

Abstract
Dynamic RNA nanotechnology based on programmable hybridization cascades with small conditional RNAs (scRNAs) offers a promising conceptual framework for engineering programmable conditional regulation in vivo. While single-base substitution (SBS) somatic mutations and single-nucleotide polymorphisms (SNPs) are important markers and drivers of disease, it is unclear whether synthetic RNA signal transducers are sufficiently programmable to accept a cognate RNA input while rejecting single-nucleotide sequence variants. Here, we explore the limits of scRNA programmability, demonstrating isothermal, enzyme-free genotyping of RNA SBS cancer markers and SNPs using scRNAs that execute a conditional hybridization cascade in the presence of a cognate RNA target. Kinetic discrimination can be engineered on a time scale of choice from minutes to days. To discriminate even the most challenging single-nucleotide sequence variants, including those that lead to nearly isoenergetic RNA wobble pairs, competitive inhibition with an unstructured scavenger strand or with other scRNAs provides a simple and effective principle for achieving exquisite sequence selectivity.
Keywords: Dynamic RNA nanotechnology, molecular programming, small conditional RNAs, hybridization chain reaction, scavenger, competitive inhibition
The programmable chemistry of nucleic acid base-pairing plays central roles in the biological circuitry within living organisms and provides a rich design space for the emerging discipline of dynamic nucleic acid nanotechnology. Nucleic acid molecules can be engineered to interact via prescribed hybridization cascades to execute diverse functions including catalysis, amplification, logic, and locomotion.1,2 To date, these efforts have been primarily directed at engineering DNA devices and circuits that operate in vitro.1,2 By contrast, synthetic RNA hybridization cascades have been relatively little-explored, yet hold great promise for engineering programmable signal transduction in vivo.3,4 Because biological RNAs interface with diverse endogenous pathways, small conditional RNAs (scRNAs) that interact and change conformation to transduce between detection of programmable RNA inputs and production of biologically active, programmable RNA outputs provide a conceptually appealing framework for introducing synthetic regulatory links into living organisms.
In nature, even single-nucleotide changes to the molecular programs encoded in a genome can have profound biological implications: single-base substitution (SBS) somatic mutations serve as markers and drivers for cancers,5,6 and single-nucleotide polymorphisms (SNPs) are associated with susceptibility to diverse classes of disease including cancers, gastrointestinal disorders, cardiovascular conditions, neuropsychiatric conditions, autoimmune diseases, and infectious diseases7,8 as well as with drug resistance in pathogenic microbial populations.9,10 Nonetheless, from an engineering perspective the metaphor of programmability is apt but imperfect. Base-pairing is not simply an informatic phenomenon dependent on the presence or absence of perfect Watson–Crick complementarity (A paired with U and G paired with C for RNA), but a physical phenomenon in which sequences with varying degrees of complementarity sample an ensemble of competing base-pairing states with differing free energies. For RNA, the programmability of base-pairing is further complicated by the fact that U can form not only Watson–Crick pair, U·A, but also nearly isoenergetic wobble pair, U·G.11 To program the function of a synthetic RNA hybridization cascade, the sequences of the constituent molecules must be designed so that the molecules predominantly execute the desired self-assembly pathway while avoiding off-pathway alternatives. It is unclear a priori whether synthetic RNA signal transducers are sufficiently programmable to accept a cognate RNA input while rejecting SBS or SNP sequence variants. Here, we explore the limits of RNA programmability in the context of dynamic RNA nanotechnology.
For this purpose, we examine the sequence selectivity of synthetic RNA hybridization cascades in which metastable scRNAs execute conditional self-assembly via the mechanism of hybridization chain reaction (HCR; Figure 1).12 Each HCR system consists of two scRNAs (h1 and h2 in Figure 1) that are designed to coexist metastably in the absence of a cognate RNA target (X) but upon arrival of the target undergo a chain reaction in which scRNAs sequentially nucleate and open to assemble into a nicked dsRNA polymer. Each scRNA comprises an input domain with an exposed single-stranded toehold and an output domain with a toehold sequestered in the hairpin loop. Hybridization of X to the input domain of h1 (labeled “a-b” in Figure 1) opens the hairpin loop to expose the output domain of h1 (“c*-b*”). Hybridization of the output domain of h1 to the input domain of h2 (“b-c”) opens the hairpin loop of h2 to expose an output domain (“b*-a*”) identical in sequence to X. Regeneration of the target sequence provides the basis for a cascade of alternating h1 and h2 polymerization steps. Each assembly operation in the HCR cascade occurs via toehold-mediated branch migration,13,14 a mode of molecular interaction demonstrated to have broad utility for engineering dynamic DNA nanotechnology.1,2 Over the past decade, DNA HCR has been widely exploited as a programmable, isothermal, enzyme-free, amplifying signal transducer for detection of nucleic acid, protein, and small molecule targets in vitro and in situ.15−17 Here, we employ RNA HCR18 as a model system to explore and surmount the limits of programmability for scRNA hybridization cascades.
Figure 1.
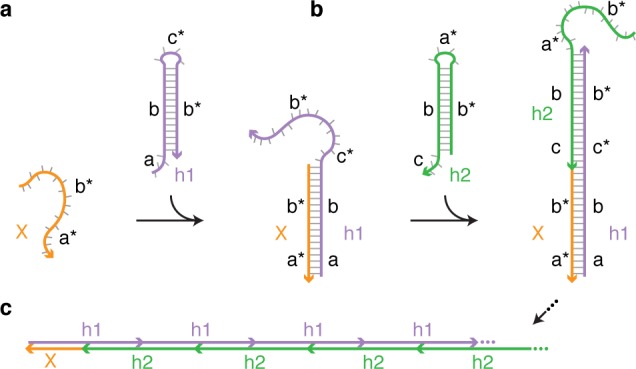
HCR mechanism.12 Metastable scRNAs (h1 and h2) self-assemble into polymers upon detection of a cognate RNA target (X). (a) X nucleates with h1 by base-pairing to single-stranded toehold “a”, mediating a branch migration that opens h1 to form complex X·h1 with single-stranded domain “c*-b*”. (b) Complex X·h1 nucleates with h2 by base-pairing to toehold “c”, mediating a branch migration that opens h2 to form complex X·h1·h2 with single-stranded domain “b*-a*”, identical in sequence to X. (c) This provides the basis for a chain reaction of alternating h1 and h2 polymerization steps.
With HCR, metastable scRNAs store the energy that drives the hybridization cascade, but are kinetically trapped to inhibit initiation of the cascade in the absence of the cognate RNA target. The cognate target functions as a programmable key that unlocks the kinetic trap for the first scRNA and initiates the ON state of the hybridization cascade. Closely related off-targets may succeed in unlocking the kinetic trap but will necessarily have one or more mismatches with the input domain of the scRNA, creating a discrimination energy gap that provides the basis for selectivity; the larger the discrimination energy gap, the cleaner the OFF state of the cascade. The discrimination energy gap is smallest for 1-nt sequence variants, making them the most challenging to detect selectively, and hence the sternest test of RNA programmability. HCR provides kinetic discrimination of sequence variants on a time scale where the cognate target has initiated substantial polymerization (ON state) but before spontaneous leakage and off-targets have caused substantial polymerization (OFF state). The timing of this selectivity window can be adjusted by altering the affinity between the input and output domains of the two species of HCR scRNAs (noting that the output domain of h2 is identical to the detected subsequence of the cognate target): increasing the energetic driving force for polymerization leads to selectivity at an earlier time point, and decreasing the energetic driving force for polymerization leads to selectivity at a later time point.
HCR cascades can be engineered to discriminate a cognate RNA target from 1- and 2-nt sequence variants on a time scale of choice from minutes to days (Figure 2). For the HCR system, Hfast, selectivity is established by the 6 min time point (Figure 2a, lanes 1 and 2) and lost by the 1 h time point (Figure 2a, lanes 5 and 6); spontaneous leakage occurs on a slower time scale with substantial polymerization becoming visible at the 100 h time point (Figure 2a, lane 16). For the HCR system, Hslow, selectivity is established by the 10 h time point (Figure 2c; lanes 9 and 10) and is still preserved at the 100 h time point (Figure 2c, lanes 13 and 14).
Figure 2.
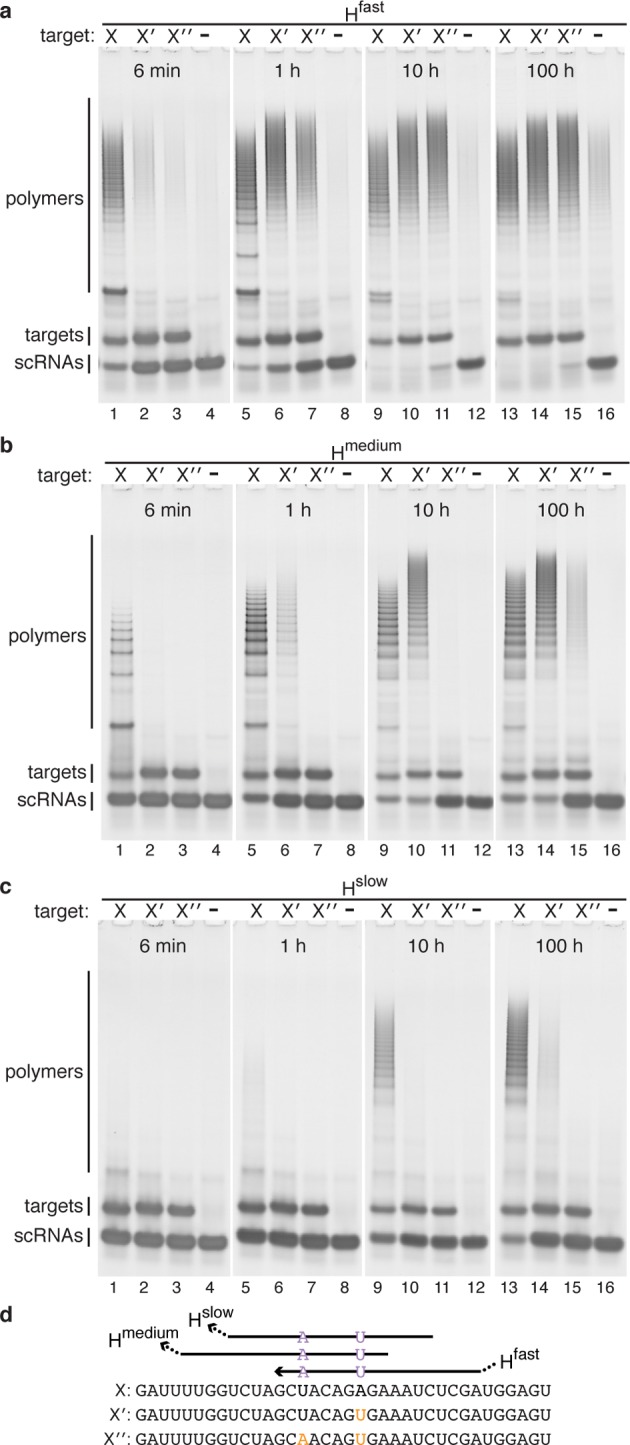
Kinetic discrimination of RNA sequence variants on a time scale of choice from minutes to days. Each reaction contains one HCR system: (a) Hfast, (b) Hmedium, (c) Hslow. ON state: cognate RNA target X. OFF state: 1-nt sequence variant X′, 2-nt sequence variant X″, or no target. Time points: 6 min, 1 h, 10 h, and 100 h. Native PAGE poststained with SYBR Gold. Targets and scRNAs at 1 μM. Reactions run in 1× PKR at 37 °C. (d) Target sequences depicted 5′ to 3′ with nucleotide variants in orange. For each HCR system, the input domain of the detecting scRNA is depicted and the location of the output domain is indicated by an ellipsis. See Supporting Information Table S3 for scRNA sequences.
To explore the sequence selectivity of scRNA hybridization cascades for targets of biological interest, we studied three SBS cancer markers: BRAF U1799A, JAK2 G1849U, and PTEN C388G.5,6 For each cancer marker, we engineered two HCR systems: one to detect the mutant target and one to detect the corresponding wildtype target. Introduction of either target into a mixture of mutant- and wildtype-detecting HCR systems led to selective activation of the cognate scRNA hybridization cascade in all cases, typically achieving an ON/OFF ratio of an order of magnitude or more (Figure 3).
Figure 3.
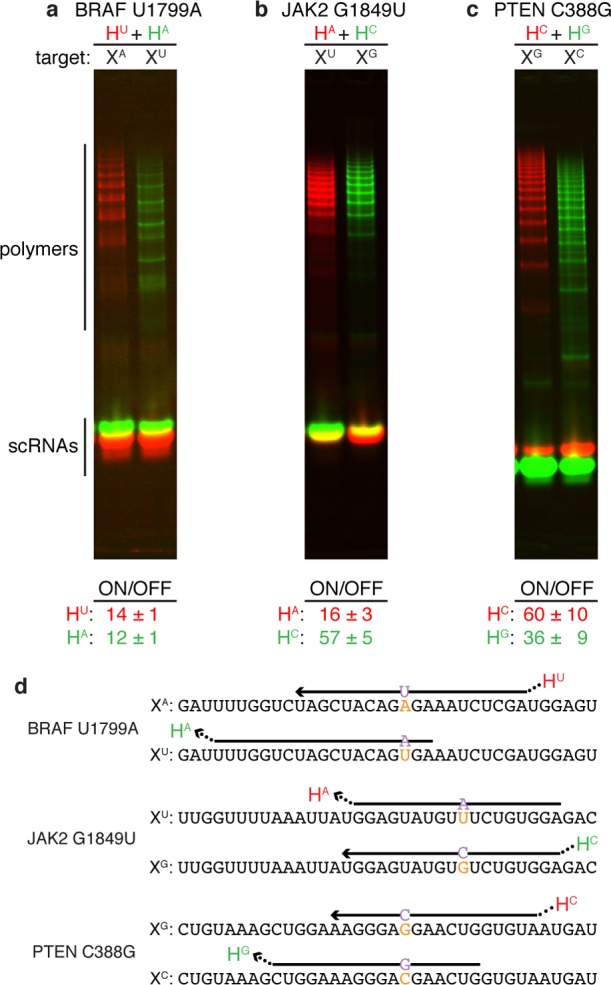
Discrimination of RNA SBS cancer markers and wildtype sequences: (a) BRAF U1799A, (b) JAK2 G1849U, (c) PTEN C388G. Each reaction contains two HCR systems labeled with spectrally distinct fluorophores, one targeting the SBS cancer marker (red channel) and one targeting the corresponding wildtype sequence (green channel): (a) HU labeled with Cy3, HA labeled with Cy5. (b) HA labeled with Alexa647, HC labeled with Alexa488. (c) HC labeled with Cy5, HG labeled with Cy3. Top: Native PAGE. Bottom: Typical ON/OFF ratio for each HCR system (median ± median absolute deviation for N = 5 experiments). All targets and scRNAs at 1 μM except scRNAs of system HA of panel (a) at 2 μM to shift the selectivity window earlier in time. Reactions run for 2 h (panel a) or 1 h (panels b and c) in 1× PKR at 37 °C. (d) Target sequences depicted 5′ to 3′ with nucleotide variants in orange. For each target, the input domain of the cognate detecting scRNA is depicted with the location of the output domain indicated by an ellipsis. See Supporting Information Table S3 for scRNA sequences and label locations; see Supporting Information Section S2 for quantification details and additional data.
The most challenging RNA SBS mutation to discriminate selectively is expected to be G → A, because an HCR system, HU, that forms Watson–Crick pair U·A with the mutant will form a nearly isoenergetic U·G wobble pair with the wildtype sequence, leading to a small discrimination energy gap and poor selectivity. Indeed, HU is not able to discriminate between the cognate target XA and the off-target XG (Figure 4c, lanes 1 and 2). In some sense, we have encountered a limit to the programmability of RNA base-pairing. However, this limit can be surmounted.
Figure 4.
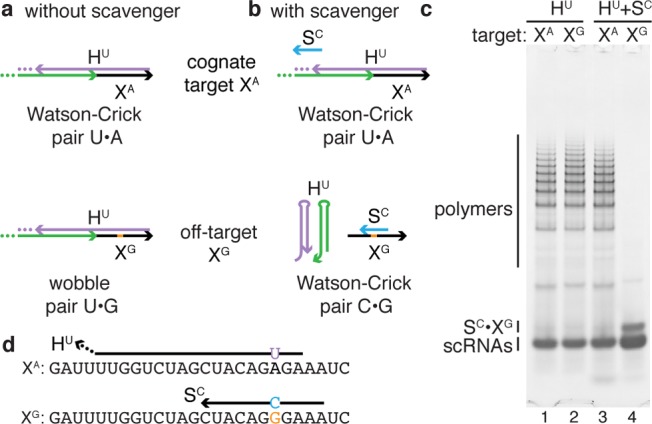
Enhancing selectivity via competitive inhibition using an unstructured scavenger strand. (a) Without scavenger, HCR system HU is not selective for cognate target XA (forming Watson–Crick pair U·A) over 1-nt sequence variant XG (forming nearly isoenergetic wobble pair U·G) (panel c: lanes 1 and 2). (b) Scavenger SC is selective for XG, restoring HU selectivity for XA via competitive inhibition (panel c: lanes 3 and 4). (c) Native PAGE poststained with SYBR Gold. Targets and scRNAs at 1 μM and scavenger at 2 μM. Reactions run for 1 h in 1× PKR at 37 °C. (d) Target sequences depicted 5′ to 3′ with nucleotide variant in orange. For cognate target XA, the input domain of the cognate detecting scRNA is depicted with the location of the output domain indicated by an ellipsis. For off-target XG, the scavenger SC is depicted. See Supporting Information Tables S2 and S3 for scavenger and scRNA sequences; see Supporting Information Section S3 for additional data.
To discriminate even the most challenging 1-nt sequence variants, we employ a short unstructured scavenger strand that selectively hybridizes to the off-target, competitively inhibiting off-target initiation of HCR to restore HCR selectivity for the cognate target. To illustrate this conceptual approach, consider again the challenging G → A mutation and HCR system HU (Figure 4ab). Scavenger SC forms a C·G base pair with off-target XG but has a mismatch with cognate target XA, establishing a discrimination energy gap for the scavenger that favors the off-target. Hence, while HU is not selective for the cognate target XA, the scavenger SC is selective for the off-target XG, restoring the selectivity of HCR via competitive inhibition (Figure 4c, lanes 3 and 4).
To explore the general utility of the scavenger concept, we tested six HCR systems designed to selectively detect the BRAF U1799A, JAK2 G1849U, and PTEN C388G mutant and wildtype sequences against all four possible sequence variants at each mutation position. These 24 case studies (one cognate target and three off-targets for each of six HCR systems) turned up six 1-nt sequence variants that challenged the selectivity of HCR cascades; in each case, HCR selectivity was restored via competitive inhibition by the appropriate scavenger (Supporting Information Section S3.2).
Surprisingly, a cocktail of four HCR systems is able to genotype any of the four possible SNPs at a given position (Figure 5a). By construction, this experiment necessitates discrimination of all possible 1-nt sequence variants, including variants that lead to nearly isoenergetic wobble pairs. How do we account for this performance given the expectation (recall Figure 4) that HU should exhibit poor selectivity for cognate target XA over off-target XG? Indeed, used in isolation, HU is spuriously initiated by off-target XG (Figure 5b, lane 2). However, using a cocktail of HU and HC (for which XG is the cognate target), spurious initiation of HU by XG is inhibited with HC playing the role of scavenger (Figure 5b, lane 3). Similar benefits are observed using a mixture of mutant- and wildtype-detecting HCR systems to genotype SBS cancer markers (Supporting Information Section S2.2). Hence, exploiting the same principle as the unstructured scavenger strand, mutual competitive inhibition between scRNAs can also meaningfully enhance selectivity for 1-nt sequence variants.
Figure 5.
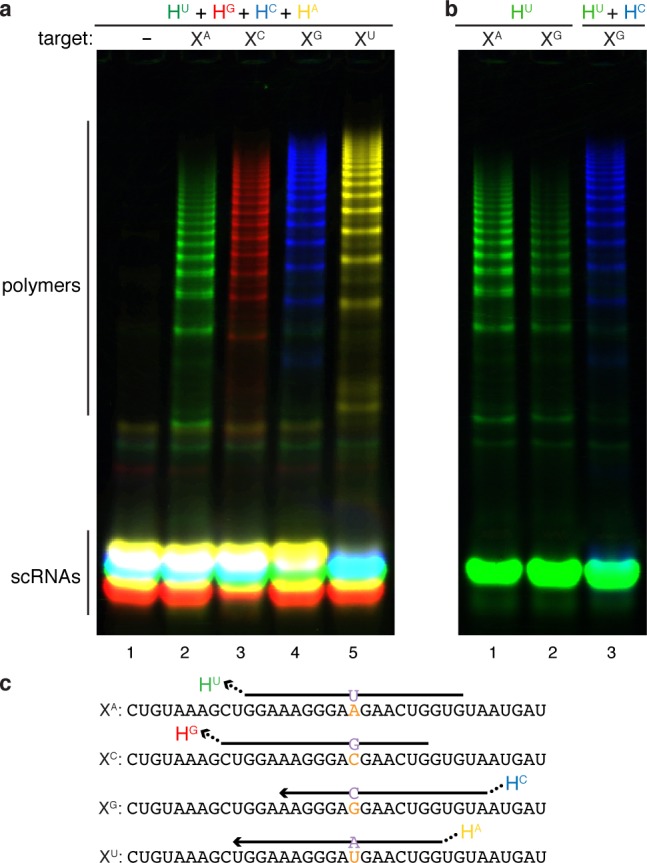
Genotyping SNPs. (a) Positive identification of any of four SNP targets (XA, XC, XG, XU) by a mixture of four HCR systems labeled with spectrally distinct fluorophores. Native PAGE: green channel, HU labeled with Alexa488; red channel, HG labeled with Cy3; blue channel, HC labeled with Cy5; yellow channel, HA labeled with Alexa750. (b) In isolation, HU is not selective for cognate target XA (forming Watson–Crick pair U·A) over off-target XG (forming nearly isoenergetic wobble pair U·G). The selectivity of HU is restored by competitive inhibition from HC, which is selective for XG. Targets at 2 μM, scRNAs for systems HU and HA at 1 μM, scRNAs for systems HC and HG at 2 μM to shift the selectivity window earlier in time. Reactions run for 1 h in 1× PKR at 37 °C. (c) Target sequences depicted 5′ to 3′ with nucleotide variants in orange. For each target, the input domain of the cognate detecting scRNA is depicted with the location of the output domain indicated by an ellipsis. See Supporting Information Table S3 for scRNA sequences and label locations; see Supporting Information Section S4 for additional data.
Recent work explored diverse design principles for performing shape and sequence transduction with scRNAs,19 demonstrating that approaches to strand nucleation, strand displacement, and motif metastability that have paced progress in the field of dynamic DNA nanotechnology are also applicable to dynamic RNA nanotechnology. The present work explores and surmounts the limits of scRNA programmability, demonstrating that scRNA hybridization cascades are sufficiently programmable to genotype RNA SBS mutations and SNPs, two classes of 1-nt sequence variants of biological significance. For the most challenging 1-nt sequence variants, competitive inhibition with an unstructured scavenger strand or with other scRNAs provides a simple and effective principle for achieving exquisite sequence selectivity. To establish a robust platform for scRNA signal transduction within living cells, considerable challenges remain to be addressed, including delivery or expression of scRNAs in biologically relevant concentrations, use of chemical modifications that prevent scRNA degradation while retaining scRNA function, and avoidance of off-pathway interactions, including with pathways that are not yet well-characterized. If these challenges can be overcome, dynamic RNA nanotechnology offers an enticing programmable framework for engineering diverse modes of conditional regulation in vivo.
Acknowledgments
We thank V. A. Beck for helpful discussions. This work was funded by the National Institutes of Health (NIH 5R01CA140759), the National Science Foundation via the Molecular Programming Project (NSF-CCF-0832824 and NSF-CCF-1317694), the Gordon and Betty Moore Foundation (GBMF2809), and the Elsa U. Pardee Foundation.
Supporting Information Available
Methods and materials, additional experimental studies. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare the following competing financial interest(s): Patents and pending patent applications..
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Bath J.; Turberfield A. J. Nat. Nanotechnol. 2007, 2, 275–284. [DOI] [PubMed] [Google Scholar]
- Zhang D. Y.; Seelig G. Nature Chem. 2011, 3, 103–113. [DOI] [PubMed] [Google Scholar]
- Liang J. C.; Bloom R. J.; Smolke C. D. Mol. Cell 2011, 43, 915–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benenson Y. Curr. Opin. Chem. Biol. 2012, 16, 278–284. [DOI] [PubMed] [Google Scholar]
- Stratton M. R. Science 2011, 331, 1553–1558. [DOI] [PubMed] [Google Scholar]
- Kurman R. J.; Shih I. M. Hum. Pathol. 2011, 42, 918–931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manolio T. A.; Brooks L. D.; Collins F. S. J. Clin. Invest. 2008, 118, 1590–1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trejo-de la O A.; Hernandez-Sancen P.; Maldonado-Bernal C. Genes Immun. 2014, 15, 199–209. [DOI] [PubMed] [Google Scholar]
- Veigas B.; Pedrosa P.; Couto I.; Viveiros M.; Baptista P. V. J. Nanobiotechnol. 2013, 11, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trembizki E.; Smith H.; Lahra M. M.; Chen M.; Donovan B.; Fairley C. K.; Guy R.; Kaldor J.; Regan D.; Ward J.; Nissen M. D.; Sloots T. P.; Whiley D. M. J. Antimicrob. Chemother. 2014, 69, 1526–1532. [DOI] [PubMed] [Google Scholar]
- Mathews D. H.; Sabina J.; Zuker M.; Turner D. H. J. Mol. Biol. 1999, 288, 911–940. [DOI] [PubMed] [Google Scholar]
- Dirks R. M.; Pierce N. A. Proc. Natl. Acad. Sci. U.S.A. 2004, 101, 15275–15278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quartin R. S.; Plewinska M.; Wetmur J. G. Biochemistry 1989, 28, 8676–8682. [DOI] [PubMed] [Google Scholar]
- Yurke B.; Turberfield A. J.; Mills A. P. Jr.; Simmel F. C.; Neumann J. L. Nature 2000, 406, 605–608. [DOI] [PubMed] [Google Scholar]
- Zhang H. Q.; Li F.; Dever B.; Li X. F.; Le X. C. Chem. Rev. 2013, 113, 2812–2841. [DOI] [PubMed] [Google Scholar]
- Wang F.; Lu C. H.; Willner I. Chem. Rev. 2014, 114, 2881–2941. [DOI] [PubMed] [Google Scholar]
- Jung C.; Ellington A. D. Acc. Chem. Res. 2014, 47, 1825–1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi H. M. T.; Chang J.; Trinh L. A.; Padilla J.; Fraser S. E.; Pierce N. A. Nat. Biotechnol. 2010, 28, 1208–1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochrein L. M.; Schwarzkopf M.; Shahgholi M.; Yin P.; Pierce N. A. J. Am. Chem. Soc. 2013, 135, 17322–17330. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.