

Abstract
Transmissible spongiform encephalopathies (TSEs) are fatal neurodegenerative disorders caused by misfolding of a cellular protein PrPC into an infectious conformation PrPSc. Previously our group demonstrated induction of PrPSc-specific antibodies with a SN6b vaccine that targets regions of the protein that are exposed upon misfolding. There are concerns that these antibodies could function as templates to promote misfolding and cause disease. To evaluate the consequences of prolonged exposure to PrPSc-specific antibodies in a prion sensitized animal, tga20 mice were vaccinated with the SN6b vaccine. No clinical signs of disease were detected up to 255 d post-vaccination, and postmortem assay of brains and spleens revealed no proteinase-K resistant PrP. These results suggest that vaccinating against TSEs with the SN6b antigen is safe from the standpoint of prion disease induction.
Keywords: disease specific epitope, immunotherapy, misfolding, prion, safety, vaccine
Introduction
Misfolding of normal cellular PrPC from a primarily α-helical structure to a β-sheet rich conformer is associated with invariably fatal neurodegenerative transmissible spongiform encephalopathies (TSEs).1 Normal cellular PrPC is most highly expressed in the central nervous system (CNS), and transmission can occur via ingestion of contaminated food,2 tissue transplants, contaminated surgical instruments, and blood transfusions.3 Progression varies among species, but β-sheet rich Proteinase-K (PK) resistant PrPSc is generally first detectable in the spleen and lymphatic system before the obex and cerebellum of the brain.4 TSEs of various strains have been observed in humans5 and an increasing number of domesticated6 and wild animals.7 Transmission from cattle to humans through consumption of contaminated meat products in Britain in the late 1980s fueled tight regulations prohibiting the inclusion of ruminant CNS tissues in feed and meat products. These prevention strategies appear to be effective because the incidence of bovine spongiform encephalopathy (BSE) has drastically declined. However, epidemiologists are concerned that long incubation periods, that vary depending on susceptibility, could mean that additional waves of human infection may still be observed.8 Further alarm has come from the spread of the TSE chronic wasting disease (CWD), in wild and captive cervid populations, particularly in the central United States and Canada.9 While transmission of CWD is evidently very efficient in the wild, transmission to humans is unlikely due to species barriers, although no conclusive experiments have been done to rule out direct transmission from cervids to humans.10 Feed restrictions and quarantine or culling of infected herds are the only control strategies currently available, and these have little influence on the spread of CWD in wild populations, or their potential to contaminate areas shared with domestic herds.
An effective vaccine for TSEs could provide a practical method to curb the spread of these diseases, but this strategy poses several challenges.11 The most obvious challenges are inducing strong immune responses against a self-protein while avoiding potential side-effects of auto-immune reactions. Therapeutic targeting of PrPC—a virtually ubiquitous, cell surface protein—could have adverse consequences. Circulating antibodies against PrPC may trigger complement-dependent lysis of cells or induce autoimmune disease by breaking tolerance to this molecule. Alternatively, antibodies against PrPC may impair or alter the function of this normal cellular protein by triggering apoptosis in neurons,12 superoxide mediated cytotoxicity,13 or activation of inappropriate cell signaling cascades.14-17 Specificity of the antibodies for the PrPSc isoform provides a way to preferentially target the removal of the pathological species of the protein, while minimizing auto-immune side-effects that might be associated with targeting a self protein. To address this challenge, we identified a region of the protein that is specifically exposed upon misfolding (disease-specific epitopes) for the induction of antibody responses that specifically bind the misfolded conformer PrPSc,18 and translated this epitope into a vaccine that generates IgG antibodies in a number of species (sheep, mice, cattle, elk, and deer) that maintain specificity for PrPSc.19
One concern that arises with the disease-specific approach is that antibodies that bind PrPSc may induce misfolding of PrPC to a pathological conformation. We have previously attempted immunological mimicry of PrPC-PrPSc interactions to induce misfolding of PrPC to PrPSc.20 While antibodies that bind to putative binding and conversion domains were able to enhance exposure of YYR motifs of PrP, the antibodies were unable to generate proteinase K resistant PrPSc or recruiting activity in vitro.20 These observations agree with relatively stringent constraints on successful template-directed misfolding across species in vivo. However, serial PMCA assays or the products of these assays are able to more readily surmount species barriers,21,22 and different prion strains vary in their cross-species transmissibility,23 suggesting that the minimum requirements for template directed misfolding can be less stringent. Furthermore, the recent finding that lymphoid tissues are more permissive to replication of CWD prions than brain,24 suggests that the safety of any immunotherapy approach should be carefully considered. Using a single molecule binding technique and immunoprecipitations, we recently discovered that PrPSc-specific antibodies generated against the expanded YYR epitope (SN6b) bind to the bovine equivalent of human T183A prion protein associated with a rare form of early onset familial dementia.25 Thus, the PrPSc-specific polyclonal antibodies generated in response to the vaccine can recognize subtle conformational differences and may interact with partially folded PrP. While such interactions are unlikely to elicit a misfolding event, the process of template-directed misfolding remains poorly understood, and any therapeutic approach that involves binding of PrP and potential perturbation of its structure, should be carefully tested for safety in vivo.
The expanded YYR epitope comprises a 14-mer (QVYYRPVDQYSNQN) (Fig. 1) that is identical in bovine, ovine, and murine PrP. Here, we vaccinated homozygous tga20 mice with a standard immunization schedule designed to generate high-titer PrPSc-specific responses.19 The animals were monitored until they reached 311 d of age (255 d after immunization). These vaccinated, prion disease-sensitized mice did not show any abnormal behavior, and the animals remained healthy. We further tested their brains and spleens for PK-resistant PrPSc, and were unable to detect any increases in PK-resistance compared with age-matched control tga20 mice. These results suggest that the vaccination strategy using this disease-specific epitope is safe in the context of an aggressive in vivo model of TSE.

Figure 1. (A) Cartoon representation of the X-ray crystal structure of ovine PrPC 128–233 (pdb 1UW3) with YYR in red and expanded SN6b epitope sequence highlighted in yellow (B) multiple sequence alignment of ovine, bovine, and murine PrP sequences (138–192, based on ovine sequence) with SN6b sequence highlighted as in panel A.
Results
To test the safety of vaccinating against the pathological conformation of PrPSc, tga20 homozygous mice (n = 6) were vaccinated with a standard three-dose schedule (3 vaccinations at 3 wk intervals) of the SN6b vaccine antigen formulation. We previously showed that this antigen and vaccine formulation can induce PrPSc-specific IgG responses in sheep.19 Vaccination of tga20 mice elicited significant serum antibody titers against the SN6b antigen within 3 weeks after the first vaccination, and this response was further elevated after the second vaccination. For all mice, strong and consistent peptide-specific responses were observed, typically peaking at titers of ~4000 and maintaining significant titers for the duration of the trial up to 255 d post-vaccination (36 wk) (Table 1). Each mouse was monitored daily, and no clinical signs of disease were observed throughout the course of the experiment.
Table 1. SN6b-specific antibody titers for tga20 mice vaccinated at 0, 3, and 6 weeks with 10 μg of SN6b vaccine antigen (n = 6).
| Week | |||||
|---|---|---|---|---|---|
| Mouse | 0 | 3 | 6 | 10 | 36 |
| A | 1 | 4907 | 4479 | 2641 | 925 |
| B | 1 | 1059 | 3723 | 3227 | 483 |
| C | 1 | 2372 | 3780 | 3519 | 215 |
| D | 1 | 3489 | 10586 | 3982 | 775 |
| E | 1 | 2475 | 3759 | 3806 | 132 |
| F | 1 | 872 | 3907 | 3603 | 407 |
ELISA titers are expressed as the reciprocal of the highest serum dilution resulting in a reading exceeding 2 standard deviations above the negative control (pre-immune).
To determine if any biochemical indications of pre-clinical disease could be detected despite the lack of symptoms, brains and spleens of all mice were collected 255 d after receiving their first vaccination when the mice were 311 d old. Homogenates were digested with increasing concentrations of PK, followed by western blotting to detect remaining PrP in comparison to age-matched tga20 controls, younger tga20 mice, and infected tga20 mice (Figs. 2 and 3).

Figure 2. PK digests and western blots of combined brain homogenates from 6 SN6b-vaccinated, 4 age-matched, and 6 young tga20 mice. PK digests of an IP-infected (100 μl of 1% RML prion) mouse brain are shown for comparison. Error bars indicate mean and standard deviation of band intensities relative to samples not treated with PK for separate individual animal blots (not all blots shown).
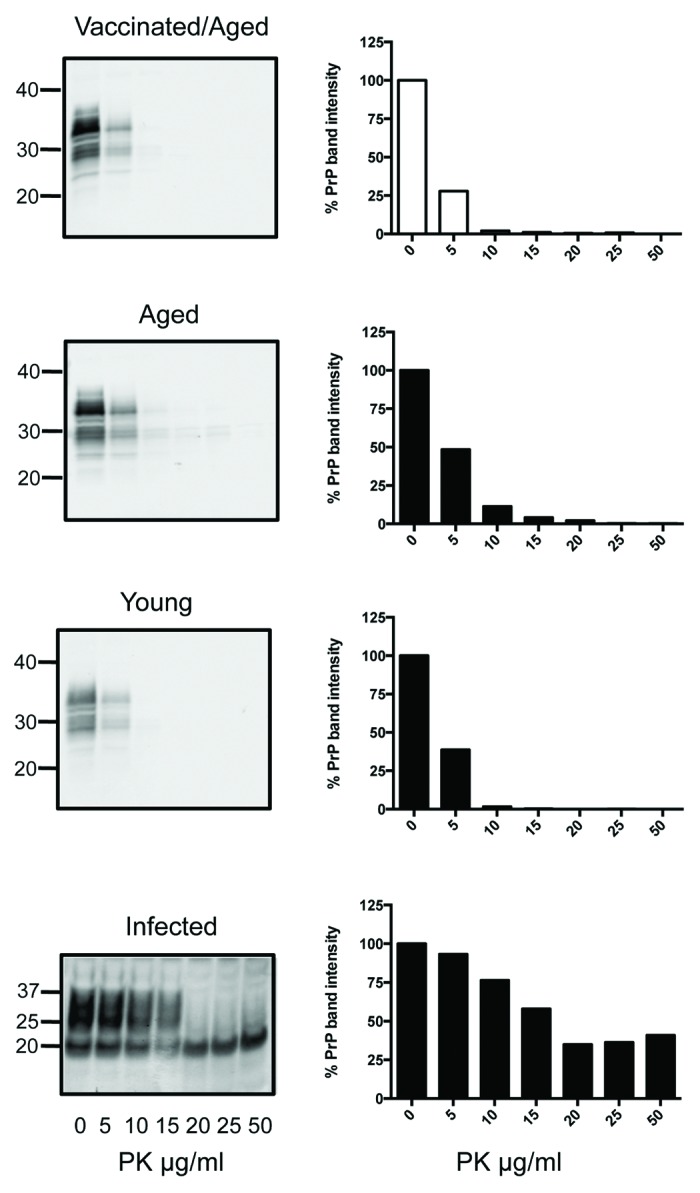
Figure 3. PK digests and western blots of combined spleen homogenates from 6 SN6b-vaccinated, 4 age-matched, and 6 young tga20 mice. PK digests of the spleen from one IP infected (100 μl of 1% RML prion) tga20 mouse are shown for comparison (bottom panel).
No significant accumulation of PrPSc could be detected in the vaccinated tga20 brains compared with the age-matched or young tga20 mice (Holm-Sidak, α = 5%). (Fig. 2, left panel). To determine if a single vaccinated animal accumulated PrPSc in the brain that was difficult to detect in the combined homogenate digests, individual brain homogenates were digested separately. The PrP detected after treatment of vaccinated mouse brains with increasing concentrations of PK for the individual brain homogenate digests was not significantly different from the age-matched, unvaccinated controls, or young tga20 mouse brains (individual blots not shown) (Mann-Whitney test, P > 0.2 in all cases). The mean PrP band intensities for all 6 animals are plotted as % compared with PrP from the same brain homogenates not exposed to PK with error bars indicating standard deviation from the mean (Fig. 2, right panel). Positive control RML infected tga20 mice developed symptoms of scrapie at 100 ± 4 d and were sacrificed 3–28 d after onset of symptoms. The RML-infected tga20 mice showed significant accumulation of PK resistant PrPSc in their brains in comparison to the SN6b vaccinated mice, apparent when digested with 20, 25, and 50 ug/ml of PK (Holm-Sidak, P < 0.0005) (Fig. 2, bottom panel).
PK resistant PrPSc is often detectable in lymphoid tissues before accumulation in the brain, and can also be detected prior to onset of symptoms.26 To determine if there was any accumulation of PrPSc in the spleens of the vaccinated mice, despite lack of symptoms or detectable PrPSc in the brains, PK titrations of combined spleen homogenates were performed, again compared with age-matched and young controls (Fig. 3). The digests revealed that sensitivity of PrP to PK digest was similar in the spleens of the SN6b-vaccinated mice, age-matched controls, and young tga20 mice (Fig. 3) (Mann-Whitney, P = 0.5594). Positive control RML-infected tga20 mice showed significant accumulation of PK resistant PrPSc in their spleens in comparison to the vaccinated tga20 mice (Mann-Whitney, P = 0.0181) (Fig. 3, bottom panel).
Discussion
Vaccination of tga20 mice with the SN6b vaccine, that generates antibodies specific for PrPSc, did not cause any clinical or biochemical signs of disease in PrP overexpressing tga20 mice. Homozygous tga20 mice carry 60 copies of the PRNP gene and express ~10 fold more PrP than wild-type mice and are thus more susceptible to development of prion disease.27 Tga20/tga20 mice develop prion disease within 60 ± 2 d and succumb to disease at 62 ± 4 d when inoculated intracranially with 30 μl of 1% brain from terminally sick mice inoculated with mouse-adapted (RML) prions.27 Wild-type mice take greater than twice as long to develop the disease under the same conditions (131 ± 9), and the disease is terminal at 166 ± 8 d.27 Our positive control tga20 mice, inoculated intra-peritoneally, developed symptoms ~100 d after infection. We aged the vaccinated tga20/tga20 mice 2.5 × longer (to 255 d after vaccination), and observed no signs of disease or significant increases in PK resistant protein in their brains or spleens by immunoblot analysis. We cannot rule out the presence of minute quantities of PrPSc given the limits of immunoblot detection, and future studies will utilize PMCA or other sensitive methods to detect PrPSc with greater sensitivity. However, the lack of any clinical symptoms suggests that no pathological prion protein was generated in the vaccinated mice (whether PK resistant or not).
Given the barriers known to exist for prion infection from one species to another,6 and the difficult task of inducing misfolding using antibodies that bind putative binding and conversion domains,20 it is unlikely that binding of antibodies to a short peptide epitope such as SN6b buried in natively folded PrP could elicit misfolding. However, the finding that antibodies for binding and conversion domains can expose, at least transiently, epitopes that are cryptic in PrPC 20, and given the mystery that still surrounds template-directed misfolding, it seems appropriate to be cautious and carefully address potential risks. A recent extensive study found that monoclonal antibodies binding to the globular domain of PrPC elicit cytotoxic signaling mediated by the N-terminal octapeptide repeat region.13 The antibody that caused the most dramatic neuronal loss ex vivo and when injected into the brains of tga20 mice was one that binds a region of native PrPC comprising residues 138–147 and 204, 208, and 212.13 The SN6b antigenic region we targeted is distinct from this sequence (159–174, by the same numbering), comprising a region that is buried in PrPC. These recent results highlight the importance of directing antibodies specifically against misfolded PrPSc, although other studies have not reported such adverse responses to prion immunotherapies.28 Here we observed no signs of clinical disease in tga20 mice vaccinated with the SN6b vaccine, suggesting that this vaccine is safe in a context that heavily favors disease. Similarly, sheep that received high doses of the SN6b vaccine (5 doses of 150 μg/animal, n = 10) and were first vaccinated almost 2 y ago, have not shown any signs of TSE (data not shown). In addition, elk vaccinated with the corresponding cervidized SN6b epitope (3 doses of 25, 50, 100, or 200 μg, n = 35) did not develop disease and postmortem analysis of obex tissues 1 y after vaccination revealed no detectable PrPSc using the BioRad ELISA method (data not shown).
The ability of the antibodies generated in response to the vaccine to discriminate subtle structural differences between mutant and wild-type proteins,25 suggests that care may be necessary in applying this vaccination approach to disease susceptible individuals. While tga20 mice expressing wild-type PrP do not show any clinical signs of disease, would mice overexpressing a disease-specific mutant respond differently? From a more general perspective, in vivo studies of vaccines like SN6b that generate antibodies that bind disease-susceptible PrP-mutants could be applied to tg mice expressing these mutants, to determine if antibody binding inhibits or enhances onset of disease. Such studies could be used to selectively perturb different aspects of PrP structure independent of infecting with an exogenous source of PrPSc, and might provide insight into the most prevalent forms of prion diseases that arise spontaneously.
Materials and Methods
Vaccine formulation and delivery
Antigens used in the vaccine were constructed and expressed fused to leukotoxin as previously described.19 Briefly, the antigenic sequence (QVYYRPVDQYSNQN) of ovine PrP was presented as (forward-back-back) × 4 fused to the C-terminus of leukotoxin. The vaccine was formulated in Emulsigen D, which contains the immunostimulant dimethyl dioctadecyl ammonium bromide. Six homozygous female tga20/tga20 mice (Strain: B6; 129S7-Prnptm1CweTg[Prnp]a20CweCnrm), that carry multiple copies of the PRNP gene and express ~10-fold more PrP than wild-type mice, were immunized at 8 weeks of age with 10 μg of antigenic protein, and given the equivalent dose at 3 week intervals for a total of 3 doses. Negative control age-matched female tga20 mice (n = 4) were aged to 311 d without vaccination, and young tga20 homozygous mice (n = 6), 3 male, 3 female were aged to 150 d. Positive control female mice (3 male, 3 female) were infected intra-peritoneally with 1% RML scrapie prions in a total volume of 100 μl of PBS.
Sample collection
Blood was collected and serum isolated from each mouse prior to vaccination, at 3-week intervals during the vaccination schedule, and just prior to euthanasia. Health of the mice was monitored daily by VIDO animal care staff. Brains and spleens were collected 255 d after the first vaccination when mice were 311 d old, and flash frozen in liquid nitrogen and stored at −80 °C.
ELISAs
Epitope-specific antibody responses were quantified by ELISA. 96-well polystyrene microtiter plates (Immulon 2HB, Dynatech, 227) were coated overnight with 0.5 μg per well of synthetic peptide antigen19 solubilized in coating buffer (1:3 of 0.05M Na2CO3, 0.05M NaHCO3, pH 9.6). Plates were washed 6 times in ddH20, blocked for 1 h with 1% skim milk powder (SMP) in Tris-Buffered Saline with 0.05% Tween-20 (TBST) and washed as above in preparation for serum addition. Serum samples were initially diluted 1:100 and serial diluted 4 fold in 1% SMP TBST.19 Serially diluted serum samples were incubated with SN6b peptide for 1 h at room temperature, washed 6 times with ddH20, and antigen-antibody complexes were probed with alkaline phosphatase-conjugated goat anti-mouse IgG (H+L) antibody (Kirkegaard and Perry Laboratories, 0751-1803) diluted 1:4000 for 1 h. Plates were washed as above and developed using ρ-nitrophenyl phosphate (PNPP), and scanned at 595 nm using a Model 3550 Microplate Reader (BioRad) following approximately 30 min incubation at room temperature. ELISA titers are expressed as the reciprocal of the highest serum dilution resulting in a reading exceeding two standard deviations above the negative control (pre-immune).
Proteinase K digest titrations
Brains were thawed on ice and homogenized to 10% in ice-cold brain lysis buffer (100 mM NaCl, 10 mM EDTA, 0.5% Nonidet P-40, 0.5% sodium deoxycholate, 10 mM Tris-HCl, pH 7.5), as previously described.20 Spleens were homogenized in 20 mM Tris, pH 7.5, 300 mM NaCl, 2% w/v Na-deoxycholate, 2% v/v Igepal, and DNaseI (150 KU/ml final concentration) by bead beating with a Mini-beadbeaterTM (Biospec Products, 3110 BX), and centrifuged at 16 000 × g for 15 min at 4 °C to remove cellular debris. Supernatant protein concentrations were determined using the Bradford method. Individual or combined homogenates were digested with Proteinase K (Sigma, P6556) at final concentrations of 0, 5, 10, 15, 20, 25, 50 μg/ml for 1 h at 37 °C. Digests were stopped by adding PMSF to a final concentration of 2 mM to all samples. Each homogenate sample was analyzed by SDS-PAGE (50 μg/lane) and western blotting to detect remaining PrP after digests. Primary antibody 6H4 (Prionics, 01-010) and secondary antibody AP-conjugated goat anti mouse IgG (H+L) (Kirkegaard and Perry Laboratories, 0751-1803) were used to detect brain PrP, while spleen PrP was detected with the primary antibody 6D11 (Santa Cruz, sc-58581) and secondary antibody Thermo-Fisher AP-conjugated Clean-Blot™ IP Detection Reagent, 21233. AP conjugated antibodies were reacted with SIGMAFAST-BCIP/NBT substrate (Sigma, B5655) for 15–60 min, air-dried and scanned with an HP Scanjet G4050 at 600 dpi (grayscale). Percent PrP band intensities for PK digests compared with the undigested control were determined using Image J 1.40 g (http://imagej.nih.gov/ij/).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
Published with permission of the Director of VIDO as journal series number 677. This research was supported by the Alberta Prion Research Institute (APRI), PrioNET Canada, and the Pan-Provincial Vaccine Enterprise (PREVENT). The authors would like to thank VIDO animal care for immunizations, veterinary care, and sample collection.
References
- 1.Pan KM, Baldwin M, Nguyen J, Gasset M, Serban A, Groth D, Mehlhorn I, Huang Z, Fletterick RJ, Cohen FE, et al. Conversion of alpha-helices into beta-sheets features in the formation of the scrapie prion proteins. Proc Natl Acad Sci U S A. 1993;90:10962–6. doi: 10.1073/pnas.90.23.10962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ward HJ, Everington D, Cousens SN, Smith-Bathgate B, Leitch M, Cooper S, Heath C, Knight RS, Smith PG, Will RG. Risk factors for variant Creutzfeldt-Jakob disease: a case-control study. Ann Neurol. 2006;59:111–20. doi: 10.1002/ana.20708. [DOI] [PubMed] [Google Scholar]
- 3.Hamaguchi T, Noguchi-Shinohara M, Nozaki I, Nakamura Y, Sato T, Kitamoto T, Mizusawa H, Yamada M. The risk of iatrogenic Creutzfeldt-Jakob disease through medical and surgical procedures. Neuropathology. 2009;29:625–31. doi: 10.1111/j.1440-1789.2009.01023.x. [DOI] [PubMed] [Google Scholar]
- 4.Daude N. Prion diseases and the spleen. Viral Immunol. 2004;17:334–49. doi: 10.1089/vim.2004.17.334. [DOI] [PubMed] [Google Scholar]
- 5.Kretzschmar H, Tatzelt J. Prion disease: a tale of folds and strains. Brain Pathol. 2013;23:321–32. doi: 10.1111/bpa.12045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Béringue V, Vilotte JL, Laude H. Prion agent diversity and species barrier. Vet Res. 2008;39:47. doi: 10.1051/vetres:2008024. [DOI] [PubMed] [Google Scholar]
- 7.Williams ES, Miller MW. Transmissible spongiform encephalopathies in non-domestic animals: origin, transmission and risk factors. Rev Sci Tech. 2003;22:145–56. doi: 10.20506/rst.22.1.1385. [DOI] [PubMed] [Google Scholar]
- 8.Holmes D. vCJD remains an enigma as UK deaths fall to zero for 2012. Lancet Infect Dis. 2012;12:914–5. doi: 10.1016/S1473-3099(12)70304-3. [DOI] [PubMed] [Google Scholar]
- 9.Saunders SE, Bartelt-Hunt SL, Bartz JC. Occurrence, transmission, and zoonotic potential of chronic wasting disease. Emerg Infect Dis. 2012;18:369–76. doi: 10.3201/eid1803.110685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gilch S, Chitoor N, Taguchi Y, Stuart M, Jewell JE, Schätzl HM. Chronic wasting disease. Top Curr Chem. 2011;305:51–77. doi: 10.1007/128_2011_159. [DOI] [PubMed] [Google Scholar]
- 11.Li L, Napper S, Cashman NR. Immunotherapy for prion diseases: opportunities and obstacles. Immunotherapy. 2010;2:269–82. doi: 10.2217/imt.10.3. [DOI] [PubMed] [Google Scholar]
- 12.White AR, Collins SJ, Maher F, Jobling MF, Stewart LR, Thyer JM, Beyreuther K, Masters CL, Cappai R. Prion protein-deficient neurons reveal lower glutathione reductase activity and increased susceptibility to hydrogen peroxide toxicity. Am J Pathol. 1999;155:1723–30. doi: 10.1016/S0002-9440(10)65487-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sonati T, Reimann RR, Falsig J, Baral PK, O’Connor T, Hornemann S, Yaganoglu S, Li B, Herrmann US, Wieland B, et al. The toxicity of antiprion antibodies is mediated by the flexible tail of the prion protein. Nature. 2013;501:102–6. doi: 10.1038/nature12402. [DOI] [PubMed] [Google Scholar]
- 14.Cashman NR, Loertscher R, Nalbantoglu J, Shaw I, Kascsak RJ, Bolton DC, Bendheim PE. Cellular isoform of the scrapie agent protein participates in lymphocyte activation. Cell. 1990;61:185–92. doi: 10.1016/0092-8674(90)90225-4. [DOI] [PubMed] [Google Scholar]
- 15.Mouillet-Richard S, Ermonval M, Chebassier C, Laplanche JL, Lehmann S, Launay JM, Kellermann O. Signal transduction through prion protein. Science. 2000;289:1925–8. doi: 10.1126/science.289.5486.1925. [DOI] [PubMed] [Google Scholar]
- 16.Schneider B, Pietri M, Pradines E, Loubet D, Launay JM, Kellermann O, Mouillet-Richard S. Understanding the neurospecificity of Prion protein signaling. Front Biosci (Landmark Ed) 2011;16:169–86. doi: 10.2741/3682. [DOI] [PubMed] [Google Scholar]
- 17.Arsenault RJ, Li Y, Potter A, Griebel PJ, Kusalik A, Napper S. Induction of ligand-specific PrP (C) signaling in human neuronal cells. Prion. 2012;6:477–88. doi: 10.4161/pri.21914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Paramithiotis E, Pinard M, Lawton T, LaBoissiere S, Leathers VL, Zou WQ, Estey LA, Lamontagne J, Lehto MT, Kondejewski LH, et al. A prion protein epitope selective for the pathologically misfolded conformation. Nat Med. 2003;9:893–9. doi: 10.1038/nm883. [DOI] [PubMed] [Google Scholar]
- 19.Hedlin PD, Cashman NR, Li L, Gupta J, Babiuk LA, Potter AA, Griebel P, Napper S. Design and delivery of a cryptic PrP(C) epitope for induction of PrP(Sc)-specific antibody responses. Vaccine. 2010;28:981–8. doi: 10.1016/j.vaccine.2009.10.134. [DOI] [PubMed] [Google Scholar]
- 20.Li L, Guest W, Huang A, Plotkin SS, Cashman NR. Immunological mimicry of PrPC-PrPSc interactions: antibody-induced PrP misfolding. Protein Eng Des Sel. 2009;22:523–9. doi: 10.1093/protein/gzp038. [DOI] [PubMed] [Google Scholar]
- 21.Kurt TD, Seelig DM, Schneider JR, Johnson CJ, Telling GC, Heisey DM, Hoover EA. Alteration of the chronic wasting disease species barrier by in vitro prion amplification. J Virol. 2011;85:8528–37. doi: 10.1128/JVI.00809-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Barria MA, Telling GC, Gambetti P, Mastrianni JA, Soto C. Generation of a new form of human PrP(Sc) in vitro by interspecies transmission from cervid prions. J Biol Chem. 2011;286:7490–5. doi: 10.1074/jbc.M110.198465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Legname G, Nguyen HO, Peretz D, Cohen FE, DeArmond SJ, Prusiner SB. Continuum of prion protein structures enciphers a multitude of prion isolate-specified phenotypes. Proc Natl Acad Sci U S A. 2006;103:19105–10. doi: 10.1073/pnas.0608970103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Béringue V, Herzog L, Jaumain E, Reine F, Sibille P, Le Dur A, Vilotte JL, Laude H. Facilitated cross-species transmission of prions in extraneural tissue. Science. 2012;335:472–5. doi: 10.1126/science.1215659. [DOI] [PubMed] [Google Scholar]
- 25.Madampage CA, Maattanen P, Marciniuk K, Brownlie R, Andrievskaia O, Potter A, Cashman N, Lee JS, Napper S. Binding of bovine T194A PrP (C) by PrP (Sc) -specific antibodies: Potential implications for immunotherapy of familial prion diseases. Prion. 2013;7:301–11. doi: 10.4161/pri.25148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Race RE, Ernst D. Detection of proteinase K-resistant prion protein and infectivity in mouse spleen by 2 weeks after scrapie agent inoculation. J Gen Virol. 1992;73:3319–23. doi: 10.1099/0022-1317-73-12-3319. [DOI] [PubMed] [Google Scholar]
- 27.Fischer M, Rülicke T, Raeber A, Sailer A, Moser M, Oesch B, Brandner S, Aguzzi A, Weissmann C. Prion protein (PrP) with amino-proximal deletions restoring susceptibility of PrP knockout mice to scrapie. EMBO J. 1996;15:1255–64. [PMC free article] [PubMed] [Google Scholar]
- 28.Roettger Y, Du Y, Bacher M, Zerr I, Dodel R, Bach JP. Immunotherapy in prion disease. Nat Rev Neurol. 2013;9:98–105. doi: 10.1038/nrneurol.2012.258. [DOI] [PubMed] [Google Scholar]