

Abstract
The second generation of Bcr-Abl inhibitors nilotinib, dasatinib, and bosutinib developed to override imatinib resistance are not active against the T315I ‘gatekeeper’ mutation. Here we describe a Type-II T315I inhibitor GNF-7, based upon a 3,4-dihydropyrimido[4,5-d]pyrimidin-2(1H)-one scaffold which is capable of potently inhibiting wild-type and T315I Bcr-Abl as well as other clinically relevant Bcr-Abl mutants such as G250E, E255V, F317L and M351T in biochemical and cellular assays. In addition, GNF-7 displayed significant in vivo efficacy against T315I-Bcr-Abl without appreciable toxicity in a bioluminescent xenograft mouse model using a transformed T315I-Bcr-Abl-Ba/F3 cell line that has a stable luciferase expression. GNF-7 is amongst the first type II inhibitors capable of inhibiting T315I to be described and will serve as a valuable lead to design next generation Bcr-Abl kinase inhibitors.
Keywords: Bcr-Abl kinase, Chronic Myelogenous Leukemia (CML), T315I gatekeeper mutation, Type-II T315I-Bcr-Abl inhibitor
INTRODUCTION
The successful development of the Bcr-Abl inhibitor imatinib for the treatment of Chronic Myelogenous Leukemia (CML) has provided the paradigm for the development of a host of other small molecule inhibitors targeting kinase whose activity become deregulated in cancer. One major problem facing the development of selective protein kinase inhibitors is the emergence of drug resistance caused by mutations in the kinase domain. Extensive in vitro and clinical work has elucidated a large number of mutations that confer resistance to imatinib either by directly influencing the drug binding site or by disfavoring the conformational rearrangements required for imatinib to bind. Several second generation Bcr-Abl inhibitors have been developed including nilotinib (Tasigna®, AMN107) (Weisberg et al., 2005), bosutinib (SKI-606) (Puttini et al., 2006), dasatinib (Sprycell®, BMS-354825) (Shah et al., 2004) that are capable of inhibiting most of the known Bcr-Abl mutants with the exception of the so-called ‘gatekeeper’ mutation T315I (O’Hare et al., 2005).
Several small molecules capable of inhibiting the T315I mutant in biochemical and cellular assays have been reported. AG-490, an inhibitor of Jak2 which is a kinase implicated in signal transduction downstream of Bcr-Abl, was shown to induce apoptosis in Ba/F3-Bcr-Abl-T315I cell line (Samanta et al., 2006). AP23846, originally developed as a Src kinase inhibits T315I Bcr-Abl dependent cellular proliferation (IC50 of 297 nM) but also inhibits parental Ba/F3 cell lines suggesting it possesses additional intracellular targets. VX-680 (MK-0457), originally developed as an aurora kinase inhibitor exhibits potent enzymatic inhibition of T315I-Abl (IC50 of 30 nM) but only modestly inhibited cellular auto-phosphorylation (IC50 of ca. 5 uM) of Ba/F3 transformed with T315I Bcr-Abl (Carter et al., 2005). Another Aurora kinase inhibitor, PHA-739358 currently being investigated in a phase II clinical trial for patients with relapsed chronic myelogeneous leukemia exhibited potent inhibition of T315I-Abl enzyme (IC50 of 5 nM). Crystallographic analysis of PHA-739358 in complex with T315I-Abl reveals (Modugno et al., 2007) that the isoleucine side chain of T315I mutant does not cause a steric clash with PHA-739358 in contrast to imatinib. TG101114 (Quintás-Cardama et al., 2007), a thiazole–based inhibitor also exhibited good potency (IC50 of 50 nM) against T315I mutant enzyme and exhibited reasonable in vivo efficacy in a xenograft mouse model harboring T315I. SGX393 (O’Hare et al., 2008) derived from pyrrolo[2,3-b]pyridine scaffold class was originally identified using a crystallographic fragment-based screening approach, displayed excellent activity (IC50 of 7.3 nM) against T315I-Bcr-Abl-Ba/F3. Interestingly, SGX393 is considerably less potent against P-loop mutations such as E255V compared with T315I. Another reported class of Bcr-Abl inhibitors is exemplified by ON012380 which is claimed to be a non-ATP-competitive Bcr-Abl inhibitor potently inhibits imatinib-resistant Bcr-Abl mutants such as T315I in cellular and biochemical assays with IC50 values below 10 nM (Gumireddy et al., 2005). ON012380 appears to target substrate binding site of Abl kinase domain but numerous other cellular kinases are inhibited by this compound. It should be noted that most T315I inhibitors disclosed to date except ON012380 are categorized as Type-I kinase inhibitors which bind exclusively to the ATP binding site of kinase with the kinase in an otherwise catalytically competent state. Recently, several compounds from the Type-II class that recognize the “DFG-out” conformation have rarely been reported to inhibit T315I. These include the 9-(arenethenyl)purine analogue, AP24163 (Huang et al., 2009), and the biaryl urea-derived inhibitors NVP-BBT594 and NVP-BGG463 (Chimia 2008; 62; 579) and DSA series compounds(Seeliger et al., 2009). AP24163 exhibited modest potency (IC50 400 nM) against T315I M351T in biochemical and cellular assays and DSA8 showed great potency (IC50 33 nM) on T315I enzyme along with moderate anti-proliferatve activity (IC50 500 nM) evaluated using T315I Bcr-abl transformed Ba/F3 cells. A co-crystal structure with wild-type and gatekeeper mutant of Src with a PP1-based type II inhibitor revealed how the inhibitor could leave ample space for an enlarged gatekeeper residue (Dar et al., 2008)
RESULTS AND DISCUSSION
Here we describe a Type-II T315I inhibitor, based upon a 3,4-dihydropyrimido[4,5-d]pyrimidin-2(1H)-one scaffold which occupies the ATP binding cleft as well as an immediately adjacent hydrophobic pocket of Abl kinase domain. A representative member of this class, GNF-6 (Okram et al., 2006) was crystallized with Abl and shown to bind in the Type-II conformation. The pyrimidopyrimidinone inhibitor descried here, GNF-7 is capable of inhibiting wild-type and T315I Bcr-Abl activity in biochemical and cellular assays and also inhibits other clinically relevant Bcr-Abl mutants such as G250E, E255V, F317L and M351T. We also demonstrate that GNF-7 is capable of inhibiting T315I-Bcr-Abl dependent tumor growth in a murine model of CML (Weisberg et al., 2005).
Design Rationale for a Type-II T315I Bcr-Abl Inhibitor
An examination of the co-crystal structures of imatinib (Nagar et al., 2002) nilotinib (Weisberg et al., 2005) and dasatinib (Tokarski et al., 2006) with Abl demonstrate that all three inhibitors form a critical hydrogen bond with the side-chain hydroxyl group of T315I and would also require a significant rearrangement of their binding conformation to accommodate a larger residue at the gatekeeper position. Mutation of this gatekeeper position appears to be a general theme for resistance to kinase inhibitors as exemplified clinically by resistance of T790M EGFR to Erolotinib (Pao et al., 2005) and experimentally by the resistance of G697R FLT3 to PKC412 and SU5614 (Cools et al., 2004), T341M c-Src to the pyrido[2,3-d]pyridinone (PP58), T681I PDGFRβto the pyrido[2,3-d]pyridinone (PP58) and imatinib, and V561M FGFR1 to pyrido[2,3-d]pyridinones (PP58) (Blencke et al., 2004; Cools et al., 2003).
It is well known that a variety of potent inhibitors of the Src-family such as AZD0530 (Hennequin et al., 2006) and bosutinib also show potent inhibition of Abl kinase. Indeed the 2-amino-5-carboxamidothiazoles, dasatinib compound class was originally developed as an inhibitor of the Src-family kinase Lck to inhibit T-cell activation with potential utility as an immunosuppressant (Wityak et al., 2003). Likewise pyrimidopyrimidones such as PD173955 were originally developed as inhibitors of Src kinase (Kraker et al., 2000) and receptor tyrosine kinase inhibitors such a EGFR, FGFR, PDGFR and c-Kit (Panek et al., 1997; Klutchko et al., 1998) and were only later demonstrated to possess potent cellular and enzymatic activity on wild-type and mutant forms of Bcr-Abl (Wisniewski et al., 2002; Dorsey et al., 2000). Co-crystal structures of PD173955 with Abl (PBD:1m52) demonstrate that these compounds bind to the ATP-binding site with the kinase otherwise assuming a conformation normally utilized to bind ATP (Type I binding) (Nagar et al., 2002). Interestingly PD173955 exhibits approximately 30-fold more potent cellular activity against Bcr-Abl relative to imatinib presumably as a consequence of possessing a much higher affinity to the ATP-binding pocket (Wisniewski et al., 2002). In contrast, imatinib only exhibits cellular activity against Bcr-Abl and some receptor kinases, such as KIT, PDGFR and DDR, but is not known to inhibit the cellular activity of any Src-family kinases. The crystal structure of imatinib bound to the kinase domain of c-Abl (Nagar et al., 2002) demonstrated that the compound binds to a conformation of the kinase where the activation loop is in a so-called “in-active” conformation first visualized in the inactive conformation of the insulin-receptor tyrosine kinase (Type II binding). This binding conformation allows the piperazine-benzamide moiety of imatinib to access an additional hydrophobic pocket directly adjacent to the ATP binding site. By superimposing the bound conformation of PD173955 and nilotinib as depicted in Figure 1, it is clear that additional functionality can be appended to the dichlorophenyl ring of PD173955 to access this adjacent hydrophobic pocket to create a new “hybrid” structure (Okram et al., 2006; Cowan-Jacob et al., 2004). We hypothesized that the resulting hybrid compound might be able to inhibit the T315I mutation due to enhanced affinity both to the hinge-binding region and to the hydrophobic back-pocket, in addition to not forming a hydrogen bond to the gatekeeper position.
Figure 1.

Superimposed structure of PD173955 (green sticks) bound to Abl (PDB;1m52) and AMN107 (yellow sticks) bound to Abl (PBD:3cs9).
In vitro Potency of GNF-7
To assess the cellular activity of the compounds, we tested them against wild-type and mutant Bcr-Abl transformed Ba/F3 cells. Wild-type Ba/F3 cell survives in the presence of interlukin-3 (IL-3) but Ba/F3 cell transformed by oncogenic kinase such as Bcr-Abl becomes capable of growing in the absence of IL-3 and provides a robust and commonly used assay for selective kinase inhibition (Melnick et al., 2006; Warmuth et al., 2007). The first hybrid compound we made, GNF-6 exhibited an IC50 of less than 5 nM on wild-type Bcr-Abl and an IC50 of 303 nM on T315I while exhibiting non-specific inhibition of untransformed Ba/F3 cells with an IC50 of 1.7 uM (Table 1). GNF-6 also effectively inhibited cellular kinase autophosphorylation of T315I-Bcr-Abl-Ba/F3 with an IC50 of 243 nM further demonstrating that the antiproliferative activity against this mutant correlates with direct inhibition of the T315I-Abl enzyme. A co-crystal structure (Okram et al., 2006) of GNF-6 with Abl revealed that the compound bound to a conformation that was virtually indistinguishable from that used by imatinib which clearly validates the design strategy (Liu and Gray, 2006). We next evaluated how altering the structure in the hinge binding region and hydrophobic back pocket might change the potency of cellular inhibition. We replaced the amino-pyrimidine hinge binding motif of GNF-6 with the phenylaminopyrimidine motif which is known to form a stronger interaction with the kinase hinge residues as shown for GNF-7 and GNF-8. These modifications improved absolute cellular potency against T315I as well as increase the ratio for selectivity relative to untransformed Ba/F3 cells. In addition, these modifications resulted in improved inhibitory potency on T315I enzyme as well as against T315I cellular autophosphorylation. We next incorporated a 3-methylimidazole by analogy to nilotinib and the resulting compound GNF-9 also demonstrated excellent activity against wild-type and mutant Bcr-Abl compared with absence of this methylimidazole (Table 1). In particular, this methylimidazole of GNF-9 significantly contributes to excellent potency of GNF-9 against T315I enzyme.
Table 1.
In vitro potency profiling for 3,4-dihydropyrimido[4,5-d]pyrimidin-2(1H)-one on Bcr-Abl
| Anti-proliferative activity (IC50, nM) on Ba/F3 cell lines | Enzymatic activity (IC50, nM) | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Structure & Code | Ba/F3 | wtBcr-Abl | G250E | E255V | T315I | F317L | M351T | c-Abl | G250E | E255V | T315I | M351T |
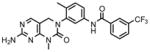 GNF-6 |
1799 | <5 | 294 | <5 | 303 (243*) | <5 | <5 | <5 | N/A | <5 | 1567 | <5 |
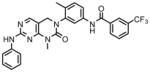 GNF-8 |
2690 | <5 | 12 | 27 | 21 (151*) | 6 | <5 | 133 | 136 | 122 | 61 | <5 |
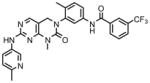 GNF-7 |
612 | <5 | <5 | 10 | 11 (40*) | <5 | <5 | 135 | 206 | <5 | 130 | <5 |
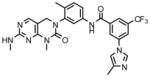 GNF-9 |
630 | 3 | 3 | 30 | 43 | 3 | 3 | <5 | 720 | <5 | <5 | N/A |
Inhibitory potency (IC50, nM) against cellular kinase autophosphorylation (Elisa)
In addition, GNF-7 displayed excellent growth inhibitory activity against human colon cancer cells (Colo205 and SW620) while a non cancer cell line, HEK293T was not particularly sensitive to inhibition by GNF-7 as described in Table 3. Due to the broad selectivity profile of GNF-7, further profiling of a larger panel of human cancer cell lines would potentially reveal additional utility for this inhibitor.
Table 3.
The growth inhibitory potency (IC50, uM) of GNF-7
| TPR-Met-Ba/F3 | NPM-ALK-Ba/F3 | JAK1-Ba/F3 | JAK2-Ba/F3 | JAK2-V617F-Ba/F3 | JAK3-Ba/F3 | FGFR3-Ba/F3 | Flt3-Ba/F3 | Flt3-ITD-Ba/F3 | PDGFRβ-Ba/F3 | TrkC-Ba/F3 | Colo205 | SW620 | U87 | HEK293T |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 3.401 | 1.466 | 0.044 | 0.127 | 0.050 | 2.018 | 0.009 | 0.359 | 0.012 | 0.011 | 0.008 | 0.005 | 0.001 | 5.956 | >10 |
Kinase Selectivity of GNF-7
We next investigated the kinase selectivity of GNF-7 against a panel of 41 recombinant kinase enzymes and on a panel of Ba/F3 cell lines transformed with diverse set of tyrosine kinases as summarized in Tables 2 and 3. This analysis revealed that the compound possessed a lack of selectivity, inhibiting both tyrosine and serine/threonine kinases. In addition to the large number of kinases inhibited by PD173955, kinases such as JNK, Bmx, and c-Raf were also inhibited by GNF-7. GNF-7 however exhibited some selectivity (4 to 100-fold) for T315I Bcr-Abl (IC50 = 11 nM) relative to kinases such as TPR-Met, NPM-ALK, JAK-3, Flt-3. Although the co-crystal structure of GNF-6 with Abl demonstrates binding to the inactive conformation (type II), it is possible that an alternative binding conformation is exploited when binding to other kinases. Further work will be required to investigate to what extent broad inhibition of mutant alleles of Bcr-Abl can be achieved while still maintaining significant selectivity relative to other kinases. Considering that the gatekeeper residue is a crucial kinase selectivity determinant (Cools et al., 2004), this will likely present a significant challenge.
Table 2.
Kinase selectivity profile of GNF-7: Enzymatic activity (% inhibition) of GNF-7 at 10 uM
| 0 | MAPKAP-K2(h) | 31 | Aurora-A(h) |
| 58 | MEK1(h) | 69 | Axl(h) |
| 98 | MKK4(m) | 98 | Bmx(h) |
| 100 | MKK6(h) | 100 | c-RAF(h) |
| 92 | p70S6K(h) | 0 | CaMKIV(h) |
| 45 | PAK2(h) | 3 | CDK1/cyclinB(h) |
| 62 | PDGFRα(h) | 12 | CHK2(h) |
| 0 | PDK1(h) | 6 | CK2(h) |
| 7 | PKBα(h) | 100 | CSK(h) |
| 51 | PKCα(h)-His | 98 | Fes(h) |
| 22 | PKCθ(h) | 100 | FGFR3(h) |
| 77 | PKD2(h) | 69 | Flt3(h) |
| 2 | ROCK-II(h) | 59 | GSK3β(h) |
| 95 | Rsk1(h) | 69 | IKKα(h) |
| 95 | SAPK2a(h) | 49 | IKKβ(h) |
| 98 | SAPK2b(h) | 89 | IR(h) |
| 35 | SAPK3(h) | 97 | JNK1α1(h) |
| 0 | SGK(h) | 99 | JNK2α2(h) |
| 98 | Syk(h) | 99 | Lck(h) |
| 100 | TrkB(h) | 68 | MAPK1(h) |
| 6 | ZAP-70(h) |
In vivo Efficacy and Toxicity of GNF-7
In order to investigate whether GNF-7 could inhibit wild-type and T315I Bcr-Abl at well tolerated doses in vivo, we investigated the tumor efficacy in luciferase xenograft model. One short coming of the original PD173955 compound was a poor pharmacokinetic profile in mice. GNF-7 exhibited excellent pharmacokinetic parameters in mice as depicted in Table 4, with good systemic exposure (AUC = 26656 hrs*nM, Cmax = 3.6 uM) along with reasonable half life (t1/2=3.2 hrs) and favorable oral bioavailability (BAV=36%) being observed following oral administration of a single dose of 20 mg/kg. GNF-7 displayed significant in vivo efficacy against T315I-Bcr-Abl in the bioluminescent xenograft mouse model using a transformed T315I-Bcr-Abl-Ba/F3 cell line that has a stable luciferase expression. As illustrated in the Figures 2, light emission from mice that were administered an oral dose of 10 or 20 mg/kg of GNF-7 once per day was significantly (T/C 38% and 23%, respectively) reduced compared with that from untreated control mice, indicating that GNF-7 effectively inhibits tumor growth of T315I-Bcr-Abl-Ba/F3 cells in mice at low doses (10 mg/kg). However, appreciable (> 10%) body weight loss was observed in mice treated with doses of GNF-7 of 20 mg/kg and above which is a common symptom of in vivo toxicity. However, the 10 mg/kg dosing group exhibited only a very small weight change as plotted in the Figure 3. The significant body weight loss in the 20 mg/kg group forced the dosing to be discontinued at day 6. An acceptable therapeutic index of GNF-7 would be anticipated at around 10 mg/kg dose as remarkable efficacy was observed with little body weight change at 10 mg/kg dose.
Table 4.
Pharmacokinetic properties of GNF-7 on Balb/C mouse
| Formulation | PEG300 (100%), solution | PEG300/water 1:1, solution |
|---|---|---|
| Dosing | 5 mpk, intravenous | 20 mpk, peroral |
| CLs (mL/min/kg) | 8.6 | |
| Vss (L/kg) | 1.12 | |
| AUC (hrs*nM) | 18527 | 875.71 |
| Cmax (nM) | 11707 | 3616 |
| Tmax (hrs) | 0.03 | 3 |
| Clast (nM) | 10 | 15 |
| Tlast (hrs) | 24 | 24 |
| T1/2 (hrs) | 3.8 | 3.2 |
| F (%) | 36 |
Figure 2.

Bioluminescent in vivo efficacy study (oral administration, once-daily dosing) for GNF-7 using Ba/F3-T315I-Bcr-Abl cell line that has a stable luciferase expression. Mice were imaged at day 5 and 7 after GNF-7 treatment.
Figure 3.
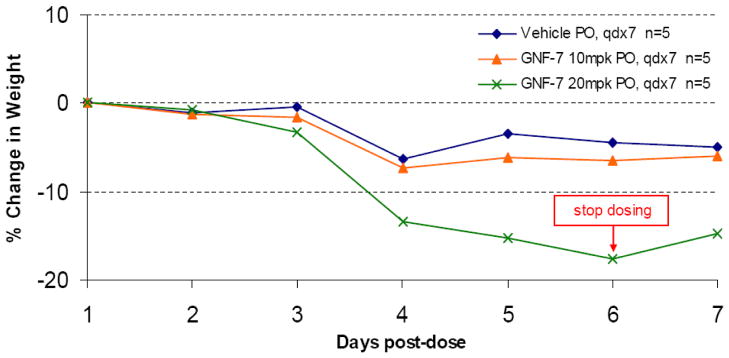
Mice body weight change during bioluminescent in vivo efficacy study (oral administration, once-daily dosing) for GNF-7
SIGNIFICANCE
Our results demonstrate that it is possible to design a Type-II inhibitor that can circumvent the T315I Bcr-Abl “gatekeeper” mutation by bridging the ATP and allosteric binding site using a linker segment that can accommodate a larger gatekeeper residue. The ability of the compounds to tolerate diverse gatekeeper amino acids results in structures with broad kinase selectivity profiles. This demonstrates that Type-II inhibitors as a class are not necessarily more selective than Type-I inhibitors. GNF-7 is likely to serve as a valuable starting point for developing Type–II inhibitors for a variety of kinases.
EXPERIMENTAL PROCEDURES
Detailed experimental procedures for Bcr-Abl kinase assay, Ba/F3 cell proliferation assay, and phosphotyrosine analysis such as auto-phosphorylation estimation are described in our previous publications (Okram et al., 2006; Adrian et al., 2006).
Supplementary Material
Acknowledgments
We would like to thank Sungjoon Kim, Xiang-ju Gu for Ba/F3 cell proliferation assay and acknowledge scientific guidance from Peter G. Schultz.
Footnotes
Supplemental Data include detailed experimental descriptions for chemical synthesis and spectral analysis data of GNF-7, in vivo efficacy evaluation, pharmacokinetics estimation.
References
- Adrian FJ, Ding Q, Sim T, Velentza A, Sloan C, Liu Y, Zhang G, Hur W, Ding S, Manley P, et al. Allosterc inhibitors of Bcr-abl-dependent cell proliferation. Nat Chem Biol. 2006;2:95–102. doi: 10.1038/nchembio760. [DOI] [PubMed] [Google Scholar]
- Blencke S, Zech B, Engkvist O, Greff Z, Orfi L, Horváth Z, Kéri G, Ullrich A, Daub H. Characterization of a conserved structural determinant controlling protein kinase sensitivity to selective inhibitors. Chem Biol. 2004;11:691–701. doi: 10.1016/j.chembiol.2004.02.029. [DOI] [PubMed] [Google Scholar]
- Carter TA, Wodicka LM, Shah NP, Velasco AM, Fabian MA, Treiber DK, Milanov ZV, Atteridge CE, Biggs WH, 3rd, Edeen PT, et al. Inhibition of drug-resistant mutants of ABL, KIT, and EGF receptor kinases. Proc Natl Acad Sci USA. 2005;102:11011–11016. doi: 10.1073/pnas.0504952102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cools J, DeAngelo DJ, Gotlib J, Stover EH, Legare RD, Cortes J, Kutok J, Clark J, Galinsky I, Griffin JD, Cross NC, et al. A tyrosine kinase created by fusion of the PDGFRA and FIP1L1 genes as a therapeutic target of imatinib in idiopathic hypereosinophilic syndrome. N Engl J Med. 2003;13:1201–1214. doi: 10.1056/NEJMoa025217. [DOI] [PubMed] [Google Scholar]
- Cools J, Mentens N, Furet P, Fabbro D, Clark JJ, Griffin JD, Marynen P, Gilliland DG. Prediction of resistance to small molecule FLT3 inhibitors: implications for molecularly targeted therapy of acute leukemia. Cancer Res. 2004;64:6385–6389. doi: 10.1158/0008-5472.CAN-04-2148. [DOI] [PubMed] [Google Scholar]
- Cowan-Jacob SW, Guez V, Fendrich G, Griffin JD, Fabbro D, Furet P, Liebetanz J, Mestan J, Manley PW. Imatinib (STI571) resistance in chronic myelogenous leukemia: molecular basis of the underlying mechanisms and potential strategies for treatment. Mini Rev Med Chem. 2004;3:285–299. doi: 10.2174/1389557043487321. [DOI] [PubMed] [Google Scholar]
- Dar AC, Lopez MS, Shokat KM. Small molecule recognition of c-Src via the Imatinib-binding conformation. Chem Biol. 2008;10:1015–1022. doi: 10.1016/j.chembiol.2008.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorsey JF, Jove R, Kraker AJ, Wu J. The pyrido[2,3-d]pyrimidine derivative PD180970 inhibits p210Bcr-Abl tyrosine kinase and induces apoptosis of K562 leukemic cells. Cancer Res. 2000;60:3127–3131. [PubMed] [Google Scholar]
- Gumireddy K, Baker SJ, Cosenza SC, John P, Kang AD, Robell KA, Reddy MV, Reddy EP. A non-ATP-competitive inhibitor of BCR-ABL overrides imatinib resistance. Proc Natl Acad Sci USA. 2005;102:1992–1997. doi: 10.1073/pnas.0408283102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hennequin LF, Allen J, Breed J, Curwen J, Fennell M, Green TP, Lambert-van der Brempt C, Morgentin R, Norman RA, Olivier A, et al. N-(5-chloro-1,3-benzodioxol-4-yl)-7-[2-(4-methylpiperazin-1-yl)ethoxy]-5- (tetrahydro-2H-pyran-4-yloxy)quinazolin-4-amine, a novel, highly selective, orally available, dual-specific c-Src/Abl kinase inhibitor. J Med Chem. 2006;49:6465–6488. doi: 10.1021/jm060434q. [DOI] [PubMed] [Google Scholar]
- Huang WS, Zhu X, Wang Y, Azam M, Wen D, Sundaramoorthi R, Thomas RM, Liu S, Banda G, Lentini SP, et al. 9-(Arenethenyl)purines as Dual Src/Abl Kinase Inhibitors Targeting the Inactive Conformation: Design, Synthesis, and Biological Evaluation. J Med Chem. 2009;52:4743–4756. doi: 10.1021/jm900166t. [DOI] [PubMed] [Google Scholar]
- Klutchko SR, Hamby JM, Boschelli DH, Wu Z, Kraker AJ, Amar AM, Hartl BG, Shen C, Klohs WD, Steinkampf RW, et al. 2-Substituted aminopyrido[2,3-d]pyrimidin-7(8H)-ones. structure-activity relationships against selected tyrosine kinases and in vitro and in vivo anticancer activity. J Med Chem. 1998;41:3276–3292. doi: 10.1021/jm9802259. [DOI] [PubMed] [Google Scholar]
- Kraker AJ, Hartl BG, Amar AM, Barvian MR, Showalter HD, Moore CW. Biochemical and cellular effects of c-Src kinase-selective pyrido[2, 3-d]pyrimidine tyrosine kinase inhibitors. Biochem Pharmacol. 2000;60:885–898. doi: 10.1016/s0006-2952(00)00405-6. [DOI] [PubMed] [Google Scholar]
- Liu Y, Gray NS. Rational design of inhibitors that bind to inactive kinase conformations. Nat Chem Biol. 2006;2:358–364. doi: 10.1038/nchembio799. [DOI] [PubMed] [Google Scholar]
- Melnick JS, Janes J, Kim S, Chang JY, Sipes DG, Gunderson D, Jarnes L, Matzen JT, Garcia ME, Hood TL, et al. An efficient rapid system for profiling the cellular activities of molecular libraries. Proc Natl Acad Sci USA. 2006;103:3153–3158. doi: 10.1073/pnas.0511292103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modugno M, Casale E, Soncini C, Rosettani P, Colombo R, Lupi R, Rusconi L, Fancelli D, Carpinelli P, Cameron AD, et al. Crystal structure of the T315I Abl mutant in complex with the aurora kinases inhibitor PHA-739358. Cancer Res. 2007;67:7987–7990. doi: 10.1158/0008-5472.CAN-07-1825. [DOI] [PubMed] [Google Scholar]
- Nagar B, Bornmann WG, Pellicena P, Schindler T, Veach DR, Miller WT, Clarkson B, Kuriyan J. Crystal structures of the kinase domain of c-Abl in complex with the small molecule inhibitors PD173955 and imatinib (STI-571) Cancer Res. 2002;62:4236–4243. [PubMed] [Google Scholar]
- O’Hare T, Eide CA, Tyner JW, Corbin AS, Wong MJ, Buchanan S, Holme K, Jessen KA, Tang C, Lewis HA, et al. SGX393 inhibits the CML mutant Bcr-AblT315I and preempts in vitro resistance when combined with nilotinib or dasatinib. Proc Natl Acad Sci USA. 2008;105:5507–5512. doi: 10.1073/pnas.0800587105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Hare T, Walters DK, Deininger MW, Druker BJ. AMN107: Tightening the grip of imatinib. Cancer Cell. 2005;7:117–119. doi: 10.1016/j.ccr.2005.01.020. [DOI] [PubMed] [Google Scholar]
- Okram B, Nagle A, Adrián FJ, Lee C, Ren P, Wang X, Sim T, Xie Y, Wang X, Xia G, et al. A general strategy for creating “inactive-conformation” abl inhibitors. Chem Biol. 2006;13:779–786. doi: 10.1016/j.chembiol.2006.05.015. [DOI] [PubMed] [Google Scholar]
- Pao W, Miller VA, Politi KA, Riely GJ, Somwar R, Zakowski MF, Kris MG, Varmus H. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PloS Med. 2005;2:e73. doi: 10.1371/journal.pmed.0020073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panek RL, Lu GH, Klutchko SR, Batley BL, Dahring TK, Hamby JM, Hallak H, Doherty AM, Keiser JA. In vitro pharmacological characterization of PD 166285, a new nanomolar potent and broadly active protein tyrosine kinase inhibitor. J Pharmacol Exp Ther. 1997;283:1433–1444. [PubMed] [Google Scholar]
- Puttini M, Coluccia AM, Boschelli F, Cleris L, Marchesi E, Donella-Deana A, Ahmed S, Redaelli S, Piazza R, Magistroni V, et al. In vitro and in vivo activity of SKI-606, a novel Src-Abl inhibitor, against imatinib-resistant Bcr-Abl+ neoplastic cells. Cancer Res. 2006;66:11314–11322. doi: 10.1158/0008-5472.CAN-06-1199. [DOI] [PubMed] [Google Scholar]
- Quintás-Cardama A, Kantarjian H, Cortes J. Flying under the radar: the new wave of BCR-ABL inhibitors. Nature Rev Drug Disc. 2007;6:834–848. doi: 10.1038/nrd2324. [DOI] [PubMed] [Google Scholar]
- Shah NP, Tran C, Lee FY, Chen P, Norris D, Sawyers CL. Overriding imatinib resistance with a novel ABL kinase inhibitor. Science. 2004;305:399–401. doi: 10.1126/science.1099480. [DOI] [PubMed] [Google Scholar]
- Samanta AK, Lin H, Sun T, Kantarjian H, Arlinghaus RB. Janus kinase 2: a critical target in chronic myelogenous leukemia. Cancer Res. 2006;66:6468–6472. doi: 10.1158/0008-5472.CAN-06-0025. [DOI] [PubMed] [Google Scholar]
- Seeliger MA, Ranjitkar P, Kasap C, Shan Y, Shaw DE, Shah NP, Kuriyan J, Maly DJ. Equally potent inhibition of c-Src and Abl by compounds that recognize inactive kinase conformations. Cancer Res. 2009;69:2384–2392. doi: 10.1158/0008-5472.CAN-08-3953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokarski JS, Newitt JA, Chang CY, Cheng JD, Wittekind M, Kiefer SE, Kish K, Lee FY, Borzillerri R, Lombardo LJ, et al. The structure of Dasatinib (BMS-354825) bound to activated ABL kinase domain elucidates its inhibitory activity against imatinib-resistant ABL mutants. Cancer Res. 2006;66:5790–5797. doi: 10.1158/0008-5472.CAN-05-4187. [DOI] [PubMed] [Google Scholar]
- Warmuth M, Kim S, Gu XJ, Xia G, Adrián F. Ba/F3 cells and their use in kinase drug discovery. Curr Opin Oncol. 2007;19:55–60. doi: 10.1097/CCO.0b013e328011a25f. [DOI] [PubMed] [Google Scholar]
- Weisberg E, Manley PW, Breitenstein W, Brüggen J, Cowan-Jacob SW, Ray A, Huntly B, Fabbro D, Fendrich G, Hall-Meyers E, et al. Characterization of AMN107, a selective inhibitor of native and mutant Bcr-Abl. Cancer Cell. 2005;7:129–141. doi: 10.1016/j.ccr.2005.01.007. [DOI] [PubMed] [Google Scholar]
- Wisniewski D, Lambek CL, Liu C, Strife A, Veach DR, Nagar B, Young MA, Schindler T, Bornmann WG, Bertino JR, et al. Characterization of potent inhibitors of the Bcr-Abl and the c-kit receptor tyrosine kinases. Cancer Res. 2002;62:4244–4255. [PubMed] [Google Scholar]
- Wityak J, Das J, Moquin RV, Shen Z, Lin J, Chen P, Doweyko AM, Pitt S, Pang S, Shen DR, et al. Discovery and initial SAR of 2-amino-5-carboxamidothiazoles as inhibitors of the Src-family kinase p56Lck. Bioorg Med Chem Lett. 2003;13:4007–4010. doi: 10.1016/j.bmcl.2003.08.054. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.