

Abstract
Oxygen-induced contraction of the ductus arteriosus (DA) involves a mitochondrial oxygen-sensor, which signals pO2 in the DA smooth muscle cell (DASMC) by increasing production of diffusible hydrogen peroxide (H2O2). H2O2 stimulates vasoconstriction by regulating ion channels and rho kinase, leading to calcium influx and calcium sensitization. Because epidermal growth factor receptor (EGFR) signaling is also redox regulated and participates in oxygen sensing and vasoconstriction in other systems, we explored the role of the EGFR and its signaling cascade (p38 and JNK) in DA contraction.
Experiments were performed in DA rings isolated from full-term New Zealand White rabbits and human DASMC. In human DASMCs increasing pO2 from hypoxia to normoxia (40 to 100 mmHg) significantly increased cytosolic calcium, p<0.01. This normoxic rise in intracellular calcium was mimicked by EGF and inhibited by EGFR siRNA. In DA rings, EGF caused contraction whilst the specific EGFR inhibitor (AG1478) and the tyrosine kinase inhibitors (genistein or tyrphostin A23) selectively attenuated oxygen-induced contraction (p <0.01). Conversely, orthovanadate, a tyrosine phosphatase inhibitor known to activate EGFR signaling, caused dose-dependent contraction of hypoxic DA and superimposed increases in oxygen caused minimal additional contraction. Ansomycin, an activator of EGFR’s downstream kinases, p38 and JNK, caused DA contraction; conversely, oxygen-induced DA contraction was blocked by inhibitors of p38 MAPK (SB203580) or JNK (JNK inhibitor II). O2-induced phosphorylation of EGFR occurred within 5-minutes of increasing pO2 and was inhibited by mitochondrial-targeted overexpression of catalase. AG1478 prevented the oxygen-induced p38 and JNK phosphorylation. In conclusion, O2-induced EGFR transactivation initiates p38/JNK-mediated increases in cytosolic calcium and contributes to DA contraction. The EGFR/p38/JNK pathway is regulated by mitochondrial redox signaling and is a promising therapeutic target for modulation of the patent ductus arteriosus.
Keywords: Oxygen sensing, tyrosine kinase inhibitors, c-Jun N-amino-terminal kinase (JNK), Patent ductus arteriosus (PDA), protein tyrosine phosphatases, Mitochondrial hydrogen peroxide
Introduction
The ductus arteriosus (DA) plays an essential role in fetal life, diverting blood away from the unventilated lungs. At birth, the increase in arterial pO2 causes DA contraction, thus diverting blood flow to the newly expanded lungs. Although vasodilatory effects of prostaglandin E2 and constrictor substances such as endothelin-11 modulate this contraction, oxygen-induced contraction is an intrinsic property of DA smooth muscle (DASMC)2; 3. Persistent patent ductus arteriosus (PDA) is a common abnormality in the newborn, affecting 55% of low birth weight, premature infants4, resulting in heart failure, and failure to thrive. Conversely, in some congenital heart disease conditions, it is critical to maintain DA patency to provide pulmonary or systemic blood flow for the newborn. Thus, understanding the mechanism by which O2 constricts the DA is an important physiologic question and has practical translational applications in the treatment of PDA.
Rapid increases in pO2 in DASMC are detected by a mitochondrial redox sensor. In response to a physiologic rise in pO2, the DASMC’s mitochondria elaborate more reactive oxygen species (ROS), including H2O25; 6. These ROS constitute a redox signal and initiate several vasoconstrictive events, including: (1) inhibition of the redox-sensitive, voltage-gated, potassium channels, resulting in membrane depolarization and calcium entry through large conductance calcium (L-type) channels3, (2) direct redox-mediated activation of L-type calcium channels7, (3) release of calcium from the sarcoplasmic reticulum and activation of store-operated channels8; 9, and (4) calcium-sensitization, primarily reflecting Rho kinase activation8; 9; 10.
Mitochondrial-derived H2O2 production is dynamically modulated in proportion to pO25 and is involved in initiating DASMC contraction11. H2O2 is a diffusible signal that changes vascular tone by oxidizing sulfhydryl moieties in amino acids, such as cysteine, that regulate the conformation and function of enzymes and ion channels. By oxidizing catalytic cysteine residues H2O2 also inactivates protein-tyrosine phosphatases, lipid phosphatases and tumor suppressor phosphatase and tension homolog (PTEN), which modulates protein phosphorylation12. Tyrosine phosphorylation regulates many cellular responses, including contraction13; 14. In vivo, tyrosine phosphorylation is reversible and dynamic. The phosphorylation states are controlled by the coordinated action of protein tyrosine kinases (PTK) and phosphatases (PTP). Phosphorylation of tyrosine residues is involved in Ca2+ influx, intracellular Ca2+ release, and Ca2+ sensitization of the contractile apparatus15. Clyman et al have reported that the tyrosine kinase inhibitor, genistein (100 μM) inhibited both Ca2+ -dependent and Ca2+-independent contraction in DA rings9; however, the specific tyrosine kinase involved was not identified.
Epidermal growth factor receptor (EGFR) is a member of the ErbB family of receptor tyrosine kinases. They are transmembrane proteins that are activated upon binding peptide growth factors and are involved in cellular transformation in cancer16. EGFR inhibitors, such as erlotinib, are used clinically to treat various cancers17. In the A431 cancer cell line, hypoxia activates EGFR signaling and induces epithelial to mesenchymal transition18. Hypoxia prolongs EGFR activation through an hypoxia-inducible factor-dependent deceleration of endocytosis-mediated EGFR deactivation19.
Although there is little information regarding the role of EGFR in vascular oxygen sensing, EGFR transactivation augments depolarization-induced Ca2+ sensitization and vasoconstriction in chronically hypoxic pulmonary vascular SMC20. Moreover, EGFR transactivation is involved in H2O2−, endothelin-1-, and angiotensin II-signaling 21; 22; 23. EGFR mediates its effects by activation of a downstream kinase cascade that includes mitogen-activated protein kinases (MAPK), extracellular signal-regulated kinases (ERKs), c-Jun N-amino-terminal/stress-activated protein kinases (JNKs/SAPKs), and p38 MAPKs21; 24; 25.
We hypothesize that oxygen-induced increases in mitochondrial H2O2 activate EGFR and contributes to oxygen-induced DA contraction. We investigated whether oxygen activates EGFR and used pharmacological probes to determine its downstream signaling cascade in rabbit DA rings and human DASMC. We report that that H2O2-dependent EGFR activation raises cytosolic calcium and promotes DA contraction by increasing p38/JNK signaling. EGFR inhibition reduces constriction to oxygen but not KCl. We conclude that the phosphorylation status of EGFR is redox sensitive and is a key regulator of oxygen sensing in the ductus arteriosus.
Methods
All animal studies were approved by the Institutional Animal Care and Use Committee of the Minneapolis Veterans Affairs Medical Center and The University of Chicago Animal Care Committee and conform to current National Institutes of Health and American Physiology Society guidelines for the use and care of laboratory animals. Human DAs were obtained from infants with various forms of congenital heart disease at the time of surgical correction either at the University of Nebraska Children’s Hospital or the University of Chicago, under protocols approved by the local institutional review board of both institutions. The demographics of these children, none of whom had PDA, has previosuly been described5; 6.
DA rings were isolated from pregnant New Zealand white rabbits at 30 –31 days of gestation (term), as previously described6; 8. Briefly, the rabbits were anesthetized with ketamine (75 mg) and xylazine (20 mg) intramuscularly and pentobarbital (50 mg) intravenously. The fetal pups were delivered by cesarean section and a midline sternotomy was performed, before initiation of respiration. The heart, lungs, and great vessels were excised en bloc and placed in deoxygenated Earle’s solution. The DA was carefully dissected free from adventitia under a dissecting microscope and then excised, excluding its attachments to the left pulmonary artery and descending thoracic aorta.
DA ring tension measurement
The isolated rabbit DA ring was connected to strain gauge transducers by two stainless steel wires. The ring was suspended in a 3-ml bath containing Earle’s solution. The ring was equilibrated with an hypoxic gas mixture (0 % O2, 5 % CO2; pO2 = 22 ±1 mmHg). The optimal resting tension (1000 mg) was determined in hypoxic medium by measuring the maximum contractile response to KCl (60 mM) at varying basal tensions (200–2,000 mg). Indomethacin (3 μM) and L-NAME (100 μM) were present throughout all experiments. O2 tension was recorded continuously with an M1-730 oxygen electrode (Microelectrode, Bedford, NH). A normoxic gas mixture (20% O2, 5% CO2, pO2=133 ±1 mmHg) was used to bubble the chamber solution creating normoxia. Data were recorded using an analog-digital computer system (MatLab, AD Instruments Inc., Medford, MA).
Human DASMC culture
DASMCs from human neonates were isolated from freshly isolated human ductus arteriosus. The identity of the cells as DASMCs was confirmed by confirming positive immunostaining for SM α-actin but not vonWillebrand’s factor. A primary culture of DASMCs was established and then the cells were harvested with trypsin, frozen in Freezing Media (10% DMSO, 50% FBS and 40% DMEM) and stored in liquid nitrogen for later use. DASMC were grown in 100 mm culture dishes and used within the first 5 passages in culture. DASMC were maintained in hypoxia (pO2 40 mmHg, pH 7.35–7.45, pCO2 30–40 mmHg using an environmentally controlled, Tri-Gas CO2 incubator (Thermo Scientific) until the protocol called for exposure to normoxia.
Exposure of hypoxic human DASMC to acute normoxia
The experiment was performed in a chamber flushed with nitrogen. Inside the hypoxic chamber, 2 small glass containers (500 mL) were filled with Earle’s solution (37°C). They were bubbled either with an hypoxic (0% O2, 5% CO2; pO2=28 ± 2 mmHg) or normoxic (20% O2, 5% CO2; pO2=134 ± 2 mmHg) gas mixture. DASMCs were immersed in the hypoxic solution for 30 minutes and then either maintained in hypoxia for 1, 3, 5, 10, or 20 minutes or quickly switched to the normoxia solution for 1, 3, 5, 10, or 20 minutes. At the end of the experiment, cells were flash frozen in liquid nitrogen and later lysed at 4°C under hypoxic conditions using the phosphoSafe extraction reagent (Calbiochem, Billerica, MA) and a phosphatase inhibitor cocktail (Calbiochem, 1:100, Billerica, MA).
Real-time PCR
Total RNA was isolated from cultured cells or harvested tissues using the PureLink RNA Mini Kit (Invitrogen, Carlsbad, CA). Total RNA (0.5 μg) was reverse-transcribed using the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA) according to the manufacturer’s instructions. Equal volumes of the resultant cDNA were used to evaluate the expression level of mRNAs among treatments and controls by real-time PCR. The quantitative real-time PCR was performed on an Applied Biosystems 7900HT (Life Technologies, Grand Island, NY) as previously described6. The primers used were purchased from Applied Biosystems (Probe ID: EGFR: Hs01076087_m1; 18S: Hs99999901_s1).
SiRNA for EGFR
To achieve specific molecular knockdown of EGFR, we used several small interfering RNA (siRNA) as previously described6. SiRNA for EGFR were: 5′GGCUGGUUA UGUCCUCAUUGCCCTC-3′; 5′GAGGGCAAUGAGGACAUAACCAGCCAC-3′; 5′-GGCG GGACAUAGUCAGCAGUGACTT-3′ and 5′-AAGUCACUGCUGACUAUGUCCCGCCAC-3′. A scrambled siRNA control was used: 5′-CUUCCUCUCUUUCUCUCCCUUGUGA-3′. siRNA was purchased from Integrated DNA Technologies (Coralville, IA, United States).
Immunoblotting
Equal amounts of protein (20 μg) were subjected to SDS/PAGE gradient gels (4–15%), transferred to polyvinylidene difluoride membranes, blocked with 5 % (w/v) BSA in Tris buffered saline with Tween 20 (TBST; 25 mM Tris-HCl, pH 7.4, 137 mM NaCl and 0.1 % Tween-20) for 1-hour, and incubated with primary antibodies in 5 % (w/v) BSA in TBST for 2-hours at room temperature. The membranes were washed at least three times with TBST at 15-minute intervals and then incubated with either mouse or rabbit or goat horseradish peroxidase-conjugated secondary antibody (1:2000) for 1-hour at room temperature. The membranes were developed with an enhanced chemiluminescence detection system according to the manufacturer’s instructions. The following antibodies were obtained from Santa Cruz Biotechnology (Dallas, TX): rabbit anti-EGFR (sc-003, used for immunoblotting), mouse anti-L-caldesmon (1:200), goat anti- phospho –caldesmon (tyr27, 1:400). The following antibodies were from Cell Signaling Technology, Inc. (Boston, MA): rabbit anti-p38 MAPK (1:1000), rabbit anti-phospho-p38 MAPK (thr180/tyr182, 1:1000), rabbit anti- SAPK/JNK (1:1000), rabbit anti-phospho-SAPK/JNK (thr183/tyr185, 1:1000). The following antibodies were from EMD (Gibbstown, NJ): ST1030 PhosphoDetect Anti-Hsp27 (pSer82) mouse mAb (1:400), mouse anti-Hsp27 (1:1000). Mouse anti-caldesmon was from Sigma (1:1000).
Immunoprecipitation
A total of 400 μg of protein from human DASMCs was used for immunoprecipitation with mouse anti-EGFR monoclonal antibody (IgG2a, sc-120). Following gel electrophoresis, tyrosine-phosphorylated EGFR was detected by immunoblotting with monoclonal anti-phosphotyrosine (SC-508, Santa Cruz biotechnology, Dallas, TX). Total EGFR was analyzed by re-probing of the same filter with anti-EGFR antibody (sc-003, 1:400, Santa Cruz biotechnology, Dallas, TX).
Calcium imaging
Dual-excitation imaging with fura-2 was used to measure the cytosolic Ca2+ response to changes in oxygen tension in cultured human DASMC, as described6; 26. The human DASMC were transferred to imaging dishes (Molecular Probes, Eugene, OR), and incubated in low-Ca2+ Hanks’ solution with the cell-permeable acetoxymethyl ester form of fura 2 (0.1 μM) and Pluronic F-127 (0.8 μM) for 15 minutes at room temperature. The plates were then washed with HEPES buffer containing 1.5 mM Ca2+ and incubated at room temperature for a further 20 minutes. The plates were then washed again and placed on the microscope stage. The drugs were added directly to the cells as a bolus by microinjection. All drugs were given in 10 μl volumes to reduce potential volume-induced artifacts. Saline injections of 10 μl had no effect on [Ca2+]i. Changes in [Ca2+]i were recorded in individual cells using a 340/380 nm MetaFluor filter-based imaging system (Universal Imaging, West Chester, PA) and a cooled charge-coupled device camera (Photometrics, Tucson, AZ).
Statistical Analysis
Data are expressed as mean±SEM. In all figures, the SEM is indicated, although in some cases it did not exceed the symbol size. Differences between groups were calculated using an unpaired Student’s t-test or simple ANOVA with post hoc analysis (Fisher’s least-significant-difference test), as appropriate. A p<0.05 was considered statistically significant.
Results
Gs proteins are involved in O2-induced DA contraction
EGFR is known to signal via G-proteins27. Consequently we assessed the effects of cholera toxin (a Gs inhibitor that is known to block the effects of EGFR28) and pertussis toxin (a Gi inhibitor) on O2-induced contraction. Cholera toxin (1 mg/ml, 20–60 minutes incubation) attenuated the O2-induced DA contraction by ~ 80% (supplemental fig. 1 A–B) without affecting the hypoxic baseline tone or the contraction to KCl (60 mM), data not shown. Pertussis toxin (1 mg/ml, administered 20–60 minutes prior to increasing pO2) had no effect on O2-induced DA contraction (supplemental fig. 1C). Because substantial published evidence suggests that endothelin may also contribute to DA contraction, we assessed the effects of endothelin receptor antagonists on DA contraction to oxygen29. BQ-123 (selective ETA receptor antagonist, 3 μM) and BQ-788 (selective ETB receptor antagonist, 3 μM) minimally reduced O2-induced contraction (supplemental fig. 1D), consistent with our previous findings30. These findings support the contention that oxygen-induced DA contraction involves Gs proteins and is not primarily dependent on endothelin-1.
Tyrosine kinases and tyrosine phosphatases are involved in O2-induced DA contraction
Two structurally and functionally distinct tyrosine kinase inhibitors, genistein (fig. 1A and 1C) and tyrphostin A23 (fig. 1B and 1C), markedly reduced O2-induced DA contraction. Daidzein, an inactive control for genistein had no effect on O2-induced DA contraction (fig. 1C). We next assessed whether tyrosine kinase inhibition targeted the upstream oxygen sensing or just the downstream effectors (activation of L-type calcium channels or store operated channels, SOCs). Genistein had no effect on 60 mM KCl-induced contraction (Fig 1D). This suggests that genistein affects an oxygen-sensing mechanism in the DA, rather than acting directly on downstream regulators of membrane potential, such as voltage-gated (Kv) channels, or L-type calcium channels (which together mediate KCl-induced contraction). Since SOCs have been implicated in DA contraction, we evaluated the possible effects of tyrosine kinase on SOCs by exposing DA rings to cyclopiazonic acid (CPA) in a calcium-free bath solution, to empty intracellular calcium and activate SOCs. A subsequent switch of the extracellular calcium from 0 to 2 mM (in the presence of nifedipine to block voltage-gated calcium channels) induced contraction, reflecting calcium entry through SOCs8. The finding that SOC-mediated DA contraction was also unaffected by genistein (fig. 1E) indicates that the SOCs are not direct targets of tyrosine kinase in the DA.
Fig 1. Tyrosine kinase inhibitors reduce the oxygen-induced DA contraction.

Different tyrosine kinase inhibitors (genistein and tyrphostin) inhibited oxygen-induced DA contraction (A and B representative traces; C: Summary data. Daizein is negative control for genistein; n=4–7, ** P<0.01. D: Genistein had no effect on KCl-induced contraction, n=6. E: Genistein had no effect on calcium switch from 0 mM to 2 mM induced contraction in the presence of CPA and nifedipine, n=5.
The selective inhibitory effects of genistein on oxygen-induced contraction but not on contraction resulting from membrane potential depolarization or SOC-activation supports the interpretation that tyrosine kinase activity contributes to the upstream mechanisms of O2-sensing mechanism in the DA.
Tyrosine phosphatase inhibition mimics O2-induced DA contraction
Since inhibition of tyrosine phosphorylation inhibited O2 constriction we next assessed whether increasing phosphorylation would cause DA constriction. Sodium orthovanadate, a non-specific tyrosine phosphatase inhibitor, caused DA contraction and reduced subsequent O2-induced contraction (fig. 2A–C). When superimposed on oxygen-induced DA contraction, sodium orthovanadate causes little additional vasoconstriction (fig. 2C). The contraction which orthovanadate induced in hypoxic DA rings can be dose-dependently inhibited by the tyrosine kinase inhibitor, genistein (fig. 2D), consistent with genistein’s effects on O2-induced contraction. In a further parallel, DA rings when exposed to 12 hours O2 and 2 hours hypoxia (recovery) lost most (80%) of the acute constrictor response to oxygen (as reported31) and demonstrated a parallel impairment of contraction to sodium orthovanadate (~50%). In contrast, the response to KCl was not affected in this experiment model31 (data not shown). The parallel pharmacological profiles of orthovanadate, a putative tyrosine phosphatase inhibitor, and O2 supports the interpretation that tyrosine phosphorylation contributes to O2-sensing in the DA.
Fig. 2. Orthovanadate, a protein tyrosine phosphatase inhibitor, causes dose-dependent DA contraction in hypoxia.

Tyrosine phosphorylation causes DA contraction, dependent on oxygen condition. A:30 μM orthovanadate causes DA contraction in hypoxia. B: Orthovanadate causes dose-dependent DA contraction and subsequently prevents “additional” O2-induced DA contraction. C: Orthovanadate dose-response contraction, (n=3–5). D: Genistein reduces orthovanadate-induced DA contraction in hypoxia (n=5).
EGFR transactivation is required for O2-mediated DA contraction
We next attempted to determine the identity of the G protein receptor-coupled tyrosine kinase that is primarily responsible for DA contraction. We compared effects of inhibitors of EGFR versus platelet derived growth factor receptor (PDGFR) on DA tone. The EGFR inhibitor, AG1478 (30 μM) inhibited 80% of the O2-induced DA contraction (fig. 3A: representative trace; fig. 3D: summary data), whereas pretreatment with the PDGFR inhibitor, AG1296 (30 μM), had no effect (fig. 3D). Moreover, AG1478 was selective for oxygen-induced vasoconstriction and did not significantly reduce KCl vasoconstriction (fig. 3A & supplemental fig. 2). These results indicated that EGFR is the receptor-coupled tyrosine kinase that is primarily responsible for DA contraction and is regulated by the balance between tyrosine kinases and phosphatases.
Fig 3. EGFR inhibitor abolished O2-induced DA contraction.

Effects of (A): AG1478 (EGFR inhibitor), (30 μM AG1478 had no effect on KCl-induced contraction), (B): AG490 (EGFR and Jak family tyrosine kinase inhibitor) and (C): PP2 (Src family protein tyrosine kinases inhibitor) on oxygen-induced DA contraction. (D): summary data and n number. Note: PDGF inhibitor (AG1296) had no effect on O2-induced contraction. AICAR is an activator of AMP kinase.
Because EGFR is known to signal by means of p38 and JAK, we next explored the role of these kinases in DA contraction. AG490 (30 μM), an EGFR and JAK family tyrosine kinase inhibitor, completely inhibited O2-mediated DA contraction (fig. 3B). EGFR is also known to be phosphorylated and activated by Src32. The Src inhibitor PP2 (30 μM) also abolished O2-mediated DA contraction (fig. 3C). In contrast, the inactive Src control PP3 did not affect O2-induced DA contraction (fig. 3D). To determine whether the inhibitory effects of blocking EGFR were specific, we also evaluated the effects of a battery of inhibitors of other putative signaling pathways: LFM-A13 (a Bruton’s tyrosine kinase inhibitor), GM6001 (a matrix metalloproteinases inhibitor) and concanavalin A (an inhibitor of endocytotic processes). These did not affect the oxygen-induced DA contraction (data not shown).
p38 and JNK but not the ERK, PI3 kinase pathways are involved in oxygen-induced DA contraction
To further explore EGFR’s downstream signal pathway, we studied the effect of ansomycin (30 μM), an activator of p38 and JNK. Ansomycin pretreatment markedly increased DA tension under hypoxic conditions (increasing tone to approximately 50% of that achieved by O2-induced contraction, n=5) and reduced subsequent oxygen-induced DA contraction (fig. 4A–B). The inhibition of p38 MAP kinase (SB202190 10 μM) or the inhibition of JNK (JNK inhibitor II, 10 μM) had, as predicted, the opposite effect, significantly attenuating O2-induced DA contraction (fig 4C). In contrast, neither inhibition of ERK kinase with PD98059 (30 μM), nor inhibition of the PI3K pathway, with LY294002 (10 μM) or Wortmannin (1 μM), altered O2-induced DA contraction (fig. 4C). These experiments indicate that p38 and JNK, but not the ERK and PI3K pathways are involved in O2-induced DA contraction.
Fig 4. p38 and JNK but not the ERK pathway are involved in O2-induced DA contraction.

(A) and (B): Relationship of the effect of ansomycin (p38 and JNK activator) on DA tension change and subsequent O2-induced DA tension change (n=5). (C) Different inhibitor experiments indicate that the p38 and JNK pathways are necessary for O2-mediated DA contraction. SB203580 (p38 inhibitor); PD98059 (ERK inhibitor); LY294002 and Wortmannin (PI3-kinase inhibitor).
Proper attachment to the extracellular matrix is essential for cell survival33. Integrins are transmembrane heterodimeric adhesion receptors that mediate both cell-cell and cell-matrix interactions throughout the body34. Cooperation between integrins and EGFR plays an important role in the regulation of cell growth, differentiation, and survival, and it depends on c-Src kinase activities35. The integrin receptor ligand gly-arg-gly-asp-ser-pro (GRGDSP) was used to test the possible effect of integrins on O2-induced DA contraction. Consistent with the proposed role of Src in the activation of EGF-mediated DA contraction, GRGDSP (500 μM) and EGF (1 μg/ml) both caused significant DA contractions (36% or 50% of the maximum O2-induced contraction, fig. 5A). As observed with oxygen, human EGF, GRGDSP and orthovanadate increased cytosolic calcium in human DASMCs (fig. 5B and 5C). In addition, EGFR siRNA (but not the scrambled control) markedly reduced EGFR mRNA and attenuated O2-induced increases in cytosolic calcium in DASMC. The O2-induced fura-2 ratio change was significantly decreased from +0.11±0.01 (scrambled siRNA, n=57) to +0.03±0.0 (siEGFR, n=87); p<0.01 (supplemental fig. 3). These data support the concept that Src-mediated activation of EGFR might contribute to O2-induced DA contraction.
Fig 5. EGF causes DA tension change and calcium influx.
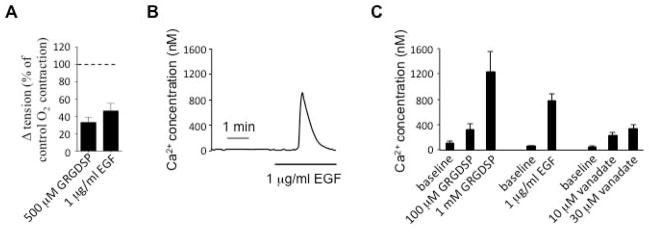
A: Integrin ligands GRGDSP and EGF cause strong DA contraction (n=4–6). B: As observed with oxygen, EGF causes calcium influx and increases cytosolic calcium in human DASMCs. C: Calcium image summary data for integrin ligands GRGDSP, EGF and vanadate (n=15–36).
Mitochondria-derived H2O2 mediates O2-induced EGFR phosphorylation in human DASMC
We next studied the acute effect O2 on EGFR in human DASMC’s. Using immunoprecipitation with anti-EGFR, we found that acute oxygen exposure for as little as 3-minutes significantly increased EGFR tyrosine phosphorylation (fig. 6A). By immunostaining technique, we found that EGFR tyrosine residue Y1173 is rapidly phosphorylated by acute (15-minutes) oxygen exposure in human DASMCs (fig. 6B–C).
Fig 6. Acute O2-induced EGFR phosphorylation in human DASMCs.

A: Consistent with the importance of EGFR in O2-contraction, a crucial EGFR tyrosine residue was phosphorylated in 3 human DA smooth muscle cell lines after 3 to 5 minute normoxia. * P<0.05, n=5, Vs hypoxia control by immunoprecipitation technique with anti-EGFR and anti-tyrosine. B: EGFR tyrosine residue Y1173 was phosphorylated by 15 minutes O2 exposure by immunostaining. Mitochondria-specific overexpression of catalase with Ad-mitoCAT abolished the effect. C: summary data for immunostaining (n=36–50). D. EGFR mRNA was significantly increased after 1 to 72 hours normoxia in cultured human DASMCs by qRt-PCR. (* P<0.05, Vs hypoxia control. * P<0.05, ** P<0.01 Vs hypoxia control, n=4).
Previous studies had shown that augmenting intracellular catalase attenuates O2 sensing in DASMC36. We further studied the compartmental specificity of the signaling H2O2 effect on EGFR phosphorylation by infecting DASMC with the adenoviral vector containing a mitochondrial-targeted catalase gene (Ad-mitoCAT6). O2-induced EGFR tyrosine Y1173 residue phosphorylation was significantly inhibited by transfection of DASMC with Ad-mitoCAT, versus Ad-empty (each at 100 MOI for 96 hours) (Fig. 6B–C). By qRT-PCR, we also found that EGFR mRNA was also significantly increased in cultured human DASMCs exposed to increased pO2 for 1 to 72 hours (fig. 6D). These findings are consistent with the interpretation that mitochondrial derived hydrogen peroxide is essential for the oxygen-induced activation (phosphorylation) of EGFR.
Acute O2 exposure causes EGFR-dependent p38, JNK phosphorylation in human DASMCs
O2 exposure caused time-dependent phosphorylation of p38 (at thr180/tyr182) and JNK (at thr183/tyr185) in cultured human DASMCs, with a peak response at 3 to 5 minutes (fig. 7A and B). The O2-induced phosphorylation of p38 and JNK was completely attenuated by the EGFR inhibitor, AG1478 (fig. 7C and D), which suggests that O2-induced EGFR transactivation is the upstream signal of p38 and JNK phosphorylation (fig. 8).
Fig 7. The O2-induced phosphorylation of p38 and JNK is completely reduced by the EGFR inhibitor, AG1478.

p38 and JNK were phosphorylated in cultured human DASMCs within 5 minutes of normoxia. (A and B) and can be significantly inhibited by EGFR inhibitor, AG1478 (C and D) (n=3–6).
Fig. 8.
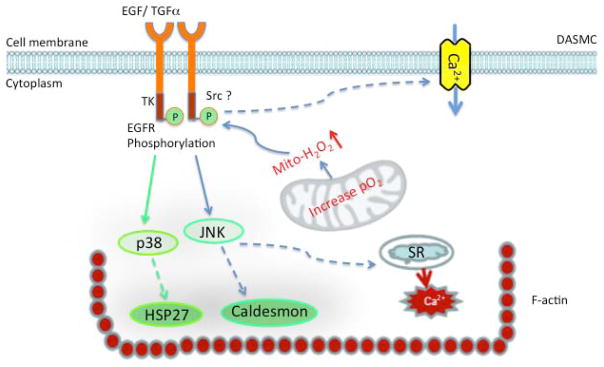
O2 induces EGFR signal pathway transactivation in DASMCs. O2 generates mitochondrial H2O2 and results in tyrosine phosphorylation of EGFR. Subsequently, the EGFR signal pathway activates p38 and JNK. This results in phosphorylation of HSP27 and caldesmon and Ca2+ release. Ultimately EGFR activation alters F-actin structural arrangement. The dotted lines indicate possible mechanisms.
Discussion
It is widely accepted that receptor tyrosine kinases play an important part in vasoconstriction elicited in response to activation of G protein coupled receptors. In this study we demonstrate that tyrosine kinases play a key role in O2-induced DA contraction (fig. 1 and fig. 2). Activation of G-protein coupled receptors (GPCRs), in particular EGFR, is involved in DA O2-sensing. We demonstrate that O2 causes EGFR transactivation and initiates signaling through a p38/JNK cascade. This signaling pathway requires mitochondrial-derived H2O2. The EGFR pathway increases calcium influx and release, resulting in DA contraction (fig. 8).
Mitochondrial-derived H2O2-mediated EGFR transactivation is involved in O2-induced DA contraction
EGF (1 μg/ml) causes about 50% of the maximal O2 -induced contraction in rabbit DA rings (fig. 5A) and stimulates a robust increase in intracellular calcium (~ 700 nM) in human DASMCs (5B–C). Conversely, the EGFR inhibitor, AG1478 abolished 80% of the O2-induced DA contraction (fig. 3A and 3D), whereas PDGFR inhibitor, AG1296 had no effect (fig. 3D). The EGFR inhibitor effect on O2-induced contraction seems specific because it had no significant effect on KCl-induced contraction (fig. 3A, supplemental fig. 2A). These data indicate that EGFR transactivation is required for O2-mediated DA contraction.
The mitochondrial electron transport chain has been proposed as the DA’s O2 sensor for more than a decade2; 37; 38. Inhibitors of either ETC complex I or complex III can fully and selectively abolish and relax O2-induced DA contraction2; 38. Recently we have found that an early step in mitochondrial-based O2 sensing involves a change in mitochondrial structure. Physiologic increases in O2 level cause mitochondrial fission. Within 5-minutes of normoxia DASMC mitochondria fragment by means of an actively regulated process (fission). Fission is required for both the pO2-induced increases in H2O2 and vasoconstriction6. Inhibition of either mitochondrial fission or mitochondria-derived H2O2 production selectively prevents O2-induced DA contraction without altering contraction to other agonists, such as KCl or phenylephrine6. Mitochondrial fission triggers a metabolic shift in the DASMCs that activates pyruvate dehydrogenase and is required for the observed increases in mitochondrial H2O2 production6. Our experiments show that EGFR tyrosine residue phosphorylation is dependent on mitochondrial H2O2 as shown by mito-catalase inhibition.
Mitochondrial-derived H2O2 signals vasoconstriction through at least three known mechanisms: 1). Calcium entry through L-type calcium channels. Roulet et al first reported that O2 caused membrane depolarization in isolated DA strips39. Approximately two thirds of the calcium required for O2-induced contraction enters the DASMC via the L-type calcium channel3. The O2-sensitive, Kv channels (Kv1.5 and Kv2.1) in human DA are inhibited by O2 and cause calcium entry through L-type calcium channels30. 2). O2 causes calcium release from the sarcoplasmic reticulum in DASMC and further induces calcium entry through store-operated channels8; 40. 3). Rho kinase-related calcium sensitization change, which permits prolonged interaction of actin and myosin at any given Ca2+ level8; 10; 40. DA contraction ultimately reflects the aggregate effect of increased cytosolic calcium in the cytosol and calcium sensitization.
EGFR regulates the intracellular effects of ligands such as EGF and transforming growth factor-α (TGFα). Upon stimulation, EGFR undergoes a transition from an inactive monomeric form to an active homodimer and stimulates its intrinsic intracellular protein-tyrosine kinase activity. Several tyrosine (Y) residues in the C-terminal domain of EGFR. including Y1173, can be autophosphorylated. EGFR has also been reported to be an H2O2-sensitive target41. The mechanism involves a direct protein sulfenylation modification of EGFR by H2O2 at a critical active site cysteine (Cys797), which enhances its tyrosine kinase activity41. In our study, the EGFR tyrosine residue Y1173, was rapidly phosphorylated by acute (3–15 minutes) O2 exposure (fig. 6A–B). By using mitochondria-specific overexpression of catalase with Ad-mitoCAT in cultured DASMCs, we proved that the mitochondria-specific H2O2 is a very important initiator of O2- induced EGFR phosphorylation (fig. 6B). The significant mRNA up-regulation of EGFR after 1 hour O2 exposure in cultured human DASMCs (fig. 6D) suggests that EGFR is O2-sensitive at the transcriptional as well as the post translational level. To confirm molecular specificity, we supplemented the findings using pharmacologic inhibitors of EGFR with siRNA targeting of EGFR in cultured human DASMC. The inhibitory effect of siRNA EGFR on the calcium increases caused by oxygen suggests that EGFR plays an important role on O2-sensing (fig. 5E). Thus EGFR is sensitive to acute oxygen change and can be quickly transactivated by mitochondria-specific H2O2.
EGFR transactivation can be induced by many stimuli including reactive oxygen species and occurs through several pathways22; 42; 43; 44. Tseng et al reported that NADPH oxidase-produced superoxide mediates EGFR transactivation by c-Src in arsenic trioxide-stimulated human keratinocytes45. Zhuang et al reported that that H2O2-induced EGFR transactivation is dependent on the activation of Src, whereas chelation of intracellular Ca2+ or inhibition of conventional and novel PKC had no effect on H2O2-induced EGFR transactivation in renal proximal tubular cells21. They also found that H2O2 induced the phosphorylation of Src tyrosine 416, that is required for Src activity, and the phosphorylation of EGFR tyrosine 845, the Src-mediated phosphorylation site. Furthermore, the inhibition of EGFR did not interfere with H2O2-induced Src activation. These data support the concept that Src acts upstream of the EGFR in H2O2-treated cells21. Our data that Src inhibitor PP2 significantly inhibited O2-induced DA contraction, whereas its inactive control PP3 had no effect, is consistent with this concept (fig. 3C–D).
Both PTKs and PTPs are sensitive to reactive oxygen species, redox status and oxygen level changes46; 47; 48. Reversible oxidation of the active site cysteine residue has recently been identified as a general mechanism for regulation of PTPs. Transient oxidation of thiols in PTPs leads to their inactivation by the formation of either an intermolecular S-S bridge or a sulfenyl-amide bond. Conversely, oxidation of PTKs leads to their activation; either by direct SH modification or, indirectly, by concomitant inhibition of PTPs that leads to sustained phosphorylation of PTKs49; 50; 51. By using a modified cysteinyl-labeling assay, we found that PTP activity is decreased after 5–10 min O2 exposure in human DASMCs (supplemental fig. 4). This may contribute to the overall increase in activity of tyrosine kinases, including EGFR, p38 and JNK (fig. 6A–C; Fig. 7).
p38 MAPK and JNK are EGFR transactivation downstream targets
The MAPK family is the main downstream target of EGFR transactivation52; 53. The family includes three subfamilies with multiple members: the extracellular signal-regulated kinases (ERKs), the c-Jun N-amino-terminal/stress-activated protein kinases (JNKs/SAPKs), and the p38 MAPKs. ERK1/2 is typically stimulated by growth factors acting via a tyrosine kinase receptor and is involved in regulation of cell proliferation, whereas p38 and JNK are preferentially activated by cellular stress and are implicated in the regulation of survival, apoptosis, differentiation and contraction, all processes which are important in DA contraction and closure. Sah et al. found that stimulation with EGF causes a biphasic activation of the ERK1/2 MAPK in human ectocervical epithelial cells. The first peak of activation is present at 20 minutes, and the second is present at 36 hours53. Kim et al also reported EGF-induced contraction mediated by the activation of ERK1/2, and regulated by the PI3K pathway, in aortic smooth muscle of DOCA-salt hypertensive rats52.
We found that p38 and JNK specific inhibitors, but not ERK1/2 inhibitor can significantly inhibit O2-induced DA contraction (fig. 4C). Activation of p38 and JNK with ansomycin increased the hypoxic DA tone and subsequently diminished further O2- induced DA contraction (fig. 4A–B). Our Western blot data showed that brief O2 exposure (within 5 minutes) caused p38 and JNK tyrosine residue phosphorylation in human DASMCs (fig. 7A and B). The oxygen-induced phosphorylation of p38 and JNK were completely inhibited by pretreatment with the EGFR inhibitor, AG1478 (fig. 7C and D). These observations suggest that p38 and JNK pathway activation, but not ERK pathway activation, are involved in O2-induced DA contraction.
p38 MAPK and JNK are involved in HSP27 and caldesmon activation
A classic vasoconstriction pathway is one in which signals activate F-actin activation and cause rearrangement of the cytoskeleton. HSP-27 and caldesmon are two major modulators involved. Activation of p38 MAPK leads to phosphorylation of HSP27 in many cell types including smooth muscle cells. The functions of HSP27 in smooth muscle are not fully defined, but there is evidence that HSP27 acts as a chaperone or binding partner for Rho and PKC; binds to and modulates interaction of actin, myosin, and tropomyosin; and regulates actin filament structure. Hsp27 and caldesmon are widely reported to be involved in cytoskeleton rearrangement and to contribute to calcium sensitization54. The latter plays a role in DA contraction8,9,10. However, there is no reported suggestion that they may be involved in oxygen-induced DA contraction. We detected phosphorylation of HSP27 (at the serine 82 residue) and caldesmon (at tyrosine 27 residues) after 3–5 minutes of normoxia in cultured human DASMCs (supplemental fig. 5A and 5B). Thus it appears that both HSP27 and caldesmon are activated by the EGFR/p38/JNK pathway.
Clinical Relevance
The merits of ligation versus the use of drugs, such as indomethacin or ibuprofen, to close the PDA in neonates are still debated55; 56. However, it is clear that both approaches have significant side-effects. Consequently, it would be helpful if a better understanding of the cell signaling that controls the normal closure of the DA leads to improved pharmacological intervention. Similarly, although prostaglandin E1 has been used to maintain DA patency in infants with congenital heart disease, such as pulmonary atresia, for over 35 years 57; 58, it too has side-effects59. Again, better understanding of the physiology may improve treatment.
Conclusions
Oxygen-induced DA contraction is mediated in part by EGFR signal transactivation. This transactivation results in p38 and JNK activation and triggers calcium influx resulting in DA contraction. This finding may offer a new opportunity for the development of improved therapies to close the abnormally patent DA in the neonate or to maintain patency in the presence of critical congenital heart disease to provide pulmonary or systemic blood flow.
Limitation
Although our study suggested that EGFR is likely transactivated by the mitochondrial-derived H2O2, it remains possible that additional G-protein coupled receptors are also involved.
Supplementary Material
Supplemental figure 1: DA ring studies suggest that G proteins are involved in normoxia- induced DA contraction. Modulators of G proteins such as pertussis toxin (PTX, an inhibitor of Gi) and cholera toxin (CTX, a persistent activator of Gs) were used to detect the possibility of a role for G proteins in DA contraction.
A: shows that CTX (1 mg/ml) can attenuate a normoxic contraction and can subsequently diminish the normoxic response of the DA without affecting the baseline hypoxic tone. B: is the summary data for CTX effect on normoxia-induced DA contraction (n=10). This suggests that normoxia-induced DA contraction involves the decrease of Gs activity. C: Shows that 20 to 60 minutes PTX pretreatment (1 mg/ml) has no effect on normoxia-induced DA contraction. 3 μM indomethacin and 100 μM l-NAME were present throughout all experiments. These inhibitors of prostaglandin and nitric oxide synthesis, respectively, were used so that a contractile mechanism independent of these mediators could be studied.
D: Representative traces: Both BQ-123 (selective ETA receptor antagonist, 3 μM) and BQ-788 (selective ETB receptor antagonist, 3 μM) minimally reduced O2-induced contraction, consistent with our previous findings.
Supplemental fig. 2: The effect of AG1478 on KCl-induced DA contraction (n=8).
Supplemental fig. 3: SiEGFR attenuates the O2 induced cytosolic calcium change. A: Effect of SiEGFR on EGFR mRNA in cultured human DASMCs (n=4–6). The IDT SiEGER1 was used in the calcium imaging study. ** P<0.01 compared to scrambled siRNA. B: SiEGFR attenuates the O2 induced cytosolic calcium change (fura-2 ratio) compared to scrambled siRNA. N=51–87, ** P<0.01 compared to Scrambled siRNA.
Supplemental fig. 4: PTP activity is decreased after 5 to 10 min O2 exposure in cultured human DASMCs by a modified cysteinyl-labeling assay from Tonks NK group.
Supplemental fig. 5: O2 caused HSP27 and caldesmon phosphorylation in human DASMCs (n=3).
Key messages.
Oxygen activates epidermal growth factor receptor (EGFR) in ductus arteriosus (DA) smooth muscle cells.
EGFR inhibition selectively attenuates O2-induced DA constriction.
pO2-induced EGFR activation is mediated by mitochondrial-derived hydrogen peroxide.
p38 MAPK and JNK mediated EGFR’s effects on oxygen-induced DA contraction.
Tyrosine kinases and phosphatases participate in oxygen-sensing in the DA.
The EGFR pathway offers new therapeutic targets to modulate patency of the ductus arteriosus.
Acknowledgments
Funding Sources:
This work was supported by Actelion Young Investigators Award (Z. Hong), NIH-RO1-HL65322 (E.K Weir) and NIH grants RO1-HL071115 and 1RC1HL099462-01 (S.L Archer).
Footnotes
Author Disclosure Information: None.
References
- 1.Coceani F, Baragatti B. Mechanisms for ductus arteriosus closure. Seminars in perinatology. 2012;36:92–7. doi: 10.1053/j.semperi.2011.09.018. [DOI] [PubMed] [Google Scholar]
- 2.Michelakis ED, Rebeyka I, Wu X, Nsair A, Thebaud B, Hashimoto K, Dyck JR, Haromy A, Harry G, Barr A, Archer SL. O2 sensing in the human ductus arteriosus: regulation of voltage-gated K+ channels in smooth muscle cells by a mitochondrial redox sensor. Circulation research. 2002;91:478–86. doi: 10.1161/01.res.0000035057.63303.d1. [DOI] [PubMed] [Google Scholar]
- 3.Tristani-Firouzi M, Reeve HL, Tolarova S, Weir EK, Archer SL. Oxygen-induced constriction of rabbit ductus arteriosus occurs via inhibition of a 4-aminopyridine-, voltage-sensitive potassium channel. The Journal of clinical investigation. 1996;98:1959–65. doi: 10.1172/JCI118999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hamrick SE, Hansmann G. Patent ductus arteriosus of the preterm infant. Pediatrics. 2010;125:1020–30. doi: 10.1542/peds.2009-3506. [DOI] [PubMed] [Google Scholar]
- 5.Weir EK, Lopez-Barneo J, Buckler KJ, Archer SL. Acute oxygen-sensing mechanisms. The New England journal of medicine. 2005;353:2042–55. doi: 10.1056/NEJMra050002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hong Z, Kutty S, Toth PT, Marsboom G, Hammel JM, Chamberlain C, Ryan JJ, Zhang HJ, Sharp WW, Morrow E, Trivedi K, Weir EK, Archer SL. Role of dynamin-related protein 1 (Drp1)-mediated mitochondrial fission in oxygen sensing and constriction of the ductus arteriosus. Circulation research. 2013;112:802–15. doi: 10.1161/CIRCRESAHA.111.300285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thebaud B, Wu XC, Kajimoto H, Bonnet S, Hashimoto K, Michelakis ED, Archer SL. Developmental absence of the O2 sensitivity of L-type calcium channels in preterm ductus arteriosus smooth muscle cells impairs O2 constriction contributing to patent ductus arteriosus. Pediatric research. 2008;63:176–81. doi: 10.1203/PDR.0b013e31815ed059. [DOI] [PubMed] [Google Scholar]
- 8.Hong Z, Hong F, Olschewski A, Cabrera JA, Varghese A, Nelson DP, Weir EK. Role of store-operated calcium channels and calcium sensitization in normoxic contraction of the ductus arteriosus. Circulation. 2006;114:1372–9. doi: 10.1161/CIRCULATIONAHA.106.641126. [DOI] [PubMed] [Google Scholar]
- 9.Clyman RI, Waleh N, Kajino H, Roman C, Mauray F. Calcium-dependent and calcium-sensitizing pathways in the mature and immature ductus arteriosus. Am J Physiol Regul Integr Comp Physiol. 2007;293:R1650–6. doi: 10.1152/ajpregu.00300.2007. [DOI] [PubMed] [Google Scholar]
- 10.Kajimoto H, Hashimoto K, Bonnet SN, Haromy A, Harry G, Moudgil R, Nakanishi T, Rebeyka I, Thebaud B, Michelakis ED, Archer SL. Oxygen activates the Rho/Rho-kinase pathway and induces RhoB and ROCK-1 expression in human and rabbit ductus arteriosus by increasing mitochondria-derived reactive oxygen species: a newly recognized mechanism for sustaining ductal constriction. Circulation. 2007;115:1777–88. doi: 10.1161/CIRCULATIONAHA.106.649566. [DOI] [PubMed] [Google Scholar]
- 11.Archer SL, Wu XC, Thebaud B, Moudgil R, Hashimoto K, Michelakis ED. O2 sensing in the human ductus arteriosus: redox-sensitive K+ channels are regulated by mitochondria-derived hydrogen peroxide. Biological chemistry. 2004;385:205–16. doi: 10.1515/BC.2004.014. [DOI] [PubMed] [Google Scholar]
- 12.Kwon J, Lee SR, Yang KS, Ahn Y, Kim YJ, Stadtman ER, Rhee SG. Reversible oxidation and inactivation of the tumor suppressor PTEN in cells stimulated with peptide growth factors. Proc Natl Acad Sci U S A. 2004;101:16419–24. doi: 10.1073/pnas.0407396101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Abe K, Toba M, Alzoubi A, Koubsky K, Ito M, Ota H, Gairhe S, Gerthoffer WT, Fagan KA, McMurtry IF, Oka M. Tyrosine kinase inhibitors are potent acute pulmonary vasodilators in rats. American journal of respiratory cell and molecular biology. 2011;45:804–8. doi: 10.1165/rcmb.2010-0371OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mita M, Tanaka H, Yanagihara H, Nakagawa J, Hishinuma S, Sutherland C, Walsh MP, Shoji M. Membrane depolarization-induced RhoA/Rho-associated kinase activation and sustained contraction of rat caudal arterial smooth muscle involves genistein-sensitive tyrosine phosphorylation. Journal of smooth muscle research = Nihon Heikatsukin Gakkai kikanshi. 2013;49:26–45. doi: 10.1540/jsmr.49.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ward JP, Knock GA, Snetkov VA, Aaronson PI. Protein kinases in vascular smooth muscle tone--role in the pulmonary vasculature and hypoxic pulmonary vasoconstriction. Pharmacol Ther. 2004;104:207–31. doi: 10.1016/j.pharmthera.2004.08.009. [DOI] [PubMed] [Google Scholar]
- 16.Normanno N, De Luca A, Bianco C, Strizzi L, Mancino M, Maiello MR, Carotenuto A, De Feo G, Caponigro F, Salomon DS. Epidermal growth factor receptor (EGFR) signaling in cancer. Gene. 2006;366:2–16. doi: 10.1016/j.gene.2005.10.018. [DOI] [PubMed] [Google Scholar]
- 17.Lee CK, Brown C, Gralla RJ, Hirsh V, Thongprasert S, Tsai CM, Tan EH, Ho JC, Chu da T, Zaatar A, et al. Impact of EGFR inhibitor in non-small cell lung cancer on progression-free and overall survival: a meta-analysis. Journal of the National Cancer Institute. 2013;105:595–605. doi: 10.1093/jnci/djt072. [DOI] [PubMed] [Google Scholar]
- 18.Misra A, Pandey C, Sze SK, Thanabalu T. Hypoxia activated EGFR signaling induces epithelial to mesenchymal transition (EMT) PloS one. 2012;7:e49766. doi: 10.1371/journal.pone.0049766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang Y, Roche O, Yan MS, Finak G, Evans AJ, Metcalf JL, Hast BE, Hanna SC, Wondergem B, Furge KA, et al. Regulation of endocytosis via the oxygen-sensing pathway. Nature medicine. 2009;15:319–24. doi: 10.1038/nm.1922. [DOI] [PubMed] [Google Scholar]
- 20.Norton CE, Broughton BR, Jernigan NL, Walker BR, Resta TC. Enhanced depolarization-induced pulmonary vasoconstriction following chronic hypoxia requires EGFR-dependent activation of NAD(P)H oxidase 2. Antioxidants & redox signaling. 2013;18:1777–88. doi: 10.1089/ars.2012.4836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhuang S, Schnellmann RG. H2O2-induced transactivation of EGF receptor requires Src and mediates ERK1/2, but not Akt, activation in renal cells. American journal of physiology. Renal physiology. 2004;286:F858–65. doi: 10.1152/ajprenal.00282.2003. [DOI] [PubMed] [Google Scholar]
- 22.Ding G, Zhang A, Huang S, Pan X, Zhen G, Chen R, Yang T. ANG II induces c-Jun NH2-terminal kinase activation and proliferation of human mesangial cells via redox-sensitive transactivation of the EGFR. American journal of physiology. Renal physiology. 2007;293:F1889–97. doi: 10.1152/ajprenal.00112.2007. [DOI] [PubMed] [Google Scholar]
- 23.Chan HL, Chou HC, Duran M, Gruenewald J, Waterfield MD, Ridley A, Timms JF. Major role of epidermal growth factor receptor and Src kinases in promoting oxidative stress-dependent loss of adhesion and apoptosis in epithelial cells. The Journal of biological chemistry. 2010;285:4307–18. doi: 10.1074/jbc.M109.047027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cuadrado A, Garcia-Fernandez LF, Gonzalez L, Suarez Y, Losada A, Alcaide V, Martinez T, Fernandez-Sousa JM, Sanchez-Puelles JM, Munoz A. Aplidin induces apoptosis in human cancer cells via glutathione depletion and sustained activation of the epidermal growth factor receptor, Src, JNK, and p38 MAPK. The Journal of biological chemistry. 2003;278:241–50. doi: 10.1074/jbc.M201010200. [DOI] [PubMed] [Google Scholar]
- 25.Chien PS, Mak OT, Huang HJ. Induction of COX-2 protein expression by vanadate in A549 human lung carcinoma cell line through EGF receptor and p38 MAPK-mediated pathway. Biochemical and biophysical research communications. 2006;339:562–8. doi: 10.1016/j.bbrc.2005.11.045. [DOI] [PubMed] [Google Scholar]
- 26.Hong Z, Olschewski A, Reeve HL, Nelson DP, Hong F, Weir EK. Nordexfenfluramine causes more severe pulmonary vasoconstriction than dexfenfluramine. American journal of physiology. Lung cellular and molecular physiology. 2004;286:L531–8. doi: 10.1152/ajplung.00247.2003. [DOI] [PubMed] [Google Scholar]
- 27.Daub H, Wallasch C, Lankenau A, Herrlich A, Ullrich A. Signal characteristics of G protein-transactivated EGF receptor. The EMBO journal. 1997;16:7032–44. doi: 10.1093/emboj/16.23.7032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hollenberg MD, Cuatrecasas P. Epidermal growth factor: receptors in human fibroblasts and modulation of action by cholera toxin. Proceedings of the National Academy of Sciences of the United States of America. 1973;70:2964–8. doi: 10.1073/pnas.70.10.2964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Coceani F, Kelsey L. Endothelin-1 release from lamb ductus arteriosus: relevance to postnatal closure of the vessel. Canadian journal of physiology and pharmacology. 1991;69:218–21. doi: 10.1139/y91-033. [DOI] [PubMed] [Google Scholar]
- 30.Michelakis E, Rebeyka I, Bateson J, Olley P, Puttagunta L, Archer S. Voltage-gated potassium channels in human ductus arteriosus. Lancet. 2000;356:134–7. doi: 10.1016/S0140-6736(00)02452-1. [DOI] [PubMed] [Google Scholar]
- 31.Thebaud B, Michelakis ED, Wu XC, Moudgil R, Kuzyk M, Dyck JR, Harry G, Hashimoto K, Haromy A, Rebeyka I, Archer SL. Oxygen-sensitive Kv channel gene transfer confers oxygen responsiveness to preterm rabbit and remodeled human ductus arteriosus: implications for infants with patent ductus arteriosus. Circulation. 2004;110:1372–9. doi: 10.1161/01.CIR.0000141292.28616.65. [DOI] [PubMed] [Google Scholar]
- 32.Biscardi JS, Maa MC, Tice DA, Cox ME, Leu TH, Parsons SJ. c-Src-mediated phosphorylation of the epidermal growth factor receptor on Tyr845 and Tyr1101 is associated with modulation of receptor function. The Journal of biological chemistry. 1999;274:8335–43. doi: 10.1074/jbc.274.12.8335. [DOI] [PubMed] [Google Scholar]
- 33.Giannoni E, Buricchi F, Grimaldi G, Parri M, Cialdai F, Taddei ML, Raugei G, Ramponi G, Chiarugi P. Redox regulation of anoikis: reactive oxygen species as essential mediators of cell survival. Cell death and differentiation. 2008;15:867–78. doi: 10.1038/cdd.2008.3. [DOI] [PubMed] [Google Scholar]
- 34.Lin B, Arai AC, Lynch G, Gall CM. Integrins regulate NMDA receptor-mediated synaptic currents. Journal of neurophysiology. 2003;89:2874–8. doi: 10.1152/jn.00783.2002. [DOI] [PubMed] [Google Scholar]
- 35.Danilkovitch-Miagkova A, Angeloni D, Skeel A, Donley S, Lerman M, Leonard EJ. Integrin-mediated RON growth factor receptor phosphorylation requires tyrosine kinase activity of both the receptor and c-Src. The Journal of biological chemistry. 2000;275:14783–6. doi: 10.1074/jbc.C000028200. [DOI] [PubMed] [Google Scholar]
- 36.Reeve HL, Tolarova S, Nelson DP, Archer S, Weir EK. Redox control of oxygen sensing in the rabbit ductus arteriosus. The Journal of physiology. 2001;533:253–61. doi: 10.1111/j.1469-7793.2001.0253b.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chandel NS, Schumacker PT. Cellular oxygen sensing by mitochondria: old questions, new insight. Journal of applied physiology. 2000;88:1880–9. doi: 10.1152/jappl.2000.88.5.1880. [DOI] [PubMed] [Google Scholar]
- 38.Cogolludo AL, Moral-Sanz J, van der Sterren S, Frazziano G, van Cleef AN, Menendez C, Zoer B, Moreno E, Roman A, Perez-Vizcaino F, Villamor E. Maturation of O2 sensing and signaling in the chicken ductus arteriosus. American journal of physiology. Lung cellular and molecular physiology. 2009;297:L619–30. doi: 10.1152/ajplung.00092.2009. [DOI] [PubMed] [Google Scholar]
- 39.Roulet MJ, Coburn RF. Oxygen-induced contraction in the guinea pig neonatal ductus arteriosus. Circ Res. 1981;49:997–1002. doi: 10.1161/01.res.49.4.997. [DOI] [PubMed] [Google Scholar]
- 40.Clyman RI, Waleh N, Kajino H, Roman C, Mauray F. Calcium-dependent and calcium-sensitizing pathways in the mature and immature ductus arteriosus. American journal of physiology. Regulatory, integrative and comparative physiology. 2007;293:R1650–6. doi: 10.1152/ajpregu.00300.2007. [DOI] [PubMed] [Google Scholar]
- 41.Paulsen CE, Truong TH, Garcia FJ, Homann A, Gupta V, Leonard SE, Carroll KS. Peroxide-dependent sulfenylation of the EGFR catalytic site enhances kinase activity. Nature chemical biology. 2012;8:57–64. doi: 10.1038/nchembio.736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Saito Y, Berk BC. Transactivation: a novel signaling pathway from angiotensin II to tyrosine kinase receptors. Journal of molecular and cellular cardiology. 2001;33:3–7. doi: 10.1006/jmcc.2000.1272. [DOI] [PubMed] [Google Scholar]
- 43.Zhang H, Chalothorn D, Jackson LF, Lee DC, Faber JE. Transactivation of epidermal growth factor receptor mediates catecholamine-induced growth of vascular smooth muscle. Circulation research. 2004;95:989–97. doi: 10.1161/01.RES.0000147962.01036.bb. [DOI] [PubMed] [Google Scholar]
- 44.Gschwind A, Zwick E, Prenzel N, Leserer M, Ullrich A. Cell communication networks: epidermal growth factor receptor transactivation as the paradigm for interreceptor signal transmission. Oncogene. 2001;20:1594–600. doi: 10.1038/sj.onc.1204192. [DOI] [PubMed] [Google Scholar]
- 45.Tseng HY, Liu ZM, Huang HS. NADPH oxidase-produced superoxide mediates EGFR transactivation by c-Src in arsenic trioxide-stimulated human keratinocytes. Archives of toxicology. 2012;86:935–45. doi: 10.1007/s00204-012-0856-9. [DOI] [PubMed] [Google Scholar]
- 46.Luo H, Wu Z, Tremblay J, Thorin E, Peng J, Lavoie JL, Hu B, Stoyanova E, Cloutier G, Qi S, Wu T, Cameron M, Wu J. Receptor tyrosine kinase Ephb6 regulates vascular smooth muscle contractility and modulates blood pressure in concert with sex hormones. J Biol Chem. 2012;287:6819–29. doi: 10.1074/jbc.M111.293365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Beitner-Johnson D, Ferguson T, Rust RT, Kobayashi S, Millhorn DE. Calcium-dependent activation of Pyk2 by hypoxia. Cell Signal. 2002;14:133–7. doi: 10.1016/s0898-6568(01)00253-4. [DOI] [PubMed] [Google Scholar]
- 48.Chiarugi P. PTPs versus PTKs: the redox side of the coin. Free Radic Res. 2005;39:353–64. doi: 10.1080/10715760400027987. [DOI] [PubMed] [Google Scholar]
- 49.Giannoni E, Buricchi F, Grimaldi G, Parri M, Cialdai F, Taddei ML, Raugei G, Ramponi G, Chiarugi P. Redox regulation of anoikis: reactive oxygen species as essential mediators of cell survival. Cell Death Differ. 2008;15:867–78. doi: 10.1038/cdd.2008.3. [DOI] [PubMed] [Google Scholar]
- 50.Chiarugi P, Buricchi F. Protein tyrosine phosphorylation and reversible oxidation: two cross-talking posttranslation modifications. Antioxid Redox Signal. 2007;9:1–24. doi: 10.1089/ars.2007.9.1. [DOI] [PubMed] [Google Scholar]
- 51.Chiarugi P, Giannoni E. Anchorage-dependent cell growth: tyrosine kinases and phosphatases meet redox regulation. Antioxid Redox Signal. 2005;7:578–92. doi: 10.1089/ars.2005.7.578. [DOI] [PubMed] [Google Scholar]
- 52.Kim J, Lee CK, Park HJ, Kim HJ, So HH, Lee KS, Lee HM, Roh HY, Choi WS, Park TK, Kim B. Epidermal growth factor induces vasoconstriction through the phosphatidylinositol 3-kinase-mediated mitogen-activated protein kinase pathway in hypertensive rats. Journal of pharmacological sciences. 2006;101:135–43. doi: 10.1254/jphs.fp0060021. [DOI] [PubMed] [Google Scholar]
- 53.Sah JF, Eckert RL, Chandraratna RA, Rorke EA. Retinoids suppress epidermal growth factor-associated cell proliferation by inhibiting epidermal growth factor receptor-dependent ERK1/2 activation. The Journal of biological chemistry. 2002;277:9728–35. doi: 10.1074/jbc.M110897200. [DOI] [PubMed] [Google Scholar]
- 54.Gerthoffer WT. Signal-transduction pathways that regulate visceral smooth muscle function. III. Coupling of muscarinic receptors to signaling kinases and effector proteins in gastrointestinal smooth muscles. American journal of physiology. Gastrointestinal and liver physiology. 2005;288:G849–53. doi: 10.1152/ajpgi.00530.2004. [DOI] [PubMed] [Google Scholar]
- 55.Clyman RI, Couto J, Murphy GM. Patent ductus arteriosus: are current neonatal treatment options better or worse than no treatment at all? Seminars in perinatology. 2012;36:123–9. doi: 10.1053/j.semperi.2011.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ohlsson A, Walia R, Shah SS. Ibuprofen for the treatment of patent ductus arteriosus in preterm and/or low birth weight infants. The Cochrane database of systematic reviews. 2013;4:CD003481. doi: 10.1002/14651858.CD003481.pub5. [DOI] [PubMed] [Google Scholar]
- 57.Heymann MA, Rudolph AM. Ductus arteriosus dilatation by prostaglandin E1 in infants with pulmonary atresia. Pediatrics. 1977;59:325–9. [PubMed] [Google Scholar]
- 58.Weir EK, Matisonn RE, Losman E, Chesler E. Prostaglandin E1 in infants with pulmonary atresia. South African medical journal = Suid-Afrikaanse tydskrif vir geneeskunde. 1978;54:700–2. [PubMed] [Google Scholar]
- 59.Huang FK, Lin CC, Huang TC, Weng KP, Liu PY, Chen YY, Wang HP, Ger LP, Hsieh KS. Reappraisal of the prostaglandin E1 dose for early newborns with patent ductus arteriosus-dependent pulmonary circulation. Pediatrics and neonatology. 2013;54:102–6. doi: 10.1016/j.pedneo.2012.10.007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental figure 1: DA ring studies suggest that G proteins are involved in normoxia- induced DA contraction. Modulators of G proteins such as pertussis toxin (PTX, an inhibitor of Gi) and cholera toxin (CTX, a persistent activator of Gs) were used to detect the possibility of a role for G proteins in DA contraction.
A: shows that CTX (1 mg/ml) can attenuate a normoxic contraction and can subsequently diminish the normoxic response of the DA without affecting the baseline hypoxic tone. B: is the summary data for CTX effect on normoxia-induced DA contraction (n=10). This suggests that normoxia-induced DA contraction involves the decrease of Gs activity. C: Shows that 20 to 60 minutes PTX pretreatment (1 mg/ml) has no effect on normoxia-induced DA contraction. 3 μM indomethacin and 100 μM l-NAME were present throughout all experiments. These inhibitors of prostaglandin and nitric oxide synthesis, respectively, were used so that a contractile mechanism independent of these mediators could be studied.
D: Representative traces: Both BQ-123 (selective ETA receptor antagonist, 3 μM) and BQ-788 (selective ETB receptor antagonist, 3 μM) minimally reduced O2-induced contraction, consistent with our previous findings.
Supplemental fig. 2: The effect of AG1478 on KCl-induced DA contraction (n=8).
Supplemental fig. 3: SiEGFR attenuates the O2 induced cytosolic calcium change. A: Effect of SiEGFR on EGFR mRNA in cultured human DASMCs (n=4–6). The IDT SiEGER1 was used in the calcium imaging study. ** P<0.01 compared to scrambled siRNA. B: SiEGFR attenuates the O2 induced cytosolic calcium change (fura-2 ratio) compared to scrambled siRNA. N=51–87, ** P<0.01 compared to Scrambled siRNA.
Supplemental fig. 4: PTP activity is decreased after 5 to 10 min O2 exposure in cultured human DASMCs by a modified cysteinyl-labeling assay from Tonks NK group.
Supplemental fig. 5: O2 caused HSP27 and caldesmon phosphorylation in human DASMCs (n=3).