 min, 50 cycles of 94°C for 30 sec, 60°C for 10 min, and storage at 10°C. No other modifications were made to the manufacturer’s protocols. The 50:50 mock mixture and environmental sample amplified with 27F/519R were run separately, but with the identical protocols.
2.7. Data Analysis
Raw data were processed using mothur (v. 1.23.1) (Schloss et al., 2009). Sequences were analyzed, allowing for no differences in the primers or MIDs, and with a minimum length of 200 bp. The data set was simplified using unique.seqs and aligned to the Silva-derived reference database (v. 102 as implemented for mothur) (Pruesse et al., 2007). Chimeras were removed using UCHIME (Edgar et al., 2011). For the first data analysis to evaluate how many chloroplast reads were present, all processed reads were kept. After the command classify.seqs to a nogap version of Silva containing bacterial, archaea, and eukaryotic sequences, each sample was assessed for the percent of chloroplast. For the second data analysis, to compare what bacteria were detected for each primer pair for the two arthropod samples, all eukaryotic reads were removed, as were rare OTUs with less than 2 reads. Weighted and unweighted Unifrac distance matrices were constructed in mothur and analyzed with non-metric multidimensional scaling (MDS) plots in PRIMER (v6) (Clarke and Gorley, 2006; Lozupone et al., 2011).
3. Results
All but three primers readily amplified bacterial 16S and produced a low-intensity band after PCR with pure chloroplast 16S. The additional modifications to 799F not used in the pyrosequencing analysis failed these initial quality controls. All primers successfully amplified the L. dispar sample, but the 3′ phosphorothioate modifications, 799F-thio and 799F-mod7thio, failed to amplify the A. constructor sample. The 50:50 mock mixture yielded a clean, single band with all primers, but the environmental samples had variable numbers of bands present, including the ~900 bp fragment from amplification of plant mitochondria 18S (Chelius and Triplett, 2001; Dams et al., 1988). Within a sample the multiple bands detected did not differ between the various forward primers.
The 454 GS Junior runs resulted in 143,260 raw reads. After removal of short reads and chimeras, 136,270 reads remained. 1364 non-chloroplast Eukaryote reads were removed from one sample, resulting in 134,906 high quality reads (Table 3). Across all primer combinations, both environmental samples had on average less than 1.0% chloroplast reads with the 50:50 mock mixture having 2.5% chloroplasts or less with any of the various versions of 799F. The primer combination 799F-tags, including the 454 B adapter, along with 1392R with the 454 A adapter and 5 bp MIDs, had the highest percentage of chloroplasts in all three cases with 2.47% for the mock mixture, 1.95% for the A. constructor sample, and 6.87% for the L. dispar sample. 799F-mod3, -mod6, and -mod7 resulted in no chloroplast reads in both environmental samples. In the L. dispar sample, 799F-thio also had no chloroplast reads. A previous 454 run with 27F/519R using the A. constructor and L. dispar environmental samples yielded 67% and 39.4% chloroplasts, respectively, while the 50:50 mock mixture resulted in 55% chloroplast reads.
Table 3.
Total number of sequence reads and percent aligning to chloroplasts with the various forward primers.
| 50:50 Mock mixture | A. constructor | L. dispar | ||||
|---|---|---|---|---|---|---|
|
| ||||||
| Primer combination | # reads | %chloroplast | # reads | %chloroplast | # reads | %chloroplast |
| 27F/519R | 661 | 55.4 | 1839 | 67 | 374 | 39.4 |
| 799F/1392R | 7966 | 1.66 | 10759 | 0.37 | 2208 | 0.36 |
| 799F-tags/1392R | 9764 | 2.47 | 3176 | 1.95 | 1675 | 6.87 |
| 799F-mod2/1392R | 4390 | 1.09 | 8586 | 0.02 | 2798 | 0.11 |
| 799F-mod3/1392R | 8657 | 0.88 | 8772 | 0 | 2307 | 0 |
| 799F-mod6/1392R | 6717 | 0.76 | 8548 | 0 | 2115 | 0 |
| 799F-mod7/1392R | 6370 | 0.63 | 6638 | 0 | 1795 | 0 |
| 799F-thio/1392R | 10133 | 2.34 | N/A | N/A | 254 | 0 |
| 799F-mod7thio/1392R | 5196 | 2.1 | N/A | N/A | 865 | 0 |
Relative abundances of taxa at the order taxonomic level were minimally impacted by the different forward primers (Figure 1, Supplemental Table 2). A MDS of weighted Unifrac also showed minimal differences in the various forward primers (Figure 2). The unweighted Unifrac MDS was most affected by the various forward primers, which was most likely due to rare reads present in the analysis (Figure 2).
Fig 1.

Relative abundances of sequence reads at the bacterial order taxonomic classification sequenced with the tested forward primers.
Fig 2.

MDS plot illustrating the differences between the bacterial communities in two arthropod samples sequenced with 8 primer pairs. Pairwise community distances determined using the unweighted Unifrac algorithm (A) and the weighted Unifrac algorithm (B). These two panels show that while there may be more bacterial taxa detected with different primers (A), when abundance is taken into account, the communities in each respective insect are nearly indistinguishable (B).
4. Discussion
Chloroplast and other plastids of bacterial origin present problems unique to certain environments in 16S pyrosequencing (Bulgarelli et al., 2012; Chelius and Triplett, 2001; Lundberg et al., 2012; Rastogi et al., 2010; Sakai et al., 2004). Although this issue is potentially widespread in insects and plant samples, it has been understudied in next-generation sequencing. We tested a series of primers to eliminate plastid contamination in herbivorous insects, and found that our primers, specifically 799F–mod 3, –mod6, and –mod7, paired with the universal primer 1392R, drastically reduced these interfering sequences. Subsequent independent experiments using these modifications reduced chloroplast contamination from as much as 99.9% in insect and plant samples to between 0–10% (CJ Mason, unpublished data; AS Hanshew, unpublished data).
799F has been used extensively in the literature to minimize chloroplast contamination in plant samples (Bulgarelli et al., 2012; Chelius and Triplett, 2001; Redford and Fierer, 2009; Redford et al., 2010). While this primer has been successful in some cases (Sun et al., 2008), it has performed poorly in others (Sagaram et al., 2009). Our results show that 799F is capable of reducing chloroplast sequence contamination in our insect samples, but three of our modified versions of 799F appear to be more effective. The differences between 799F and 799F-mod3, -mod6, and -mod7 are the positions of the 3′ mismatch between bacterial and chloroplast 16S, in addition to mismatches at the 5′ end. Shifting the position of these mismatches 1 or 2 base pairs in the primer may reduce the likelihood of chloroplast sequence amplification greater than in 799F.
Bias is a well-documented problem with all primers used in 16S PCR (Wang and Qian, 2009). 799F-mod3, -mod6, and –mod7 all maintain the ability to detect many of the bacterial phyla, but potentially bias against Chloroflexi and Verrucomicrobia (Supplemental Table 1). These phyla tend to be in minimal abundances in many animal microbial communities (Colman et al., 2012; Jones et al., 2013; Lozupone et al., 2012). In our study, we were able to detect these phyla, but at very low abundance, and they have been detected in subsequent analyses using these primers (N Davis, E Houck, K Dill-McFarland, personal communications). Cyanobacteria have a shared evolutionary history with Chloroplast that makes it challenging to have confidence in separating these two DNA sources in culture independent analyses in known plastid-abundant samples. While a biasing of community membership may occur with our primers, this is a longstanding issue that exists for all 16S primers and the reduction of chloroplast sequences in samples similar to ours greatly outweighs this shortcoming.
To our knowledge, this is the first study using these primers in insect samples. Similar to 799F use with plant samples, we found our samples produced multiple banding patterns (Bulgarelli et al., 2012; Chelius and Triplett, 2001). Unlike previous studies, banding patterns differed from plant samples in that A. constructor had 3–4 bands while L. dispar had 7–9 bands. Changes in annealing temperature and other aspects of the PCR protocol did not reduce these patterns. Thus, utilizing gel extraction between the two-step PCR was necessary and effective in targeting our band of interest. As universal bacterial primers are capable of amplifying non-bacterial sequences, it is not surprising that these complex samples resulted in multiple bands from PCR. These other bands may represent plant mitochondrial 18S, which also has homology to 799F and 1392R, but produces a band roughly 1.5 times as long as that for 16S. It is unlikely this primer amplifies arthropod 18S or plant nuclear 18S, but there may be other non-target sequences amplified by these primers. Indeed, the 1364 eukaryotic reads present in one sample were likely a result from including an additional band during gel extraction by mistake. The two-step PCR separated by a gel extraction also circumvents the concern of primer length causing non-specific amplification and biases (Berry et al., 2011). In our results, the primer pairs including the 454 adapters and MIDs were less efficacious as opposed to a two-step PCR method. The two-step PCR method produced results with fewer unwanted sequences, as well as increased consistency.
Throughout our experiments, we tested a number of polymerases with different exonuclease activities, many of which are used extensively in the literature (data not shown). With few exceptions, many were unable to produce amplicons from our samples. Our samples contain a myriad of sources of DNA, including at a minimum bacterial, insect, and plant DNA. Therefore, we think that the complexity of our samples may have reduced the effectiveness of amplification. In order to minimize primer degradation by polymerase exonuclease activities, we incorporated 3′ phosphorothioate modifications into four versions of our primers to attempt to preserve the mismatches to chloroplast 16S on the 3′ end of the primers. We found that the phosphorothioate primers performed inconsistently between samples. These modifications worked as well as the unmodified versions in the L. dispar sample, but failed to amplify in the A. constructor sample. Therefore, this primer modification should be used on a case-by-case basis.
Recently, 16S amplicon sequencing on high-throughput platforms has had increased use across a number of environmental samples, including insects and plants. Investigating bacterial communities in these systems with standard protocols can present challenges that previous studies have not yet encountered. The methods outlined in this study provide a much-needed methodological framework for addressing issues pertaining to unwanted sequence contamination. Our methods enable comparison of microbial communities in systems that were previously intractable to bacterial community analyses that can be modified to other PCR-based platforms. Many systems, including herbivorous hosts, can be chloroplast-laden and our methods establish a way of contending with this issue.
Supplementary Material
Highlights.
Homology between chloroplast 16S rRNA and bacterial primers is problematic.
Many primers used in 454 bacterial profiling perform poorly in folivorous insects.
We show 799F effectively reduces chloroplast reads in insect samples.
This contamination is eliminated by shifting 799F one or two basepairs.
Acknowledgments
We thank Rembrant Haft, Kelsea Jewell, Frank Aylward, and Adam Book for technical suggestions and Kirk Grubbs for helpful comments on the manuscript. ASH was partially funded under National Institutes of Health T32 GM07215-37. Other funding sources included Hatch WIS01598 awarded to KFR and CRC and National Science Foundation MCB-0702025 awarded to CRC. Funding sources had no involvement in conducting this research or preparing this publication.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ahn JH, Kim BY, Song J, Weon HY. Effects of PCR cycle number and DNA polymerase type on the 16S rRNA gene pyrosequencing analysis of bacterial communities. Journal of Microbiology. 2012;50:1071–1074. doi: 10.1007/s12275-012-2642-z. [DOI] [PubMed] [Google Scholar]
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic Local Alignment Search Tool. Journal of Molecular Biology. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Andreotti R, Perez de Leon AA, Dowd SE, Guerrero FD, Bendele KG, Scoles GA. Assessment of bacterial diversity in the cattle tick Rhipicephalus (Boophilus) microplus through tag-encoded pyrosequencing. BMC Microbiology. 2011;11:6. doi: 10.1186/1471-2180-11-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker GC, Smith JJ, Cowan DA. Review and re-analysis of domain-specific 16S primers. Journal of Microbiological Methods. 2003;55:541–555. doi: 10.1016/j.mimet.2003.08.009. [DOI] [PubMed] [Google Scholar]
- Bengtsson MM, Sjøtun K, Lanzén A, Ovreås L. Bacterial diversity in relation to secondary production and succession on surfaces of the kelp Laminaria hyperborea. The ISME Journal. 2012;6:2188–2198. doi: 10.1038/ismej.2012.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry D, Ben Mahfoudh K, Wagner M, Loy A. Barcoded primers used in multiplex amplicon pyrosequencing bias amplification. Applied and Environmental Microbiology. 2011;77:7846–7849. doi: 10.1128/AEM.05220-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodenhausen N, Horton MW, Bergelson J. Bacterial communities associated with the leaves and the roots of Arabidopsis thaliana. PLoS ONE. 2013;8:e56329. doi: 10.1371/journal.pone.0056329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulgarelli D, Rott M, Schlaeppi K, van Themaat EVL, Ahmadinejad N, Assenza F, Rauf P, Huettel B, Reinhardt R, Schmelzer E, Peplies J, Gloeckner FO, Amann R, Eickhorst T, Schulze-Lefert P. Revealing structure and assembly cues for Arabidopsis root-inhabiting bacterial microbiota. Nature. 2012;488:91–95. doi: 10.1038/nature11336. [DOI] [PubMed] [Google Scholar]
- Chelius MK, Triplett EW. The diversity of archaea and bacteria in association with the roots of Zea mays L. Microbial Ecology. 2001;41:252–263. doi: 10.1007/s002480000087. [DOI] [PubMed] [Google Scholar]
- Clarke K, Gorley R. PRIMER v6: User Manual/Tutorial. PRIMER-E; Plymouth, UK: 2006. [Google Scholar]
- Cole JR, Wang Q, Cardenas E, Fish J, Chai B, Farris RJ, Kulam-Syed-Mohideen AS, McGarrell DM, Marsh T, Garrity GM, Tiedje JM. The Ribosomal Database Project: improved alignments and new tools for rRNA analysis. Nucleic Acids Research. 2009;37:D141–145. doi: 10.1093/nar/gkn879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colman DR, Toolson EC, Takacs-Vesbach CD. Do diet and taxonomy influence insect gut bacterial communities? Molecular ecology. 2012:5124–5137. doi: 10.1111/j.1365-294X.2012.05752.x. [DOI] [PubMed] [Google Scholar]
- Dams E, Hendriks L, Van De Peer Y, Neefs JM, Smits G, Vandenbempt I, De Wachter R. Compilation of small ribosomal subunit RNA sequences. Nucleic Acids Research. 1988;16:r87–r173. doi: 10.1093/nar/16.suppl.r87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics. 2011;27:2194–2200. doi: 10.1093/bioinformatics/btr381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards JE, Huws SA, Kim EJ, Kingston-Smith AH. Characterization of the dynamics of initial bacterial colonization of nonconserved forage in the bovine rumen. FEMS Microbiology Ecology. 2007;62:323–335. doi: 10.1111/j.1574-6941.2007.00392.x. [DOI] [PubMed] [Google Scholar]
- Fagen JR, Giongo A, Brown CT, Davis-Richardson AG, Gano KA, Triplett EW. Characterization of the relative abundance of the citrus pathogen Ca. Liberibacter asiaticus in the microbiome of its insect vector, Diaphorina citri, using high throughput 16S rRNA sequencing. Open Microbiology Journal. 2012;6:29–33. doi: 10.2174/1874285801206010029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottel NR, Castro HF, Kerley M, Yang Z, Pelletier DA, Podar M, Karpinets T, Uberbacher E, Tuskan GA, Vilgalys R, Doktycz MJ, Schadt CW. Distinct microbial communities within the endosphere and rhizosphere of Populus deltoides roots across contrasting soil types. Applied and Environmental Microbiology. 2011;77:5934–5944. doi: 10.1128/AEM.05255-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hail D, Dowd SE, Bextine B. Identification and location of symbionts associated with potato psyllid (Bactericera cockerelli) lifestages. Environmental Entomology. 2012;41:98–107. doi: 10.1603/EN11198. [DOI] [PubMed] [Google Scholar]
- Hail D, Lauzi I, Dowd SE, Bextine B, Lauzìere I. Culture independent survey of the microbiota of the glassy-winged sharpshooter (Homalodisca vitripennis) using 454 pyrosequencing. Environmental Entomology. 2011;40:23–29. doi: 10.1603/EN10115. [DOI] [PubMed] [Google Scholar]
- Hamady M, Walker JJ, Harris JK, Gold NJ, Knight R. Error-correcting barcoded primers for pyrosequencing hundreds of samples in multiplex. Nature Methods. 2008;5:235–237. doi: 10.1038/nmeth.1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulcr J, Rountree NR, Diamond SE, Stelinski LL, Fierer N, Dunn RR. Mycangia of ambrosia beetles host communities of bacteria. Microbial Ecology. 2012;64:784–793. doi: 10.1007/s00248-012-0055-5. [DOI] [PubMed] [Google Scholar]
- Jones RT, Bressan A, Greenwell AM, Fierer N. Bacterial communities of two parthenogenetic aphid species cocolonizing two host plants across the Hawaiian Islands. Applied and Environmental Microbiology. 2011;77:8345–8349. doi: 10.1128/AEM.05974-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones RT, Knight R, Martin AP. Bacterial communities of disease vectors sampled across time, space, and species. The ISME Journal. 2010;4:223–231. doi: 10.1038/ismej.2009.111. [DOI] [PubMed] [Google Scholar]
- Jones RT, Sanchez LG, Fierer N. A cross-taxon analysis of insect-associated bacterial diversity. PLoS ONE. 2013;8:e61218. doi: 10.1371/journal.pone.0061218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley ST, Dobler S. Comparative analysis of microbial diversity in Longitarsus flea beetles (Coleoptera: Chrysomelidae) Genetica. 2011;139:541–50. doi: 10.1007/s10709-010-9498-0. [DOI] [PubMed] [Google Scholar]
- Klindworth A, Pruesse E, Schweer T, Peplies J, Quast C, Horn M, Glöckner FO. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Research. 2013;41:e1. doi: 10.1093/nar/gks808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köhler T, Dietrich C, Scheffrahn RH, Brune A. High-resolution analysis of gut environment and bacterial microbiota reveals functional compartmentation in the gut of wood-feeding higher termites (Nasutitermes spp.) Applied and Environmental Microbiology. 2012;78:4691–4701. doi: 10.1128/AEM.00683-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koopman MM, Fuselier DM, Hird S, Carstens BC. The carnivorous pale pitcher plant harbors diverse, distinct, and time-dependent bacterial communities. Applied and Environmental Microbiology. 2010;76:1851–1860. doi: 10.1128/AEM.02440-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar PS, Brooker MR, Dowd SE, Camerlengo T. Target region selection is a critical determinant of community fingerprints generated by 16S pyrosequencing. PLoS ONE. 2011;6:e20956. doi: 10.1371/journal.pone.0020956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalzar I, Harrus S, Mumcuoglu KY, Gottlieb Y. Composition and seasonal variation of Rhipicephalus turanicus and Rhipicephalus sanguineus bacterial communities. Applied and Environmental Microbiology. 2012;78:4110–4116. doi: 10.1128/AEM.00323-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane DJ. 16S/23S rRNA Sequencing. In: Stackebrandt E, Goodfellow M, editors. Nucleic Acid Techniques in Bacterial Systematics. John Wiley & Sons, Ltd; New York: 1991. pp. 115–175. [Google Scholar]
- Leveau JHJ, Tech JJ. Grapevine microbiomics: bacterial diversity on grape leaves and berries revealed by high-throughput sequence analysis of 16S rRNA amplicons. Acta Horticulturae. 2011;905:31–42. [Google Scholar]
- Lozupone C, Lladser ME, Knights D, Stombaugh J, Knight R. UniFrac: an effective distance metric for microbial community comparison. The ISME Journal. 2011;5:169–172. doi: 10.1038/ismej.2010.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozupone CA, Stombaugh JI, Gordon JI, Jansson JK, Knight R. Diversity, stability and resilience of the human gut microbiota. Nature. 2012;489:220–230. doi: 10.1038/nature11550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundberg DS, Lebeis SL, Paredes SH, Yourstone S, Gehring J, Malfatti S, Tremblay J, Engelbrektson A, Kunin V, Del Rio TG, Edgar RC, Eickhorst T, Ley RE, Hugenholtz P, Tringe SG, Dangl JL. Defining the core Arabidopsis thaliana root microbiome. Nature. 2012;488:86–90. doi: 10.1038/nature11237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran NA, Hansen AK, Powell JE, Sabree ZL. Distinctive gut microbiota of honey bees assessed using deep sampling from individual worker bees. PLoS ONE. 2012;7:e36393. doi: 10.1371/journal.pone.0036393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nossa CW, Oberdorf WE, Yang L, Aas JA, Paster BJ, DeSantis TZ, Brodie EL, Malamud D, Poles MA, Pei Z. Design of 16S rRNA gene primers for 454 pyrosequencing of the human foregut microbiome. World Journal of Gastroenterology. 2010;16:4135–4144. doi: 10.3748/wjg.v16.i33.4135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osei-Poku J, Mbogo CM, Palmer WJ, Jiggins FM. Deep sequencing reveals extensive variation in the gut microbiota of wild mosquitoes from Kenya. Molecular Ecology. 2012;21:5138–5150. doi: 10.1111/j.1365-294X.2012.05759.x. [DOI] [PubMed] [Google Scholar]
- Palavesam A, Guerrero FD, Heekin AM, Wang J, Dowd SE, Sun Y, Foil LD, Pérez de León AA. Pyrosequencing-based analysis of the microbiome associated with the horn fly, Haematobia irritans. PLoS ONE. 2012;7:e44390. doi: 10.1371/journal.pone.0044390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruesse E, Quast C, Knittel K, Fuchs BM, Ludwig W, Peplies J, Glöckner FO. SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Research. 2007;35:7188–7196. doi: 10.1093/nar/gkm864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rastogi G, Sbodio A, Tech JJ, Suslow TV, Coaker GL, Leveau JHJ. Leaf microbiota in an agroecosystem: spatiotemporal variation in bacterial community composition on field-grown lettuce. The ISME Journal. 2012;6:1812–1822. doi: 10.1038/ismej.2012.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rastogi G, Tech JJ, Coaker GL, Leveau JHJ. A PCR-based toolbox for the culture-independent quantification of total bacterial abundances in plant environments. Journal of Microbiological Methods. 2010;83:127–132. doi: 10.1016/j.mimet.2010.08.006. [DOI] [PubMed] [Google Scholar]
- Redford AJ, Bowers RM, Knight R, Linhart Y, Fierer N. The ecology of the phyllosphere: geographic and phylogenetic variability in the distribution of bacteria on tree leaves. Environmental Microbiology. 2010;12:2885–2893. doi: 10.1111/j.1462-2920.2010.02258.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redford AJ, Fierer N. Bacterial succession on the leaf surface: a novel system for studying successional dynamics. Microbial Ecology. 2009;58:189–198. doi: 10.1007/s00248-009-9495-y. [DOI] [PubMed] [Google Scholar]
- Reid NM, Addison SL, Macdonald LJ, Lloyd-Jones G. Biodiversity of active and inactive bacteria in the gut flora of wood-feeding huhu beetle larvae (Prionoplus reticularis) Applied and Environmental Microbiology. 2011;77:7000–7006. doi: 10.1128/AEM.05609-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell JA, Weldon S, Smith AH, Kim KL, Hu Y, Lukasik P, Doll S, Anastopoulos I, Novin M, Oliver KM. Uncovering symbiont-driven genetic diversity across North American pea aphids. Molecular Ecology. 2013;22:2045–2059. doi: 10.1111/mec.12211. [DOI] [PubMed] [Google Scholar]
- Sagaram US, DeAngelis KM, Trivedi P, Andersen GL, Lu SE, Wang N. Bacterial diversity analysis of Huanglongbing pathogen-infected citrus, using PhyloChip arrays and 16S rRNA gene clone library sequencing. Applied and Environmental Microbiology. 2009;75:1566–1574. doi: 10.1128/AEM.02404-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai M, Matsuka A, Komura T, Kanazawa S. Application of a new PCR primer for terminal restriction fragment length polymorphism analysis of the bacterial communities in plant roots. Journal of Microbiological Methods. 2004;59:81–89. doi: 10.1016/j.mimet.2004.06.005. [DOI] [PubMed] [Google Scholar]
- Salem H, Kreutzer E, Sudakaran S, Kaltenpoth M. Actinobacteria as essential symbionts in firebugs and cotton stainers (Hemiptera, Pyrrhocoridae) Environmental Microbiology. 2013;15:1956–1968. doi: 10.1111/1462-2920.12001. [DOI] [PubMed] [Google Scholar]
- Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, Sahl JW, Stres B, Thallinger GG, Van Horn DJ, Weber CF. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Applied and Environmental Microbiology. 2009;75:7537–7541. doi: 10.1128/AEM.01541-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sergeant MJ, Constantinidou C, Cogan T, Penn CW, Pallen MJ. High-throughput sequencing of 16S rRNA gene amplicons: effects of extraction procedure, primer length and annealing temperature. PLoS ONE. 2012;7:e38094. doi: 10.1371/journal.pone.0038094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shade A, Mcmanus PS, Handelsman J. Unexpected diversity during community succession in the apple flower microbiome. mBio. 2013;4:e00602–12. doi: 10.1128/mBio.00602-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sipos R, Székely AJ, Palatinszky M, Révész S, Márialigeti K, Nikolausz M. Effect of primer mismatch, annealing temperature and PCR cycle number on 16S rRNA gene-targetting bacterial community analysis. FEMS Microbiology Ecology. 2007;60:341–350. doi: 10.1111/j.1574-6941.2007.00283.x. [DOI] [PubMed] [Google Scholar]
- Sudakaran S, Salem H, Kost C, Kaltenpoth M. Geographical and ecological stability of the symbiotic mid-gut microbiota in European firebugs, Pyrrhocoris apterus (Hemiptera, Pyrrhocoridae) Molecular Ecology. 2012;21:6134–6151. doi: 10.1111/mec.12027. [DOI] [PubMed] [Google Scholar]
- Sun L, Qiu F, Zhang X, Dai X, Dong X, Song W. Endophytic bacterial diversity in rice (Oryza sativa L.) roots estimated by 16S rDNA sequence analysis. Microbial Ecology. 2008;55:415–424. doi: 10.1007/s00248-007-9287-1. [DOI] [PubMed] [Google Scholar]
- Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Molecular Biology and Evolution. 2011;28:2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Gilbreath TM, Kukutla P, Yan G, Xu J. Dynamic gut microbiome across life history of the malaria mosquito Anopheles gambiae in Kenya. PLoS ONE. 2011;6:e24767. doi: 10.1371/journal.pone.0024767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Qian PY. Conservative fragments in bacterial 16S rRNA genes and primer design for 16S ribosomal DNA amplicons in metagenomic studies. PLoS ONE. 2009;4:e7401. doi: 10.1371/journal.pone.0007401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinstock GM. Genomic approaches to studying the human microbiota. Nature. 2012;489:250–256. doi: 10.1038/nature11553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong CNA, Ng P, Douglas AE. Low-diversity bacterial community in the gut of the fruitfly Drosophila melanogaster. Environmental Microbiology. 2011;13:1889–1900. doi: 10.1111/j.1462-2920.2011.02511.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.