

Summary
Background
Bevacizumab and erlotinib target different tumour growth pathways with little overlap in their toxic-effect profiles. On the basis of promising results from a phase 1/2 trial assessing safety and activity of erlotinib plus bevacizumab for recurrent or refractory non-small-cell lung cancer (NSCLC), we aimed to assess efficacy and safety of this combination in a phase 3 trial.
Methods
In our double-blind, placebo-controlled, randomised phase 3 trial (BeTa), we enrolled patients with recurrent or refractory NSCLC who presented to 177 study sites in 12 countries after failure of first-line treatment. Patients were randomly allocated in a one-to-one ratio to receive erlotinib plus bevacizumab (bevacizumab group) or erlotinib plus placebo (control group) according to a computer-generated randomisation sequence by use of an interactive voice response system. The primary endpoint was overall survival in all enrolled patients. Patients, study staff, and investigators were masked to treatment assignment. We assessed safety by calculation of incidence of adverse events and tissue was collected for biomarker analyses. This trial is registered with ClinicalTrials.gov, number NCT00130728.
Findings
Overall survival did not differ between 317 controls and 319 patients in the bevacizumab group (hazard ratio [HR] 0·97, 95% CI 0·80–1·18, p=0·7583). Median overall survival was 9·3 months (IQR 4·1–21·6) for patients in the bevacizumab group compared with 9·2 months (3·8–20·2) for controls. Progression-free survival seemed to be longer in the bevacizumab group (3·4 months [1·4–8·4]) than in the control group (1·7 months [1·3–4·1]; HR 0·62, 95% CI 0·52–0·75) and objective response rate suggested some clinical activity of bevacizumab and erlotinib. However, these secondary endpoint differences could not be defined as significant because the study prespecified that the primary endpoint had to be significant before testing of secondary endpoints could be done, to control type I error rate. In the bevacizumab group, 130 (42%) of 313 patients with safety data had a serious adverse event, compared with 114 (36%) controls. There were 20 (6%) grade 5 adverse events, including two arterial thromboembolic events, in the bevacizumab group, and 14 (4%) in the control group.
Interpretation
Addition of bevacizumab to erlotinib does not improve survival in patients with recurrent or refractory NSCLC.
Funding
Genentech.
Introduction
Lung cancer is the leading cause of cancer-related deaths worldwide.1–3 1·5 million people were diagnosed with the disease in 2008 and more than 1·3 million died.1 Non-small-cell lung cancers (NSCLCs) account for more than 85% of all lung cancers;1 about 75% of patients with NSCLC present with advanced-stage (unresectable or metastatic) disease.
Erlotinib is a small-molecule inhibitor of the epidermal growth factor receptor (EGFR), a tyrosine kinase receptor,4,5 which is approved by the US Food and Drug Administration for treatment of patients with locally advanced or metastatic NSCLC whose disease has not responded to more than one previous chemotherapy regimen.4,5 A phase 3 study5 showed that second-line or third-line monotherapy with erlotinib improved overall survival in patients with NSCLC.
The recombinant, anti-vascular endothelial growth factor (anti-VEGF) monoclonal antibody bevacizumab, combined with paclitaxel and carboplatin, was approved by the US Food and Drug Administration for first-line treatment of patients with unresectable, locally advanced, recurrent, or metastatic non-squamous NSCLC.6 A phase 3 study showed this combination significantly improved overall survival and progression-free survival in patients with NSCLC compared with carboplatin and paclitaxel alone.7,8 Another phase 3 trial9 showed that the addition of bevacizumab to cisplatin and gemcitabine improved progression-free survival and objective responses rates for first-line treatment of non-squamous NSCLC; however, overall survival was not improved.
Bevacizumab and erlotinib target different tumour growth pathways (angiogenesis and EGFR activity, respectively) with little overlap in their toxic-effect profiles. These two drugs have potentially complementary mechanisms to control tumour growth.10–14
The safety and activity of combination erlotinib-bevacizumab were assessed in a phase 1/2 trial15 for patients with relapsed and refractory non-squamous NSCLC. The combination dose was established at 15 mg/kg bevacizumab once every 3 weeks and 150 mg erlotinib once per day. The objective response rate in 34 patients in phase 2 was 20%, disease-control rate was 85%, and median overall survival was 12·6 months.15
In a multicentre phase 2 trial16 of patients with relapsed and refractory non-squamous NSCLC who were randomly allocated to receive erlotinib plus bevacizumab, bevacizumab and chemotherapy, or chemotherapy alone, median overall survival was better in the groups that received bevacizumab (13·7 months for erlotinib plus bevacizumab and 12·6 months for bevacizumab and chemotherapy) than it was with chemotherapy alone (8·6 months); safety data favoured the erlotinib plus bevacizumab group.
In this phase 3 trial, we aimed to further assess the efficacy of bevacizumab in combination with erlotinib compared with erlotinib and placebo in patients with recurrent or refractory advanced-stage NSCLC who had disease progression during or after first-line therapy.
Methods
Study design and participants
In our international, double-blind, placebo-controlled phase 3 trial (BeTa), we enrolled patients who presented to 177 study sites in 12 countries with cytologically or histologically confirmed advanced-stage NSCLC that was recurrent or refractory after standard first-line chemotherapy or chemoradiotherapy. Patients were eligible if they were aged 18 years or older and had Eastern Cooperative Oncology Group performance status scores of 2 or lower. Patients with squamous cell carcinoma were eligible if their disease was extrathoracic or if intrathoracic lesions were peripheral. Patients with a history of brain metastases who were treated with a minimum of whole-brain radiotherapy and with no ongoing dexamethasone requirement, patients requiring anticoagulation (low-molecular-weight heparins only), and patients who had received neoadjuvant and adjuvant therapy for stage I–IIIa disease were also eligible.
Patients were ineligible for inclusion if they had had a myocardial infarction within the previous 6 months, unstable angina, congestive heart failure, symptomatic arrhythmia, substantial peripheral vascular disease, uncontrolled hypertension, history of gross haemoptysis, presence of a cavitary lesion or tumour invading or abutting major blood vessels, bleeding diathesis or coagulopathy, abnormal haematological values, abnormal liver function tests, used warfarin or equivalents, used aspirin or non-steroidal anti-inflammatory drugs, a surgical procedure on-study or within 28 days before randomisation, previous treatment with anti-EGFR or anti-angiogenesis agents, another invasive cancer within 5 years before randomisation, neurosurgery for brain metastases within 6 months of day 0, and brain biopsy within 3 months of day 0.
All enrolled patients provided signed informed consent and consented to analysis of archival diagnostic tissue if available. Institutional review board approval was obtained at every study site.
Randomisation and masking
Patients were randomly allocated in a one-to-one ratio to receive erlotinib plus bevacizumab (bevacizumab group) or erlotinib plus placebo (control group) by use of an interactive voice-response system with a computer-generated allocation sequence. Staff at Genentech (South San Fransisco, CA, USA), investigators, and patients were masked to treatment assignment. Patients were stratified by sex, baseline Eastern Cooperative Oncology Group performance status score (0 or 1 vs 2), smoking history (never vs current or previous), and study site. Unmasked adverse events data were provided every month to an independent data and safety monitoring board.
Procedures
Patients received placebo or bevacizumab administered at 15 mg/kg by intravenous infusion on the first day of 3-week cycles (±4 days). Erlotinib was taken orally at 150 mg per day, beginning on the first day of the first cycle. Patients remained on treatment until there was documented evidence of radiographic or clinical disease progression or unmanageable toxic effects. If toxic effects were caused by bevacizumab, patients could continue on erlotinib until disease progression or unmanageable toxic effects. Patients who discontinued erlotinib received no further study treatment. Subsequent therapy was provided at the treating doctor’s discretion. All patients were followed-up for survival until death, loss to follow-up, or study termination by the sponsor, whichever occurred first.
We undertook clinical and laboratory assessments at baseline and every 6 weeks to week 24, and every 12 weeks thereafter. Tumour responses were investigator-assessed according to Response Evaluation Criteria in Solid Tumours (RECIST) 1.0.17
Adverse events were graded according to US National Cancer Institute Common Toxicity Criteria for Adverse Events version 3.0. Patients were assessed for all grades of adverse events, serious adverse events (including grade ≥3 pulmonary haemorrhage or grade ≥2 symptomatic central nervous system haemorrhage), and adverse events requiring study-drug interruption or discontinuation.
At study entry, tumour biopsy material was requested from patients to assess EGFR and Kras expression. Archival tumour tissue samples were collected with accompanying pathology reports. For those patients with data for biomarker expression, we did EGFR fluorescence in-situ hybridisation (FISH) analyses with a PathVysion kit (Abbott Molecular, Des Plaines, IL, USA). High EGFR copy number was defined as high polysomy (≥4 gene copies in ≥40% of cells) or amplification (≥2 genes or chromosomes or ≥15 gene copies in ≥10% of cells). Immunohistochemical analysis of EGFR was done with PharmDx kits (DAKO, Glostrup, Denmark); positive EGFR expression was defined as EGFR staining of more than 10% of tumour cells. Highly sensitive analyses of EGFR somatic gene mutations in exons 18–21 and Kras mutations in exons 2 and 3 were done by use of denaturing HPLC (Transgenomics, Omaha, NE, USA).
The primary efficacy endpoint was overall survival. Secondary endpoints included progression-free survival, objective response rate, duration of objective response, safety, and assessment of associations between efficacy endpoints and expression of EGFR and Kras biomarkers. Overall survival was defined as time from randomisation to death from any cause. Progression-free survival was defined as time from randomisation to documented disease progression, as determined by investigator with use of RECIST, or death on study treatment (from any cause within 30 days of last dose), whichever occurred first.
Statistical analyses
We designed the study to detect 33% improvement in median survival, which required 417 deaths to provide 83% power for final analysis of overall survival. We planned an interim efficacy analysis after 280 deaths had occurred (~67% of required deaths for final analysis); the significance level for comparisons of overall survival was established from the Lan-DeMets α spending function with an O’Brien-Fleming boundary of 0·0124 at 67% event time and 0·0462 at final analysis.
At the interim and final efficacy analyses, all patients randomly allocated to treatment groups were included in survival analyses (intention-to-treat analysis). Only patients with measurable disease at baseline were included in the analysis of objective response and the exploratory analysis of disease control.
We used stratified Cox proportional hazard models to estimate hazard ratios (HRs) and 95% CIs for overall survival and progression-free survival in the intention-to-treat population. We used a two-sided log-rank test, stratified by randomisation stratification factors apart from study site to compare overall survival and progression-free survival between trial groups. To control the overall type I error rate at the 5% level for efficacy endpoints, we used fixed-sequence testing, requiring that the primary endpoint be significant before statistical testing of key secondary endpoints.18,19 We estimated median and IQRs for overall survival and progression-free survival with Kaplan-Meier methods. IQRs are shown throughout the manuscript in parentheses after medians. 95% CIs for median time-to-events were calculated by the Brookmeyer and Crowley method20 where indicated.
We assessed overall and progression-free survival in subpopulations of patients (eg, biomarker-defined subgroups) with an unstratified Cox proportional hazards model and Kaplan-Meier method. Differences in treatment effects between subgroups were assessed by testing the interaction effect between treatment groups and characteristics of patients. Statistical analyses were done with SAS versions 9.1 and 9.2.
This trial is registered with ClinicalTrials.gov, number NCT00130728.
Role of the funding source
The study sponsor (Genentech) was involved in study design, data collection, data analysis, and interpretation of results, working closely with the investigators. All authors had full access to all the data in the study and had final responsibility for the decision to submit for publication.
Results
Between June 8, 2005, and April 16, 2008, we enrolled 636 patients at 177 study sites in 12 countries and randomly allocated 319 patients to the bevacizumab group and 317 to the control group (figure 1, table 1). 200 (63%) patients in the bevacizumab group discontinued treatment because of disease progression, compared with 243 (77%) in the control group.
Figure 1. Trial profile.
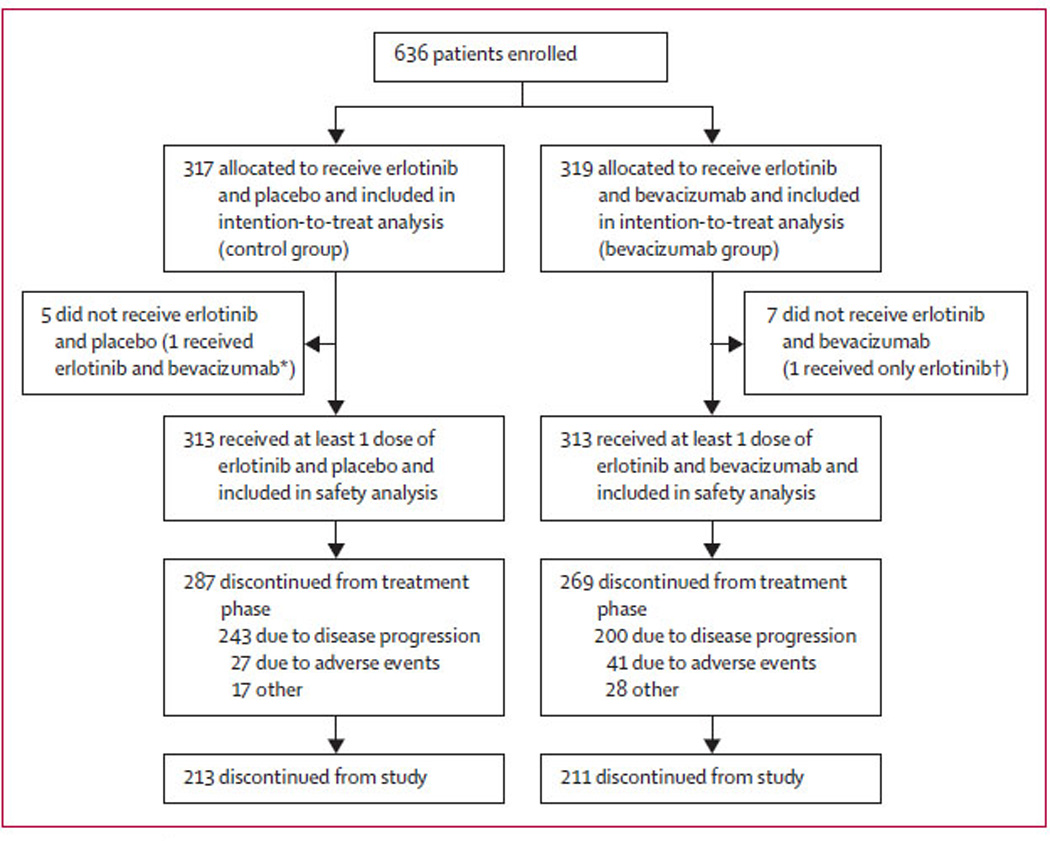
*This patient was analysed as part of the bevacizumab group for safety analyses. †This patient was analysed as part of the control group for safety analyses.
Table 1.
Demographics and baseline characteristics, including biomarkers
| Control group (n=317) | Bevacizumab group (n=319) | |
|---|---|---|
| Sex | ||
| Men | 170 (54%) | 171 (54%) |
| Women | 147 (46%) | 148 (46%) |
| Age, years | 65·0 (10·3) | 64·8 (104) |
| Race | ||
| White | 257 (81%) | 264 (83%) |
| Black | 33 (10%) | 21 (7%) |
| Asian/Pacific islander | 18 (6%) | 23 (7%) |
| Hispanic | 8 (3%) | 10 (3%) |
| American Indian/Alaskan native | 1 (<1%) | 0 |
| Other | 0 | 1 (<1%) |
| Smoking history | ||
| Never | 33 (10%) | 34 (11%) |
| Previous | 212 (67%) | 237 (74%) |
| Current | 72 (23%) | 48 (15%) |
| ECOG performance status score | ||
| 0 | 121/317 (38%) | 129/318 (41%) |
| 1 | 176/317 (56%) | 166/318 (52%) |
| 2 | 20/317 (6%) | 23/318 (7%) |
| Time since initial diagnosis | ||
| <6 months | 78/312 (25%) | 77/313 (25%) |
| 6–12 months | 122/312 (39%) | 120/313 (38%) |
| >12 months | 112/312 (36%) | 116/313 (37%) |
| Histology | ||
| Large-cell carcinoma | 25 (8%) | 23 (7%) |
| Adenocarcinoma | 235 (74%) | 242 (76%) |
| Squamous | 17 (5%) | 11 (3%) |
| Other* | 40 (13%) | 43 (13%) |
| Patients with treated brain metastases | 30 (9%) | 38 (12%) |
| EGFR status | ||
| FISH positive | 43/102 (42%) | 33/102 (32%) |
| FISH negative | 59/102 (58%) | 69/102 (68%) |
| IHC positive | 119/161 (74%) | 135/184 (73%) |
| IHC negative | 42/161 (26%) | 49/184 (27%) |
| EGFR mutant | 18/170 (11%) | 12/185 (6%) |
| EGFR wild-type | 152/170 (89%) | 173/185 (94%) |
| Kras mutation status | ||
| Mutant | 38/178 (21%) | 48/190 (25%) |
| Wild-type | 140/178 (79%) | 142/190 (75%) |
Data are n (%), mean (SD), or n/n with assessable tissue and test results (%). ECOG=Eastern Cooperative Oncology Group. EGFR=epidermal growth factor receptor. FISH=fluorescence in-situ hybridisation. IHC=immunohistochemistry.
Includes bronchoalveolar carcinoma and tumours defined as not otherwise specified or other.
Baseline characteristics were much the same between treatment groups (table 1). 448 (70%) of 636 patients had previously received carboplatin as first-line therapy and 113 (18%) had received cisplatin; other frequently used drugs included taxanes (294 [46%] patients received paclitaxel and 132 [21%] received docetaxel) and gemcitabine (148 [23%]). Rates of response to previous first-line treatment were balanced between groups. 68 (11%) of 636 patients entered the study with treated brain metastases. 613 (98%) of 626 treated patients used concomitant medications during treatment, including steroids, anti-emetics, analgesics, or other drugs.
We did the interim efficacy analysis on Feb 7, 2008, after 284 deaths (68% of 417 deaths required for final analysis), at which point the data monitoring committee recommended that the study continue. Median follow-up time for patients who were randomly allocated to treatment groups was 19 months (IQR 11–25 months; range 0·2–34 months).
Overall survival did not differ between the patients in the bevacizumab group and controls (figure 2 and figure 3). Median overall survival was about 9·2–9·3 months in both groups (figure 3). In the Kaplan-Meier analysis, 1-year survival was 42·1% in the bevacizumab group and 40·7% in the control group.
Figure 2. Kaplan-Meier curves for overall survival.

Overall survival is shown for assessable patients randomly allocated to erlotinib plus bevacizumab (bevacizumab group) or erlotinib plus placebo (control group). Thep value for overall survival is based on a stratified log-rank test; stratification factors were Eastern Cooperative Oncology Group performance status, smoking history, and sex.
Figure 3. Forest plot for subpopulations of patients defined by demographics and baseline characteristics, including biomarker expression.

Dashed line shows hazard ratio for the overall population. Overall survival hazard ratios were estimated by use of an unstratified Cox model. FISH=fluorescence in-situ hybridisation. NA=not assessable. NR=not reached. ECOG=Eastern Cooperative Oncology Group. NSCLC=non-small-cell lung cancer. EGFR=epidermal growth factor receptor.
Figure 4 shows progression-free survival and table 2 shows an analysis of objective response rate. Median progression-free survival was 3·4 months (IQR 1·4–8·4) in the bevacizumab group compared with 1·7 months (1·3–4·1) in the control group (figure 4). The objective response rate in the bevacizumab group was much higher than it was in the control group (table 2). Three patients had a complete response after treatment with bevacizumab compared with one patient in the control group (table 2). Median duration of overall response seemed to be longer in the bevacizumab group than in the control group (table 2). The disease control rate was 45% (136 patients) in the bevacizumab group and 34% (104 patients) in the control group. Because of the prespecified use of fixed-sequence testing to control the overall type I error rate, we did not compare secondary efficacy results statistically.
Figure 4. Kaplan-Meier curves for progression-free survival.

Progression-free survival is shown for assessable patients in the bevacizumab group (randomly allocated to erlotinib plus bevacizumab) and control group (randomly allocated to erlotinib plus placebo). Because of the prespecified use of fixed sequence testing to control the overall type I error rate, which required that the primary endpoint (overall survival) be significant before statistical testing of key secondary endpoints, progression-free survival results could not be defined as significant.
Table 2.
Response rates, disease control rates, and duration of objective response in patients with measurable disease at baseline
| Control group (n=306) | Bevacizumab group (n=301) | |
|---|---|---|
| Objective response | 19 (6%) | 38 (13%) |
| Complete response | 1 (<1%) | 3 (1%) |
| Partial response | 18 (6%) | 35 (12%) |
| Stable disease | 85 (28%) | 98 (33%) |
| Progressive disease | 154 (50%) | 112 (37%) |
| Disease control rate | ||
| Complete response, partial response, or stable disease |
104 (34%) | 136 (45%) |
| Median duration of objective response, months |
8·4 (3·5–14·9, 3·4–14·9) | 9·7 (6·9–19·5, 5·7–19·5) |
Data are n (%) or median (95% CI, IQR).
A subgroup analyses of 67 patients who never smoked suggested a possible improvement in overall survival in the bevacizumab group compared with the control group (HR 0·44, 95% CI 0·21–0·94; figure 3).
477 (75%) of 636 enrolled patients had tumour tissue available for biomarker analyses. 390 patients (202 in the bevacizumab group and 188 in the control group) had results for at least one of the following tests: EGFR immunohistochemistry, EGFR FISH, EGFR mutation status, and Kras mutation status. 355 (56%) patients had assessable tests for EGFR mutations and 368 (58%) had assessable tests for Kras mutations. Figure 3 shows overall survival for patients with available tissue and assessable results. Although subgroup analysis of suggested overall survival seemed to favour bevacizumab in patients with EGFR-mutated tumours compared with those with EGFR-wild-type tumours, the difference did not achieve significance (p=0·1826). Survival outcomes were similar between patients with tumours with Kras mutations and those that were Kras wild-type. Progression-free survival outcomes were much the same in patients with tumours with EGFR mutations and those that were EGFR wild-type (data not shown).
188 (60%) of 313 patients in the bevacizumab group had grade 3 or 4 adverse events, compared with 151 (48%) of 313 controls (table 3). Incidence of grade 3 arterial thromboembolic events was higher in the bevacizumab group than it was in the control group, but was much the same as previously reported rates in patients with advanced NSCLC who were treated with bevacizumab.6,7 15 patients (5%) in the bevacizumab group and four (1%) in the control group had grade 3 or 4 hypertension, and no patients had grade 5 hypertension (table 3).
Table 3.
Adverse events
| Control group (n=313) | Bevacizumab group (n=313) | |
|---|---|---|
| Any adverse event | 309 (99%) | 312 (>99%) |
| Any serious adverse event | 114 (36%) | 130 (42%) |
| Any grade ≥3 adverse event | Grade 3/4: 151 (48%); grade 5: 14 (4%) |
Grade 3/4: 188 (60%); grade 5: 20 (6%) |
| Haemorrhage | Grade 3/4: 7 (2%) | Grade 3/4: 8 (3%); grade 5: 2 (1%) |
| Pulmonary haemorrhage | Grade 3/4: 1 (<1%) | Grade 3/4: 2 (1%); grade 5: 1 (<1%) |
| CNS haemorrhage | 0 | Grade 3/4: 1 (<1%) |
| Arterial thromboembolic event | Grade 3/4: 1 (<1%) | Grade 3/4: 10 (3%); grade 5: 2 (1%) |
| Hypertension | Grade 3/4: 4 (1%) | Grade 3/4: 15 (5%) |
| Interstitial lung disease-like events | Grade 3/4: 1 (<1%); grade 5: 1 (<1%) |
Grade 3/4: 1 (<1%); grade 5: 1 (<1%) |
| Rash | Grade 3/4: 19 (6%) | Grade 3/4: 49 (16%) |
Data are for the safety-assessable patient population. CNS=central nervous system.
There were 20 (6%) grade 5 adverse events in the bevacizumab group, compared with 14 (4%) in the control group, most commonly dyspnoea (three events vs two events), pneumonia (two vs four events), and pulmonary embolism (one event in each group). Two deaths due to bleeding (pulmonary haemorrhage and gastrointestinal haemorrhage) occurred in the bevacizumab group, compared with none in the control group. No bleeding events of grade 3 or higher (including central nervous system haemorrhage) were reported in 68 patients entering the study with treated brain metastases.
41 patients (13%) in the bevacizumab group discontinued treatment because of adverse events, compared with 27 (9%) in the control group (figure 1).
Patients in the bevacizumab group received a mean dose of 132·2 mg (SD 25·8) erlotinib per day for a duration of 152·2 days (173·4); controls received a mean dose of 139·3 mg (20·8) erlotinib every day for a duration of 101·5 days (119·0). Patients in the bevacizumab group received a median of 4 (IQR 2–9; range 1–47) doses of bevacizumab, compared with a median of 2 (IQR 2–6; range 0–37) doses of placebo in the control group.
In the bevacizumab group, 24 (8%) of 319 patients received bevacizumab in subsequent lines of therapy, compared with 38 (12%) of 317 patients in the control group. In the bevacizumab group, 164 (51%) of 319 patients received one or more subsequent therapies (most commonly pemetrexed or gemcitabine) after discontinuation of study treatment, compared with 193 (61%) of 317 in the control group.
Discussion
We assessed the efficacy of addition of bevacizumab to erlotinib in patients with recurrent, advanced, or metastatic NSCLC after failure of standard first-line chemotherapy, and showed that addition of bevacizumab to erlotinib did not improve overall survival (figure 2). Bevacizumab added to erlotinib seemed to prolong progression-free survival, objective response rate, and duration of response compared with erlotinib alone; median progression-free survival and objective response rates were about double those in the control group. However, because of our prespecified use of fixed-sequence testing, the secondary endpoints were not compared statistically.
Results from our subset analyses for survival were generally consistent with overall trial results. Most subpopulations of patients derived a benefit to progression-free survival but not overall survival from the addition of bevacizumab. Although a few subsets of patients in the bevacizumab group showed longer survival than did those in the control group, the sample sizes were small and CIs were wide; hence, these subgroup results should be interpreted with caution.
This trial had limitations, including the absence of accounting for crossover effects of a potentially active therapy (eg, bevacizumab) on overall survival. More patients in the control group than in the bevacizumab group received subsequent lines of therapy during follow-up, including subsequent treatment with bevacizumab, which could have confounded the comparison of overall survival between the two cohorts. Reports of an overall survival benefit in the setting of refractory NSCLC have become more difficult in recent years, because of the increased number of moderately active agents available for subsequent treatment. A predictive tissue marker was not identified in this study, although our biomarker analysis was done with samples obtained by surgery or for diagnostic intent (rather than new biopsies), potentially restricting the value of this biomarker analysis.
Several preclinical studies10–13 showed an enhanced benefit from combination EGFR and angiogenesis inhibitors (panel). A preclinical study14 in an orthotopic lung cancer model showed that inhibition of EGFR and VEGFR signalling led to profound antiangiogenic, antivascular, and antitumour effects.14 The hypothesis was therefore made that combination of erlotinib and bevacizumab would be more clinically effective than would erlotinib monotherapy, while maintaining acceptable toxic effects. Furthermore, studies21,22 have suggested that blocking of VEGFR and EGFR signalling could overcome primary or acquired resistance to EGFR inhibitors in xenograft models. Second-line NSCLC treatment with vandetanib, a drug targeting both angiogenesis and EGFR activity, has led to prolonged progression-free survival in two clinical trials,23,24 although overall survival was not improved.
EGFR expression, overexpression, and mutation have been implicated in the pathogenesis of NSCLC,25–30 suggesting that patients who are EGFR positive (by immunohistochemistry or FISH analyses) or who have EGFR mutations might derive increased benefit from EGFR-targeted therapies. In a subgroup analysis of EGFR mutation status, the HR for overall survival was apparently lower in patients with EGFR-mutant tumours than in patients with wild-type tumours. However, this result should be interpreted with caution because only 30 patients in the study had EGFR-mutated tumours and the 95% CIs for the HRs were wide (upper limits >1·0) and overlapping.
Predictive markers show responses to treatment, whereas prognostic markers show the natural history of disease. Predictive conclusions about the role of biomarkers in erlotinib treatment cannot be made because both treatment groups contained erlotinib. However, the EGFR mutation data do support EGFR mutation as a prognostic marker for NSCLC. Activation of mutations in Kras occur in 30% of NSCLCs and might indicate poor prognosis or mediate resistance to EGFR inhibitors.31,32 By contrast with previous reports, the Kras data in our study do not support Kras mutation status as an independent marker of poor prognosis for NSCLC.
Both regimens were well tolerated, although patients given bevacizumab had slightly more toxic effects than did controls, with an increased proportion of grade 5 events. The toxic effects of erlotinib and bevacizumab that we noted were consistent with the known individual drug toxic-effect profiles; no new safety signals were reported. The incidence of events related to bevacizumab, including arterial thromboembolic events, venous thromboses, and haemorrhages, was consistent with previously reported data for bevacizumab.6 Events typically associated with erlotinib, such as diarrhoea, nausea, and vomiting, were reported at much the same rates in both groups. The incidence of rash associated with erlotinib was higher in the bevacizumab group, although in most cases rash was manageable and did not lead to substantial treatment discontinuation. The increased overall incidence of adverse events in the bevacizumab group probably reflects some additive effects of the drug combination, and the greater median length of time on-study for patients receiving erlotinib and bevacizumab.
In summary, erlotinib and bevacizumab given to patients with advanced-stage NSCLC after failure of standard first-line chemotherapy did not result in improved overall survival compared with erlotinib and placebo. Although we could not assess significance, potential clinical activity of the combination is suggested by possible improvements in progression-free survival and objective response rates and the tolerable safety profile, which supports the hypothesis that the combination could be useful in treatment of NSCLC. However, the absolute improvement in median progression-free survival (6 weeks; figure 4) by this combination will need to be balanced against the slightly increased rate of toxic effects and cost, perhaps requiring more detailed analysis of patient-derived outcomes.
A trial33 testing the addition of erlotinib to bevacizumab maintenance in patients without progressive disease after four cycles of chemotherapy and bevacizumab showed improved progression free survival but not overall survival compared to bevacizumab alone. Our trial did not test a maintenance concept but instead was designed to detect a survival improvement for the combination of bevacizumab and erlotinib in a more advanced population of patients that had progressed after front-line treatment. Both studies suggest that despite improvements in several efficacy endpoints, improving survival remains a challenge in the treatment of NSCLC. Therefore, optimisation of outcomes from a combination of bevacizumab and erlotinib will probably require new techniques to prospectively identify subsets of patients who are most likely to benefit from this treatment.
Supplementary Material
{kind=link}
Acknowledgments
We thank the patients and study investigators who participated in this study, Christopher Bowden (Genentech, South San Franscisco, CA, USA) for critical review and input, and Abie Craiu (Genentech), Bich Tran (MD Anderson Cancer Center, Houston, TX, USA), and Joerg Jacoby (MD Anderson Cancer Center) for assistance with preparation of the manuscript. RSH was supported in part by the US National Institutes of Health through the MD Anderson’s Cancer Center Support Grant CA016672.
Footnotes
Contributors
RSH, RA, FB, PF, LH, GAO, GV, PO’C, and JH contributed to study design, data collection, analysis, and interpretation, and writing of the manuscript. C-HS contributed to data analysis and interpretation and writing of the manuscript.
Conflicts of interest
Genentech funded this clinical trial. RSH, LH, PF, and GAO have received funding from Genentech. RSH has consulted for Genentech, OSI, and Roche. GAO has consulted for Genentech and Abraxis. PF has consulted for Genentech, participated in speakers’ bureaux for Genentech and OSI Pharmaceuticals, and grant funding from the Metro Minnesota Community Clinical Oncology Program, a non-profit research program sponsored by NCI. GV has consulted for Genentech and participated in speakers’ bureaux for Genentech and Sanofi-Aventis. C-HS and PO’C have been employed by Genentech and were stockholders with Roche.
References
- 1.Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 2.Jemal A, Siegel R, Xu J, Ward E. Cancer statistics, 2010. CA Cancer J Clin. 2010;60:277–300. doi: 10.3322/caac.20073. [DOI] [PubMed] [Google Scholar]
- 3.Pazdur R, Coia LR, Hoskins WJ, Wagman LD. Cancer management: a multidisciplinary approach. 8th edn. Manhasset, NY, USA: CMP Healthcare Media; 2004. [Google Scholar]
- 4.Erlotinib (Tarceva) [package insert] South San Francisco, CA, USA: Genentech; [Google Scholar]
- 5.Shepherd FA, Rodrigues Pereira J, Ciuleanu , et al. Erlotinib in previously treated non-small-cell lung cancer. N Engl J Med. 2005;353:123–132. doi: 10.1056/NEJMoa050753. [DOI] [PubMed] [Google Scholar]
- 6.Bevacizumab (Avastin) [package insert] South San Francisco, CA, USA: Genentech; [Google Scholar]
- 7.Sandler A, Gray R, Perry MC, et al. Paclitaxel–carboplatin alone or with bevacizumab for non-small-cell lung cancer. N Engl J Med. 2006;355:2542–2550. doi: 10.1056/NEJMoa061884. [DOI] [PubMed] [Google Scholar]
- 8.Johnson DH, Fehrenbacher L, Novotny WF, et al. Randomized phase II trial comparing bevacizumab plus carboplatin and paclitaxel with carboplatin and paclitaxel alone in previously untreated locally advanced or metastatic non-small-cell lung cancer. J Clin Oncol. 2004;22:2184–2191. doi: 10.1200/JCO.2004.11.022. [DOI] [PubMed] [Google Scholar]
- 9.Reck M, von Pawel J, Zatloukal P, et al. Phase III trial of cisplatin plus gemcitabine with either placebo or bevacizumab as first-line therapy for nonsquamous non-small-cell lung cancer: AVAiL. J Clin Oncol. 2010;27:2415–2422. doi: 10.1200/JCO.2007.14.5466. [DOI] [PubMed] [Google Scholar]
- 10.Ciardiello F, Bianco R, Damiano V, et al. Antiangiogenic and antitumor activity of anti-epidermal growth factor receptor C225 monoclonal antibody in combination with vascular endothelial growth factor antisense oligonucleotide in human GEO colon cancer cells. Clin Cancer Res. 2000;6:3739–3747. [PubMed] [Google Scholar]
- 11.Shaheen RM, Ahmad SA, Liu W, et al. Inhibited growth of colon cancer carcinomatosis by antibodies to vascular endothelial and epidermal growth factor receptors. Br J Cancer. 2001;85:584–589. doi: 10.1054/bjoc.2001.1936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bozec A, Sudaka A, Fischel JL, et al. Combined effects of bevacizumab with erlotinib and irradiation: a preclinical study on a head and neck cancer orthotopic model. Br J Cancer. 2008;99:93–99. doi: 10.1038/sj.bjc.6604429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schicher N, Paulitschke V, Swoboda A, et al. Erlotinib and bevacizumab have synergistic activity against melanoma. Clin Cancer Res. 2009;15:3495–3502. doi: 10.1158/1078-0432.CCR-08-2407. [DOI] [PubMed] [Google Scholar]
- 14.Wu W, Onn A, Isobe T, et al. Targeted therapy of orthotopic human lung cancer by combined vascular endothelial growth factor and epidermal growth factor receptor signaling blockade. Mol Cancer Ther. 2007;6:471–483. doi: 10.1158/1535-7163.MCT-06-0416. [DOI] [PubMed] [Google Scholar]
- 15.Herbst RS, Johnson DH, Mininberg E, et al. Phase I/II trial evaluating the anti-vascular endothelial growth factor monoclonal antibody bevacizumab in combination with the HER-1/epidermal growth factor receptor tyrosine kinase inhibitor erlotinib for patients with recurrent non-small-cell lung cancer. J Clin Oncol. 2005;23:2544–2555. doi: 10.1200/JCO.2005.02.477. [DOI] [PubMed] [Google Scholar]
- 16.Herbst RS, O’Neill VJ, Fehrenbacher L, et al. Phase II study of efficacy and safety of bevacizumab in combination with chemotherapy or erlotinib compared with chemotherapy alone for treatment of recurrent or refractory non small-cell lung cancer. J Clin Oncol. 2007;25:4743–4750. doi: 10.1200/JCO.2007.12.3026. [DOI] [PubMed] [Google Scholar]
- 17.Eisenhauer EA, Therasse P, Bogaerts J, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1) Eur J Cancer. 2009;45:228–247. doi: 10.1016/j.ejca.2008.10.026. [DOI] [PubMed] [Google Scholar]
- 18.Dmitrienko A, Molenberghs G, Chuang-Stein C, Offen W. Analysis of clinical trials using SAS. Cary, NC, USA: SAS Institute; 2005. [Google Scholar]
- 19.Westfall PH, Krishen A. Optimally weighted, fixed sequence and gatekeeper multiple testing procedures. J Stat Planning Inference. 2001;99:25–40. [Google Scholar]
- 20.Brookmeyer R, Crowley J. A confidence interval for median survival time. Biometrics. 1982;38:29–41. [Google Scholar]
- 21.Ciardiello F, Bianco R, Caputo R, et al. Antitumor activity of ZD6474, a vascular endothelial growth factor receptor tyrosine kinase inhibitor, in human cancer cells with acquired resistance to antiepidermal growth factor receptor therapy. Clin Cancer Res. 2004;10:784–793. doi: 10.1158/1078-0432.ccr-1100-03. [DOI] [PubMed] [Google Scholar]
- 22.Naumov GN, Nilsson MB, Cascone T, et al. Combined vascular endothelial growth factor receptor and epidermal growth factor receptor (EGFR) blockade inhibits tumor growth in xenograft models of EGFR inhibitor resistance. Clin Cancer Res. 2009;15:3484–3494. doi: 10.1158/1078-0432.CCR-08-2904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Herbst RS, Sun Y, Eberhardt WEE, et al. Vandetanib plus docetaxel versus docetaxel as second-line treatment for patients with advanced non-small-cell lung cancer (ZODIAC): a double-blind, randomised, phase 3 trial. Lancet Oncol. 2010;11:619–626. doi: 10.1016/S1470-2045(10)70132-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Natale RB, Bodkin D, Govindan R, et al. Vandetanib versus gefitinib in patients with advanced non-small-cell lung cancer: results from a two-part, double-blind, randomized phase II study. J Clin Oncol. 2009;27:2523–2529. doi: 10.1200/JCO.2008.18.6015. [DOI] [PubMed] [Google Scholar]
- 25.Hirsch FR, Varella-Garcia M, Dziadziuszko R, et al. Fluorescence in situ hybridization subgroup analysis of TRIBUTE, a phase III trial of erlotinib plus carboplatin and paclitaxel in non-small cell lung cancer. Clin Cancer Res. 2008;14:6317–6323. doi: 10.1158/1078-0432.CCR-08-0539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Andratschke NH, Dittmann KH, Mason KA, et al. Epidermal growth factor receptor as a target to improve treatment of lung cancer. Clin Lung Cancer. 2004;5:340–352. doi: 10.3816/CLC.2004.n.012. [DOI] [PubMed] [Google Scholar]
- 27.Roskoski R., Jr. The ErbB/HER receptor protein-tyrosine kinases and cancer. Biochem Biophys Res Commun. 2004;319:1–11. doi: 10.1016/j.bbrc.2004.04.150. [DOI] [PubMed] [Google Scholar]
- 28.Lynch TJ, Bell DW, Sordella R, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004;350:2129–2139. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- 29.Paez JG, Jänne PA, Lee JC. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004;304:1497–1500. doi: 10.1126/science.1099314. [DOI] [PubMed] [Google Scholar]
- 30.Pao W, Miller V, Zakowski M, et al. EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Natl Acad Sci USA. 2004;101:13306–13311. doi: 10.1073/pnas.0405220101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Eberhard DA, Johnson BE, Amler LC, et al. Mutations in the epidermal growth factor receptor and in KRAS are predictive and prognostic indicators in patients with non-small-cell lung cancer treated with chemotherapy alone and in combination with erlotinib. J Clin Oncol. 2005;23:5900–5909. doi: 10.1200/JCO.2005.02.857. [DOI] [PubMed] [Google Scholar]
- 32.Miller VA, Riely GJ, Zakowski MF, et al. Molecular characteristics of bronchioloalveolar carcinoma and adenocarcinoma, bronchioloalveolar carcinoma subtype, predict response to erlotinib. J Clin Oncol. 2008;26:1472–1478. doi: 10.1200/JCO.2007.13.0062. [DOI] [PubMed] [Google Scholar]
- 33.Miller VA, O’Connor P, Soh C, Kabbinavar F for the ATLAS Investigators. A randomized, double-blind, placebo-controlled, phase IIIb trial (ATLAS) comparing bevacizumab (B) therapy with or without erlotinib (E) after completion of chemotherapy with B for first-line treatment of locally advanced, recurrent, or metastatic non-small cell lung cancer (NSCLC) Proc Am Soc Clin Oncol. 2009;27 doi: 10.1200/JCO.2012.47.3983. LBA8002 (abstr) [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.