

Abstract
Background
Gut barrier failure has been implicated in the progression from single organ injury to multiple organ failure. The unstirred mucus layer is a major component of the physiological gut barrier, its role in acute pancreatitis(AP) is not clearly defined.
Study Design
Rats underwent biliopancreatic duct ligation-induced AP; two controls were used: biliopancreatic duct ligation with drainage and sham duct ligation. After 4.5 hours, serum and ascitic amylase activity was measured. Mucus was analyzed for reactive nitrogen intermediates(RNI)-mediated damage, reactive oxygen species(ROS)-induced damage, and total antioxidant capacity. Mucus coverage and villous injury was assessed histologically. Ileum permeability was measured by diffusion of a fluorescent dextran probe. Histology and morphology of the mucus layer were validated in a mouse AP model (intraductal taurocholate plus caerulein).
Results
Biliopancreatic duct ligation increased serum α-amylase, ascitic volume, and ascitic α-amylase. Intestinal permeability was increased, which was associated with loss of the unstirred mucus layer but not villous injury. These changes correlated with increased ROS-and-RNI-mediated mucus damage as well as decreased mucus total antioxidant capacity but were not present in the two control groups. Using a different model of AP in mice, the finding of mucus layer disruption was recapitulated at 6 hours after AP, but by 24 hours, rebound hypersecretion of inspissated mucus was seen.
Conclusions
These results support the hypothesis that damage to the unstirred mucus layer with evidence of oxidative stress occurs during AP-induced gut barrier failure.
Introduction
The mortality rate of severe acute pancreatitis remains high and is related to the subsequent development of acute respiratory failure, sepsis, and/or the multiple organ failure syndrome (MODS) (1). Studies in both animal models and humans have documented that gut permeability is increased shortly after the onset of pancreatitis (2,3) and clinically the magnitude of gut barrier failure has been shown to correlate with the development of sepsis, MODS and an increased risk of death (4,5).
Because of the correlation between pancreatitis-induced gut injury and subsequent increases in morbidity and mortality, a number of prospective, randomized controlled clinical trials have tested the efficacy of various gut-protective strategies in patients with acute pancreatitis. Strategies tested to decrease pancreatitis-induced morbidity include the use of early enteral nutrition, probiotics, and selective antibiotic digestive decontamination (SDD) (6,7). While the use of probiotics in acute pancreatitis remains controversial (8), early enteral nutrition and SDD have been shown to be clinically beneficial. Several studies show a reduction in the incidence of sepsis and organ failure; some studies show a survival advantage (9). However, additional progress in the generation of novel gut-protective clinical strategies requires a more thorough understanding of the mechanisms underlying the pathogenesis of pancreatitis-induced gut injury.
Currently, it is well accepted that acute pancreatitis-induced gut injury involves a splanchnic ischemia-reperfusion (I/R) injury. This injury is largely related to systemic hypovolemia (3), and an increased gut inflammatory response related to an overwhelming systemic inflammatory response (4). While previous studies investigating the mechanisms of pancreatitis-induced gut injury have focused on systemic factors and injury to the enterocytes lining the intestinal villi, the mucus layer overlying these enterocytes has not been specifically studied. Yet, the loss of this mucus layer has been found to be a major factor contributing to increases in gut permeability and intestinal injury in trauma-hemorrhagic shock (10, 11). This mucus layer consists of two distinct layers; a loosely adherent outer layer, which is continuously being shed into the gut lumen and an adherent layer which is tightly bound to the underlying enterocytes. These two layers are also commonly referred to as the unstirred mucus layer.
The importance of the mucus layer as a barrier to luminal contents, including bacteria, their products, and various digestive enzymes, relates to properties of mucin. Mice engineered to lack MUC2, a major component of the mucin layer, (12) and mice with a mis-sense mutation in MUC2, develop chronic colitis (13). Mucins restrict the ability of intraluminal, water soluble factors and bacteria to come in direct contact with the underlying enterocytes (14). This barrier property of mucus was documented in pharmacologic studies showing the major barrier to the transport of a compound across the gut wall was related to the mucus layer, not the tight junctions between enterocytes or the lipid membrane of the underlying enterocytes (14-17). This concept of the major protective and barrier roles played by the intestinal mucus layer was convincingly shown in a recent proof-of-principle study of the normal intestine, in which removal of the mucus layer was associated with increased gut permeability (18).
During moderate ischaemia-reperfusion injury of the colon in human patients undergoing colonic surgery, and in an experimental model of colonic ischaemia-reperfusion in rats, ischemia (60 minutes) was associated with mucus layer disruption with a rebound goblet cell hypersecretory phase on reperfusion (19). Because splanchnic hypoperfusion during AP is profound and sustained, we sought to better understand this effect on the unstirred mucus layer and loss of gut barrier function during early AP, and whether a rebound hypersecretion occurred in the delayed phase. Thus, in this study, we tested the hypothesis that experimental AP in rats causes a free radical-mediated injury to the mucus layer associated with increased gut permeability.
Materials and Methods
Animals
Male Sprague Dawley rats weighing 320 to 400 grams were housed under barrier-sustained conditions, at a temperature of 25 °C, with 12-h light/dark cycles and acclimated for at least five days before experimentation. The rats had free access to water and chow (Teklan 22/5 Rodent Diet W-8640; Harlan Teklad; Madison, WI). All rats were maintained in accordance with the recommendations of the Guide for the Care and Use of Laboratory Animals. All animal protocols were approved by the New Jersey Medical School Animal Care Committee.
Mice were male wild-type C57BL6 from the breeding colony at the University of Edinburgh, housed under standard conditions with free access to standard chow and water. Mouse experiments were conducted with institutional ethical approval under the Use of Animals (Scientific Procedures) Act 1984.
Experimental Design
The major goal of this study was to test the hypothesis that the intestinal mucus layer is rapidly lost shortly after the onset of acute pancreatitis and that this mucus loss is associated with increased gut permeability. Additionally, we sought to characterize the chemical changes in the mucus following pancreatitis, since reactive oxygen and nitrogen species are capable of damaging and degrading mucin molecules and thereby reducing mucus viscosity, hydrophobicity, and barrier function (20,21). Three groups of rats were studied: one experimental group and two controls (Figure 1A). In our experimental AP group, pancreatitis was induced by biliopancreatic duct ligation (AP) without proximal diversion. In our first control group (PDD), rats underwent biliopancreatic duct ligation but also underwent proximal diversion of the common biliary/pancreatic secretions. In our second control group (SP), rats underwent identification of the biliopancreatitc duct, but the duct was not ligated. AP was assessed by measurement of serum α-amylase level, volume of ascitic fluid, and ascitic fluid α-amylase level. All groups of rats were sacrificed and sampled 4.5 hours following the ligation or sham ligation of the pancreatic duct. This time point was selected based on data demonstrating that structural and functional pancreatic changes, as well as α-amylase levels, had not yet peaked thereby allowing us to correlate mucus and gut permeability changes with the onset of early acute pancreatitis (22,23). Using these three groups of rats, two sets of experiments were conducted. In the first set of studies, we measured the extent of free radical induced stress to the mucus layer. 18 rats were used in this first set: 6 AP, 6 SP, and 6 PDD. Mucus oxidant stress was assessed by measuring carbonyl derivatives and nitrated tyrosine residues in the mucus, as these are markers of reactive oxygen and reactive nitrogen reactions respectively. One important physiologic role of mucus is to protect against intraluminally-generated oxidants. Thus, we measured mucus total antioxidant capacity, which is a functional assay that reflects the ability of mucus to neutralize reactant oxidant species. The second set of experimental animals, which were not subjected to the intestinal manipulation required for mucus collection, had gut injury assessed morphologically and functionally. An additional 18 rats were used in this second set: 6 AP, 6 SP, and 6 PDD. Morphological assessment included evidence of injury to the mucus layer and villous injury while functional assessment of the gut barrier involved permeability measurements using a fluorescently-labeled dextran permeability probe. A total of 36 rats were used in this set experiments.
Figure 1.
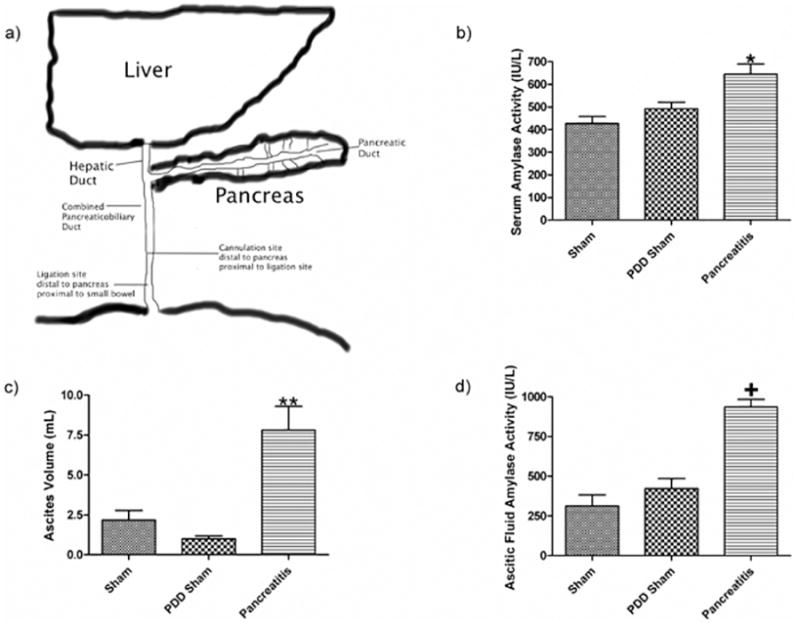
a) Schematic representation of the rodent's pancreatic and biliary systems where pancreatic duct drains directly into the biliary ductal system. Pancreatic duct ligation is performed just distal to site where the pancreatic duct enters the common bile duct and the biliary drainage catheter, when employed, is placed proximal to site of ligation. Pancreatic duct ligation causes acute pancreatitis as evidenced by b) an increase in serum amylase activity, c) an increase in ascitic fluid volume and d) an increase in the amylase activity of the ascitic fluid. Data expressed as mean ± SEM with n=6 animals per group; * p<0.05 vs. other groups, ** p<0.01 vs. other groups, and + p<0.001 vs. other groups.
Because we wished to explore the mucus barrier and any associated injury in in the delayed phase of AP, and also to validate the findings in a model of experimental AP without duct occlusion, we tested a second model of AP in mice. Four groups of mice were studied: 1) experimental AP induced by retrograde biliopancreatic duct injection of 5% sodium taurocholate with simultaneous intraperitoneal deposition of caerulein, sacrificed at 6 hours (n=6); 2) experimental AP induced by retrograde biliopancreatic duct injection of 5% sodium taurocholate with simultaneous intraperitoneal deposition of cerulein with mice sacrificed at 24 hours (n=7); 3) sham laparotomy with sacrifice at 6 hours (n=6); 4) sham laparotomy with sacrifice at 24 hours (n=6). Blood, pancreatic tissue, and terminal ileum were sampled from each animal. Serum was analyzed for α-amylase level; pancreatic tissue was assessed for histological confirmation of AP; ileum was stained with Alcian blue to define the integrity of the mucin layer.
Rat acute pancreatitis model
Rats of all groups were weighed, anesthetized with intraperitoneal sodium pentobarbital (dose, 50 mg/kg), and placed on a heating pad to maintain euthermia. Using aseptic technique, the internal jugular vein and femoral artery were isolated and cannulated with 50-gauge silicone catheter tubing or polyethylene (PE-50) tubing containing 0.1 mL heparinized saline solution (concentration 10 units/mL), respectively for blood samples. A small, 2.5 cm midline incision was made and the duodenum was identified. In the AP group, the distal biliopancreatic duct was identified and ligated with a 4-0 silk suture as previously described (22). In the PDD group, the distal biliopancreatic duct was identified and ligated with a 4-0 silk suture as in the AP group. The biliopancreatic duct was then cannulated with polyethylene (PE-10) tubing proximal to the site of ligation. This tube was then externalized and secured with a 4-0 silk suture. In the SP group, the distal biliopancreatic duct was identified but remained unmanipulated. In all groups, the abdomen was then closed with a running 4-0 silk suture. Additional intraperitoneal sodium pentobarbital boluses (5 mg/kg) were given as needed throughout the post duct-ligation experimental period. At the conclusion of the experiment, euthanasia was administered by intravenous sodium pentobarbital injection via jugular vein (dose 25 mg/kg).
Mouse acute pancreatitis model
Mice were obtained from our in-house breeding colony (University of Edinburgh, UK) housed under standard conditions with free access to standard chow and water. Mice were aged 18.7 ± 0.8 weeks and there was no difference in age between experimental groups. Mice were anesthetized with intraperitoneal ketamine and metomidate according to weight and local guidelines. At laparotomy, the biliopancreatic duct was identified and cannulated transduodenally. After temporary occlusion of the bile duct to prevent reflux, 100μL of 5% sodium taurocholate (Sigma, St. Louis, Mo) dissolved in phosphate buffered saline was injected over 1 minute into the biliopancreatic duct. The cannula was withdrawn and a purse-string 5-0 silk suture was used to close the duodenal puncture. Prior to abdominal wall closure with silk, a 500μL bolus of caerulein (30 μg/kg in saline) was injected in the peritoneal cavity in AP mice. Sham-operated control mice underwent anesthesia, laparotomy and were given an intraperitoneal saline bolus (500 μL). No intraductal injection was given to sham-operated control mice. Prior to recovery from the anesthetic, analgesia was given by a subcutaneous bolus of buprenorphine (0.02 μg/kg). Mice were transferred to a warm recovery area and monitored by clinical observation for sickness behaviour (for example, a hunched appearance, piloerection, and lack of normal inquisitive behavior). At 6 or 24 hours, based on experimental group, mice were terminally re-anesthetized using isofluorane and exsanguinated by direct cardiac puncture prior to tissue sampling. A 10 cm segment of ileum was sampled 5 cm from the cecum and placed into 10% buffered formalin prior to processing to wax. 5 μm sections were dewaxed in xylene and stained with haematoxylin and eosin or Alcian blue.
Measurement of rat serum α-amylase level
At 4.5 hours after AP induction blood was collected from the femoral artery catheter in a heparinzed syringe and centrifuged (Denville 260D; Denville Scientific; Metuchen, NJ) at 3000 rpf for 10 min at 4 °C to generate plasma. Amylase was measured by ELISA (QuantiChrom α-amylase Assay Kit, BioAssay Systems, Hayward, CA) according the manufacturer's instructions and expressed as IU/L. Mouse blood was left at room temperature for 30 minustes then microcentrifuged at 5000 × g for 10 minutes to generate serum. Serum aliquots were stored at -80 °C. Serum amylase activity was measured using a commercial kit (Alpha Laboratories Ltd., Eastleigh, UK) adapted for use on a Cobas Fara centrifugal analyser (Roche Diagnostics Ltd, Welwyn Garden City, UK.).
Ascitic fluid
All fluid was aspirated from the abdomen into a syringe with an 18-gauge needle and the volume recorded. Amylase in the collected ascites was measured as above.
Collection of mucus sample
The distal 30 mm segment of the terminal ileum was identified and excised from the intestinal tract and the underlying mesentery. The external portion of the segment was washed with phosphate buffered saline (PBS) to remove any debris. The lumen of this segment was then flushed with PBS to remove fecal matter. The intestinal mucus was then collected by gently compressing the exterior of the segment as previously described (21). The mucus samples were then homogenized (Tissue Tearor, Biospec Products, Bartlesville, OK) in 500μL of PBS over ice and then centrifuged at 14.0 rcf for 75 minutes (Denville 260D; Denville Scientific; Metuchen, NJ). The supernatant was removed and stored at -80°C until further use.
Measurement of the protein concentration of mucus samples
The concentration of protein in the mucus samples was measured by the Thermo Scientific Pierce BCA (bicinchoninic acid) Assay (Thermo Fisher Scientific; Rockford, IL) according the manufacturer's instructions, expressed as μg/ml, and used to determine the volume of mucus necessary for equal protein loading.
Measurement of nitrated tyrosine residues
Nitration of tyrosine residues, a marker for reactive nitrogen intermediate (RNI) mediated injury, was measured with a competitive ELISA (Nitrotyrosine ELISA; Millipore; Billerica, MA). Mucus samples were analyzed based on equal protein loading (10 μg) according to the manufacturer's instructions. The extent of RNI- mediated damage was expressed by comparing nitrotyrosine sample values to those obtained with the nitrated bovine serum albumin (BSA) standards and expressed as nitro-BSA equivalents as previously described (21).
Measurement of carbonyl derivatives
Carbonyl derivatives, a marker of ROS-mediated damage, were quantified by ELISA (OxiSelect Protein Carbonyl ELISA; Cell Biolabs; San Diego, CA). Mucus samples were analyzed based on equal protein loading (10 μg/mL) of the samples according to the manufacturer's instructions. ROS mediated damage was expressed as protein carbonyl nmol/mg.
Measurement of total antioxidant capacity
Mucus sample antioxidant capacity was measured via ELISA (Total Antioxidant Capacity Assay; Abcam; Cambridge, MA) based on equal protein loading (10 μg/mL), as per manufacturer's instructions. Total antioxidant capacity is expressed as trolox equivalents (per nmol) as previously described (21).
Villous injury and mucus coverage
After sacrifice, a 5 cm segment of the terminal ileum was excised, opened along the mesenteric border and pinned to a paraffin block with the mucosal side up. In order to better visualize the mucus layer, the mucosal surface was then covered with 3% Alcian blue followed by fixation in Carnoy's solution for two hours as previously described (21). After processing, 2-4 μm sections were cut and stained with hematoxylin and eosin. Samples were evaluated by light microscopy at 100× magnification. Five, random fields containing 100-250 villi per animal were analyzed in a blinded fashion. Ileal cross sections were then examined in four, random, 40× visual fields. The overall incidence of morphologic villous damage was expressed as the number of injured villi divided by the total number of villi examined. Villous injury was expressed as percentage of damaged villi. The total lengths of villi in 4 random 40× visual fields were measured in a blinded fashion and the lengths of these segments covered with mucus were also measured. Ileal mucus coverage is expressed as percent of the villi that were not covered with mucus.
In vivo gut barrier permeability
Ileal permeability was measured in vivo using a 4-kD fluorinated dextran permeability probe FD-4 (Sigma, St. Louis, Mo) as follows: At the end of the experimental period (4.5 hours following ligation), the previous laparotomy incision was opened. A 10 cm segment of ileum was measured in a retrograde fashion starting 5 cm from the ileocecal valve. This segment was isolated from the adjacent bowel, flushed with 5 cc of normal saline to remove intraluminal contents and ligated on both ends. One milliliter of FD-4 solution (25 mg/mL in 1 M PBS) was injected in a retrograde fashion into the lumen of the isolated bowel segment. A systemic venous blood sample was collected after 30 minutes of circulation. The blood sample was then centrifuged (Denville 260D; Denville Scientific; Metuchen, NJ) at 3000 rpm at 4 °C for 10 minutes to obtain plasma. The plasma sample, along with FD-4 standards, were then analyzed in a microplate fluorescence reader (FLx800, BioTek Instruments, Winooski, VT) through an excitation filter of 485/20 and an emission filter of 528/20. Gut permeability was expressed as the amount of FD-4 found in plasma in micrograms per milliliter as previously described (21).
Statistical analysis
Results are expressed as mean ± SEM. Continuous data was analyzed by means of a one-way analysis of variance using the post hoc Tukey method. Statistical significance was considered achieved at p < 0.05. Analysis was conducted using GraphPad Prism software (GraphPad Software Inc., La Jolla, CA)
Results
At the time of sacrifice, the biliopancreatic duct-ligated rats without biliary drainage (AP group) had evidence of acute pancreatitis as reflected by increased serum α-amylase levels, ascitic fluid volumes and ascitic fluid α-amylase levels; all three values were increased compared to the two control groups (Figure 1B, 1C, 1D). The observation that the PDD sham rats, whose biliopancreatic ducts were ligated but also drained, did not manifest evidence of acute pancreatitis validated this group as an important bile and pancreatic secretion diversion control group.
Ligation of the biliopancreatic duct did not yield demonstrable evidence of loss of gut barrier function when the pancreatic duct was drained, as gut permeability was not increased in the PDD sham as compared to the non-ligated sham pancreatitis group (SP) (Figure 2A). In contrast, gut permeability was increased approximately 3-fold in the pancreatitis group as compared to the two control groups (Figure 2A). This pancreatitis-induced increase in gut permeability was associated with a significant, almost 2-fold, increase in the number of intestinal villi that had lost their mucus layer coverage as compared to the sham and PDD groups (Figure 2B). In contrast to the gut permeability and mucus layer data, the level of morphologic villous injury was minimal in the pancreatitis as well as the two control groups, with all groups having comparable numbers of normal-appearing villi (Figure 2C). Thus, the major gut histomorphologic change observed in the pancreatitis group was loss of the mucus layer and this mucus loss occurred in the absence of a microscopically, morphologically visible increase in villous injury, although some of the animals showed enlargement of the submucosa of the villi. Figure 3 shows representative photomicrographs demonstrating an intact mucus layer covering intestinal villi in both sham groups. In contrast, in the acute pancreatitis group, many of the villi are not covered by the unstirred mucus layer. Additionally, although not quantified, the mucus layer between the individual villi appear to be preserved better in the sham rats (Figure 3A) than the PDD or the AP rats (Figure 3B, C).
Figure 2.
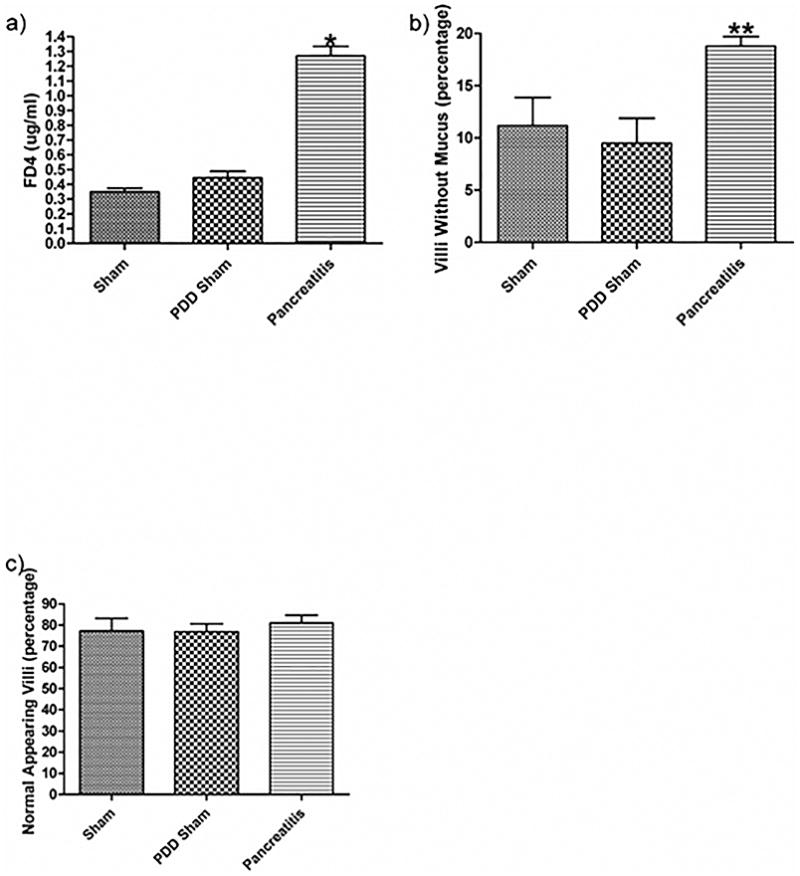
Pancreatic duct ligation-induced acute pancreatitis is associated with gut barrier failure as evidenced by a) an increase in gut permeability as manifest by the transluminal migration of a FD-4 probe and b) a decrease in percentage of mucus covering the intestinal villi. c) There was no change noted in the percentage of normal appearing villi between the groups. Data expressed as mean ± SEM with n=6 animals per group. * p<0.001 vs. other groups, ** p<0.05 vs. other groups.
Figure 3.
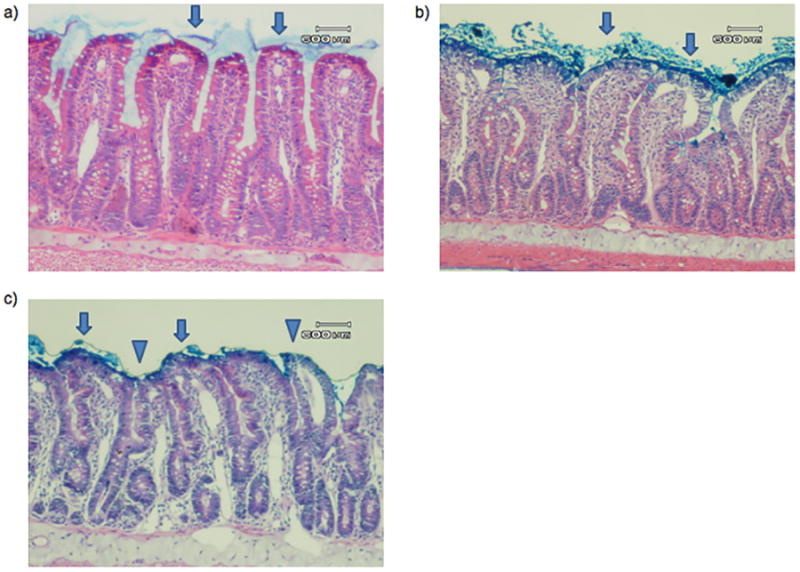
Representative photomicrograph of a) SP, b) PDD, and c) AP histology. In both of the sham pancreatitis groups, including a) sham pancreatitis and b) pancreatic duct diversion sham pancreatitis, villi are covered with an intact mucus layer (arrow). In the acute pancreatitis group, some villi are covered with an intact mucus layer (arrow), but the majority of villi demonstrate mucus sloughing (triangle). There is no significant difference noted in injured villi between groups. A 600μm reference is provided for scale.
To begin to investigate the mechanism by which acute pancreatitis leads to loss of the mucus layer, we tested the hypothesis that acute pancreatitis-induced mucus loss was related to oxidative or nitrosative modification of the mucus. This was done by measuring mucus protein carbonyl and nitrated tyrosine levels, which are markers of oxidative and nitrosive stress respectively. This hypothesis was supported by the observation that mucus from the acute pancreatitis group had a 3-fold increase in protein carbonyl levels, as compared to both PDD and sham groups (Figure 4A). Likewise, there was a 2-fold increase in nitrated tyrosine residues in the mucus of the pancreatitis group as compared to the two control groups (Figure 4B). An under-appreciated role of the intestinal mucus is as an anti-oxidant whereby it scavenges oxidants produced in the gut lumen during digestion(21). Thus, if acute pancreatitis subjected the mucus to an increased oxidative stress, a decrease in the anti-oxidant neutralizing capacity of the mucus would be expected. Using the trolox assay to measure the anti-oxidant capacity of the mucus, we found that the anti-oxidant neutralizing capacity of the mucus from the rats subjected to acute pancreatitis was reduced as compared to the sham pancreatitis and the PDD groups (Figure 4C).
Figure 4.
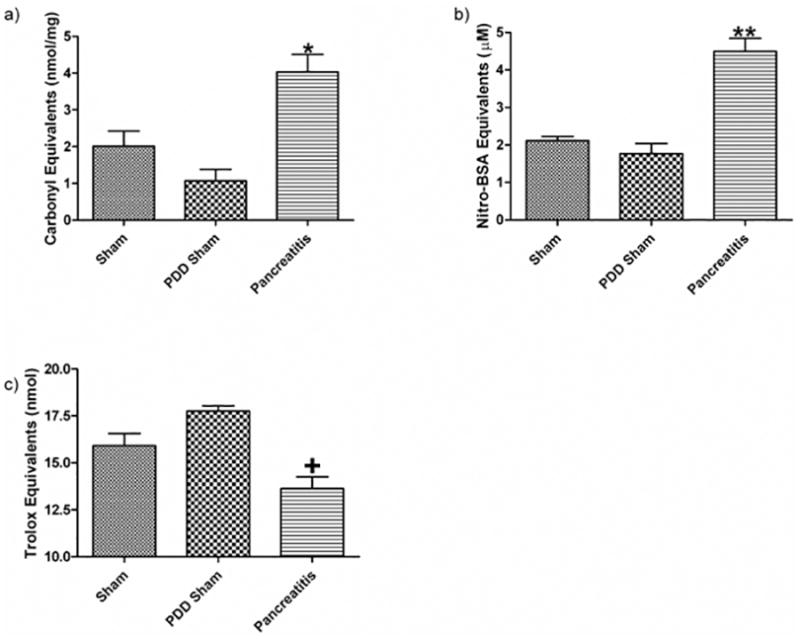
Following acute pancreatitis, gut barrier failure and mucus loss are associated with chemical changes to the mucus layer. These changes include an increase in the amount of a) carbonyl derivatives, b) decreased mucus anti-oxidant neutralizing capacity as well as c) increased nitrotyrosine residues seen in the collected mucus. Data expressed as mean ± SEM with n=6 animals per group * p<0.01 vs. other groups, ** p<0.001 vs. other groups, + p<0.05 vs. other groups.
In order to validate the concept of mucus loss in a different model of acute experimental pancreatitis, we investigated the mucus layer during AP in mice (Figures 5 and 6). When the mucus layer was examined on Alcian blue-stained fixed sections of terminal ileum, at 6 hours after the induction of AP we saw loss of integrity of the mucus layer at the villous tips when compared to the undisrupted mucus layer observed in sham-operated control mice (Fig 6A and B). In keeping with the observations of a rebound mucus hypersecretion following reperfusion in splanchnic ischaemia made by by Grootjans et al (30), we noted a profound increase in quantity of mucus in the delayed phase of AP in mice at 24 hours (Fig 6C). Mucus amounts were notably increased and mucus was located in the intervillous channels in mice with AP (Fig 6C) but not sham controls (Fig 6D). Goblet cells in AP appeared vacuolated (Fig 5F). Overall, the appearance in the delayed phase was of increased, thickened, adherent inspissated mucus.
Figure 5.
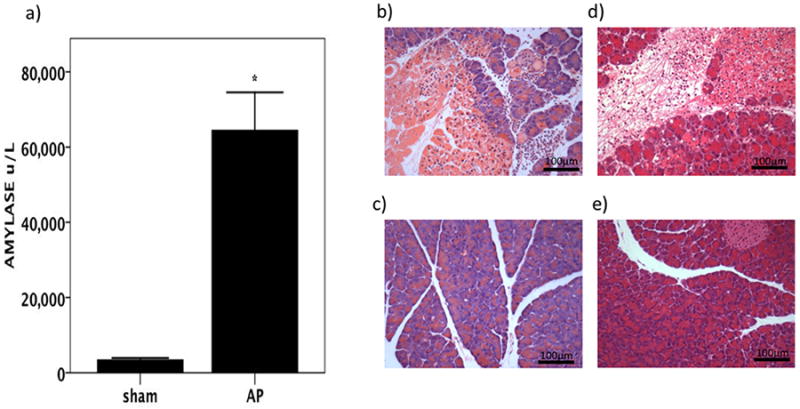
In mice, experimental AP results in biochemical acute pancreatitis which at six hours shows a) elevated serum amylase and b) oedema, necrosis, and inflammatory cell infiltration in the pancreas at compared to c) sham-operated control mice (haematoxylin and eosin stain, ×200 magnification). By 24 hours following the induction of AP, d) there is still evidence of oedema, necrosis, and inflammatory cell infiltration in the pancreas compared to e) sham-operated control mice at the same time point. Haematoxylin and eosin stain, ×200 magnification.
Figure 6.
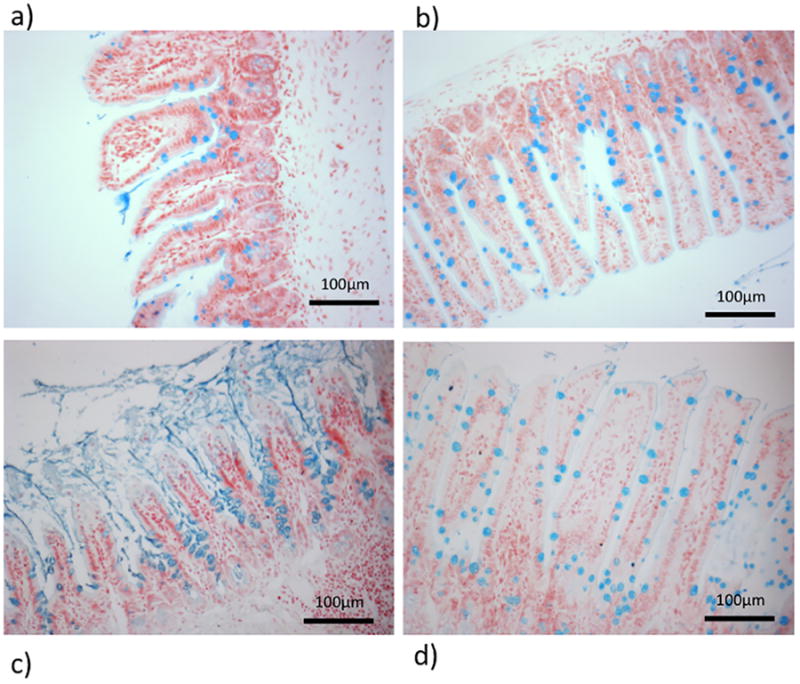
6 hours after the induction of AP, a) thinning and loss of the mucus layer at the villous tips is observed whereas the mucus layer remains intact in b) sham controls. By 24 hours c) profound rebound mucus hypersecretion can be seen with thick, inspissated mucus. d) Sham controls are unaffected. In both sham groups (panels b and d) mucus is located within the goblet cells of the villi. Alcian blue stain, ×200 magnification. Data expressed as mean ± SEM with n=6 animals per group * p<0.0001 AP vs. sham.
Together, these observations provide further support for the notion that acute pancreatitis results in oxidant stress to the mucus layer.
Discussion
Recent evidence suggests that sepsis and multi-system organ injury following severe acute pancreatitis involves loss of gut barrier function which leads to bacterial translocation, the systemic transmission of pathogenic compounds, and the generation of a hyperinflammatory state (6,24,25). Furthermore, gut-directed therapy such as early enteral nutrition and selective gut decontamination have been documented to be clinically beneficial (7). Thus, understanding the mechanisms by which acute pancreatitis injures the gut and leads to increased gut permeability are of clinical significance. The role of the unstirred mucus layer in this process is not well studied, despite the fact that the mucus layer is a principle component of the gut barrier and its loss is sufficient to significantly increase gut permeability (11,18). In this study, we show that a) the intestinal mucus layer is lost following acute pancreatitis, b) that this loss of the unstirred mucus layer is associated with gut barrier failure, c) that the loss of the unstirred mucus layer is associated with ROS- and RNI-mediated structural changes to the mucus layer and d) that loss of the unstirred mucus layer occurs before microscopic morphologic evidence of damage to the underlying mucosa.
Our data suggesting that ROS and RNI are involved in the pathogenesis of pancreatitis-induced loss of the mucus layer are consistent with studies documenting an association between ileal ROS formation and increased gut injury and permeability in a sodium taurocholate injection-induced model of acute pancreatitis (26) and with studies which show that pharmacologic inhibition of iNOS limits pancreatitis-induced gut barrier failure and bacterial translocation (27). While these, and other studies, implicate free radical generation in the pathogenesis of gut barrier failure following acute pancreatitis, more information is needed to fully elucidate the possible mechanisms whereby these free radical species cause gut barrier failure. A limitation of this work is the fact that the correlation between ROS- and RNI-modified intestinal mucus and increased intestinal permeability is only associative. Future studies testing whether anti-oxidants or nitric oxide synthesis inhibitors would preserve both the mucus layer and gut barrier would be important.
We therefore propose the following sequence of events as being involved in pancreatitis-induced loss of the mucus layer and subsequent increased intestinal permeability. First, acute pancreatitis-induced hypovolemia and a systemic inflammatory state leads to shunting of blood from the splanchnic to the systemic circulation which in turn leads to a gut ischemia-reperfusion injury (2,7,25). The oxidants and reactive nitrogen intermediates thus generated attack the mucus layer and reduce its barrier properties and lead to its loss. The underlying mucus-free, naked epithelial mucosal layer is now directly exposed to bacteria and other potentially toxic factors within the lumen, which in turn exacerbate gut injury. One limitation of this proposed scenario is the lack of significant microscopic morphologic damage seen in the rat biliopancreatic duct ligation model. However, this can be explained by the early time point, which may have been too early for the development of morphologic evidence of gut injury. Additionally, since electron microscope studies were not carried out, it remains possible that gut injury did occur but that microscopic studies were not sensitive enough to detect these injuries. To begin to address this concern, we used a delayed time point (24 hours after induction) in the mouse AP experiments, in which morphological injury became evident. In the pancreatic duct ligation model, which mimics biliary pancreatitis more than alcoholic pancreatitis (28), pancreatic proteases do not enter the gut lumen. This is in contrast to toxin-induced models of pancreatitis wherein pancreatic proteases are secreted into the gut lumen and morphologic evidence of gut injury is more pronounced (27, 29-31). The potentially toxic role of luminal pancreatic proteases is supported by studies showing that the ischemic or stressed gut is susceptible to autodigestion by luminal digestive enzymes if they come into direct contact with the epithelial barrier (10, 32, 33). Thus, the fact that the duct ligation model prevents pancreatic enzymes from reaching the gut lumen could help explain the lack of microscopic morphologic evidence of gut injury. Further support for the role of luminal pancreatic proteases in exacerbating gut injury comes from studies in a trauma-hemorrhagic shock model where pharmacologic neutralization of luminal pancreatic proteases (34) or pancreatic duct diversion (35) limited gut injury and preserved gut barrier function. On the other hand, our result suggest that the duration and magnitude of the ischemia-reperfusion insult was sufficient to cause injury to the mucus layer as well as loss of gut barrier function, despite the absence of intraluminal pancreatic proteases and evidence of microscopic morphologic gut injury.
The current study documents that the systemic effects of acute pancreatitis are sufficient to disrupt the mucus layer, and hence increase gut permeability, even in the absence of intraluminal pancreatic proteases. Thus, one advantage of the pancreatic duct ligation model is that it eliminates luminal pancreatic enzymes and serves as a proof-of-principle model testing the role of mucus disruption on pancreatitis-induced gut permeability changes. Our results also suggest that pancreatic proteases are not likely necessary for the initiation of the mucus changes observed in early acute pancreatitis, although their presence could potentiate damage to the oxidant-modified mucus layer (33).
In summary, our results document that a) the intestinal mucus layer is lost following acute pancreatitis, b) that this loss of the unstirred mucus layer is associated with gut barrier failure, c) that the loss of the unstirred mucus layer is associated with ROS- and RNI-mediated structural changes to the mucus layer, d) that loss of the unstirred mucus layer occurs before there is morphologic evidence of damage to the underlying mucosa and e) that this process appears to be largely independent of the intraluminal activity of the pancreatic proteases. Thus, in conclusion, we have demonstrated that the loss of gut barrier function is associated with oxidant (ROS and RNI)-mediated changes to the mucus gel layer and thus, damage to and loss of the unstirred mucus layer appear to be a necessary component of gut injury and gut barrier failure following acute pancreatitis. This suggests one possible mechanism whereby systemic oxidative injury causes both loss of and damage to the mucus layer triggering gut barrier failure, and in so doing, provides insight into how gut mucus protective strategies might improve outcomes in patients with pancreatitis.
Acknowledgments
Disclosure: Supported by NIH grant T32 069330 (VA)
References
- 1.Johnson CD. Organ failure in acute pancreatitis. J Org Dysfunct. 2006;2:171–5. [Google Scholar]
- 2.Rahman SH, Ammori BJ, Holmfield J, et al. Intestinal hypoperfusion contributes to gut barrier failure in severe acute pancreatitis. J Gastrointest Surg. 2003;7:26–35. doi: 10.1016/S1091-255X(02)00090-2. [DOI] [PubMed] [Google Scholar]
- 3.Andersson R, Wang XD. Gut barrier dysfunction in experimental acute pancreatitis. Ann Acad Med Singapore. 1999;28:141–6. [PubMed] [Google Scholar]
- 4.Ammori BJ, Leeder PC, King RF, et al. Early increase in intestinal permeability in patients with severe acute pancreatitis: correlation with endotoxemia, organ failure, and mortality. J Gastrointest Surg. 1999;3:252–62. doi: 10.1016/s1091-255x(99)80067-5. [DOI] [PubMed] [Google Scholar]
- 5.McNaught CE, Woodcock NP, Mitchell CJ, et al. Gastric colonisation, intestinal permeability and septic morbidity in acute pancreatitis. Pancreatology. 2002;2:463–8. doi: 10.1159/000064712. [DOI] [PubMed] [Google Scholar]
- 6.Flint RS, Windsor JA. The role of the intestine in the pathophysiology and management of severe acute pancreatitis. HPB. 2003;5:69–85. doi: 10.1080/13651820310001108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Anand N, Park JH, Wu BU. Modern management of acute pancreatitis. Gastroenterol Clin North Am. 2012;41:1–8. doi: 10.1016/j.gtc.2011.12.013. [DOI] [PubMed] [Google Scholar]
- 8.Besselink M, Timmerman HM, Buskens E. Probiotic prophylaxis in patients with predicted severe acute pancreatitis (PROPATRIA): design and rationale of a double-blind, placebo-controlled randomised multicenter trial. BMC Surg. 2004;4:12. doi: 10.1186/1471-2482-4-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Petrov MS, van Santvoort HC, Besselink MG, et al. Enteral nutrition and the risk of mortality and infectious complications in patients with severe acute pancreatitis: a meta-analysis of randomized trials. Arch Surg. 2008;143:1111–7. doi: 10.1001/archsurg.143.11.1111. [DOI] [PubMed] [Google Scholar]
- 10.Fishman JE, Levy G, Alli V, et al. Oxidative modification of the intestinal mucus layer is a critical but unrecognized component of trauma hemorrhagic shock-induced gut barrier failure. Am J Physiol Gastrointest Liver Physiol. 2013;304:G57–63. doi: 10.1152/ajpgi.00170.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Qin X, Sheth SU, Sharpe SM, et al. The Mucus Layer is Critical in Protecting Against Ischemia-Reperfusion-Mediated Gut Injury and in the Restitution of Gut Barrier Function. Shock. 2011;35:275–81. doi: 10.1097/SHK.0b013e3181f6aaf1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Van der Sluis M, De Koning BA, De Bruijn AC, et al. Muc2-deficient mice spontaneously develop colitis, indicating that MUC2 is critical for colonic protection. Gastroenterology. 2006;131:117–29. doi: 10.1053/j.gastro.2006.04.020. [DOI] [PubMed] [Google Scholar]
- 13.Heazlewood CK, Cook MC, Eri R, et al. Aberrant mucin assembly in mice causes endoplasmic reticulum stress and spontaneous inflammation resembling ulcerative colitis. PLoS Med. 2008;5:e54. doi: 10.1371/journal.pmed.0050054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sharpe SM, Doucet DR, Qin X, et al. Role of intestinal mucus and pancreatic proteases in the pathogenesis of trauma–hemorrhagic shock-induced gut barrier failure and multiple organ dysfunction syndrome. J Org Dysfunct. 2008;4:168–76. [Google Scholar]
- 15.Lugea A, Salas A, Casalot J, et al. Surface hydrophobicity of the rat colonic mucosa is a defensive barrier against macromolecules and toxins. Gut. 2000;46:515–21. doi: 10.1136/gut.46.4.515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bounous G, McArdle AH, Hodges DM, et al. Biosynthesis of intestinal mucin in shock: relationship to tryptic hemorrhagic enteritis and permeability to curare. Ann Surg. 1966;164:13–22. doi: 10.1097/00000658-196607000-00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nimmerfall F, Rosenthaler J. Significance of the goblet-cell mucin layer, the outermost luminal barrier to passage through the gut wall. Biochem Biophys Res Commun. 1980;94:960–6. doi: 10.1016/0006-291x(80)91328-5. [DOI] [PubMed] [Google Scholar]
- 18.Sharpe SM, Qin X, Lu Q, et al. Loss of the intestinal mucus layer in the normal rat causes gut injury but not toxic mesenteric lymph nor lung injury. Shock. 2010;34:475–81. doi: 10.1097/SHK.0b013e3181dc3ff5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Grootjans J, Hundscheid IH, Lenaerts K, et al. Ischemia-induced mucus barrier loss and bacterial penetration are rapidly counteracted by increased goblet cell secretory activity in human and rat colon. Gut. 2013;62:250–8. doi: 10.1136/gutjnl-2011-301956. [DOI] [PubMed] [Google Scholar]
- 20.Grisham MB, Ritter Von C, Smith BF, et al. Interaction between oxygen radicals and gastric mucin. Am J Physiol. 1987;253:G93–6. doi: 10.1152/ajpgi.1987.253.1.G93. [DOI] [PubMed] [Google Scholar]
- 21.Brownlee IA, Knight J, Dettmar PW. Action of reactive oxygen species on colonic mucus secretions. Free Radic Biol Med. 2007;43:800–8. doi: 10.1016/j.freeradbiomed.2007.05.023. [DOI] [PubMed] [Google Scholar]
- 22.Meyerholz DK, Samuel I. Morphologic characterization of early ligation-induced acute pancreatitis in rats. Am J Surg. 2007;194:652–8. doi: 10.1016/j.amjsurg.2007.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Merriam LT, Wilcockson D, Samuel I. Ligation-induced acute pancreatitis increases pancreatic and circulating trypsinogen activation peptides. J Surg Res. 1996;60:417–21. doi: 10.1006/jsre.1996.0068. [DOI] [PubMed] [Google Scholar]
- 24.Berezina TL, Zaets SB, Mole DJ, et al. Mesenteric lymph duct ligation decreases red blood cell alterations caused by acute pancreatitis. Am J Surg. 2005;190:800–4. doi: 10.1016/j.amjsurg.2005.07.023. [DOI] [PubMed] [Google Scholar]
- 25.Flint RS, Phillips ARJ, Power SE, et al. Acute pancreatitis severity is exacerbated by intestinal ischemia-reperfusion conditioned mesenteric lymph. Surgery. 2008;143:404–13. doi: 10.1016/j.surg.2007.10.005. [DOI] [PubMed] [Google Scholar]
- 26.Mikami Y, Dobschütz EV, Sommer O, et al. Matrix metalloproteinase-9 derived from polymorphonuclear neutrophils increases gut barrier dysfunction and bacterial translocation in rat severe acute pancreatitis. Surgery. 2009;145:147–56. doi: 10.1016/j.surg.2008.08.036. [DOI] [PubMed] [Google Scholar]
- 27.Simsek I, Mas MR, Yasar M, et al. Inhibition of inducible nitric oxide synthase reduces bacterial translocation in a rat model of acute pancreatitis. Pancreas. 2001;23:296–301. doi: 10.1097/00006676-200110000-00011. [DOI] [PubMed] [Google Scholar]
- 28.Foitzik T, Hotz HG, Eibl G, et al. Experimental models of acute pancreatitis: are they suitable for evaluating therapy? Int J Colorectal Dis. 2000;15:127–35. doi: 10.1007/s003840000216. [DOI] [PubMed] [Google Scholar]
- 29.Mole DJ, McFerran NV, Collett G, et al. Tryptophan catabolites in mesenteric lymph may contribute to pancreatitis-associated organ failure. Br J Surg. 2008;95:855–67. doi: 10.1002/bjs.6112. [DOI] [PubMed] [Google Scholar]
- 30.van Minnen LP, Blom M, Timmerman HM, et al. The use of animal models to study bacterial translocation during acute pancreatitis. J Gastrointest Surg. 2007;11:682–9. doi: 10.1007/s11605-007-0088-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nieuwenhuijs VB, van Dijk JE, Gooszen HG, et al. Obstructive jaundice, bacterial translocation and interdigestive small-bowel motility in rats. Digestion. 2000;62:255–61. doi: 10.1159/000007824. [DOI] [PubMed] [Google Scholar]
- 32.Chang M, Alsaigh T, Kistler EB, et al. Breakdown of mucin as barrier to digestive enzymes in the ischemic rat small intestine. PLoS ONE. 2012;7:e40087. doi: 10.1371/journal.pone.0040087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kistler EB, Alsaigh T, Chang M, Schmid-Schönbein GW. Impaired small-bowel barrier integrity in the presence of lumenal pancreatic digestive enzymes leads to circulatory shock. Shock. 2012;38:262–7. doi: 10.1097/SHK.0b013e31825b1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Deitch EA, Shi HP, Lu Q, et al. Serine proteases are involved in the pathogenesis of trauma-hemorrhagic shock-induced gut and lung injury. Shock. 2003;19:452–6. doi: 10.1097/01.shk.0000048899.46342.f6. [DOI] [PubMed] [Google Scholar]
- 35.Caputo FJ, Rupani B, Watkins AC, et al. Pancreatic duct ligation abrogates the trauma hemorrhage-induced gut barrier failure and the subsequent production of biologically active intestinal lymph. Shock. 2007;28:441–6. doi: 10.1097/shk.0b013e31804858f2. [DOI] [PubMed] [Google Scholar]