

Abstract
DEAD-box helicases play essential roles in RNA metabolism across species, but emerging data suggest that they have additional functions in immunity. Through RNAi screening we identify an evolutionarily conserved and interferon-independent role for the DEAD-box helicase DDX17 in restricting Rift Valley fever virus (RVFV), a mosquito-transmitted virus in the bunyavirus family that causes severe morbidity and mortality in humans and livestock. Loss of Drosophila DDX17 (Rm62) in cells and flies enhanced RVFV infection. Similarly, depletion of DDX17 but not the related helicase DDX5 increased RVFV replication in human cells. Using cross-linking immunoprecipitation high-throughput sequencing (CLIP-seq), we show that DDX17 binds the stem loops of host pri-miRNA to facilitate their processing, and also an essential stem loop in bunyaviral RNA to restrict infection. Thus, DDX17 has dual roles in the recognition of stem loops: in the nucleus for endogenous miRNA biogenesis and in the cytoplasm for surveillance against structured non-self elements.
INTRODUCTION
RNA helicases control nearly every facet of RNA metabolism, including transcription, splicing, miRNA biogenesis, translation and decay (Linder and Jankowsky, 2011). Comprising the largest family of helicases, the DEAD-box proteins are found in all three kingdoms of life and share 12 conserved motifs, including the DEAD motif characterized by the amino acids Asp-Glu-Ala-Asp. While DEAD-box proteins are most appreciated for their roles in RNA metabolism, some have important functions in antiviral defense. For example, mammalian retinoic acid-inducible gene 1 (RIG-I/DDX58) and myeloma differentiation-associated factor 5 (MDA-5), collectively termed RIG-I-like receptors (RLRs), recognize non-self elements in viral RNAs such as double-stranded RNA (dsRNA) and 5′-triphosphorylated RNA, leading to the transcriptional induction of Type I interferon (IFN-I) and pro-inflammatory cytokines (Loo and Gale, 2011). However, some viruses are not restricted by RLRs in some contexts or encode potent RLR antagonists, and thus additional sensors may have evolved (Bowie and Unterholzner, 2008).
Although RLRs are not strictly conserved in invertebrates such as mosquitoes and Drosophila, insects use a related helicase to combat viral infection. The DEAD-box helicase Dicer-2 (Dcr-2) is a core component of the RNA interference (RNAi) pathway that recognizes double-stranded or structured viral RNAs and cleaves them into 21-nt small-interfering RNAs (siRNAs) (Ding and Voinnet, 2007; Sabin et al., 2013). Virus-derived siRNAs are loaded into an Argonaute-2 (Ago2)-containing RNA-induced silencing complex that cleaves viral RNA. Additionally, during Drosophila C virus (DCV) infection, Dcr-2 controls induction of the antiviral gene Vago (Deddouche et al., 2008).
More recently, several other DEAD-box proteins have been implicated in sensing viral nucleic acids or regulating downstream signaling. For example, DDX41 recognizes intracellular DNA and bacterial cyclic dinucleotides (Parvatiyar et al., 2012; Zhang et al., 2011b), while a complex of DDX1, DDX21 and DHX36 senses viral dsRNA specifically in dendritic cells (Zhang et al., 2011a). Other recently identified helicase sensors or components of antiviral signaling pathways include DDX3, DHX9 and DDX60 (Kim et al., 2010; Miyashita et al., 2011). Thus, the landscape of DEAD-box helicases in innate immunity is more diverse than previously appreciated, and many antiviral helicases likely remain obscure.
As many aspects of innate immunity are conserved in flies as well as many DEAD-box helicases, we performed an RNAi screen to identify novel antiviral helicases. We focused on the arthropod-borne virus (arbovirus) Rift Valley fever virus (RVFV), a tri-segmented negative sense RNA virus in the bunyavirus family (Ikegami and Makino, 2011). In humans, RVFV infection typically causes an acute febrile illness but can progress to more severe manifestations such as encephalitis and hemorrhagic fever with 1–3% mortality. In livestock, infection is particularly lethal with 100% abortion rates and near 100% fatality in neonates (Ikegami and Makino, 2011). No effective vaccines or therapeutics exist for RVFV infection, and therefore additional targets for pharmacologic intervention are needed. Furthermore, we have shown that RVFV is not restricted by RLRs in some contexts including fibroblasts, suggesting other sensors may restrict this pathogen (Moy et al., 2014).
We identified Drosophila Rm62 as a novel host factor that restricts RVFV infection in vitro and in vivo. This restriction was specific for bunyaviruses, as Rm62 also controlled the replication of the distantly related bunyavirus La Crosse virus (LACV), but not viruses from the three other families tested. Remarkably, the antiviral role of Rm62 was conserved in human cells, as the human homolog DDX17 restricted RVFV infection. DDX17 was identified in a high molecular weight complex with Drosha and later shown to regulate the Microprocessor complex that mediates pri-miRNA processing and miRNA biogenesis, but its direct RNA targets are not fully known (Fukuda et al., 2007; Gregory et al., 2004; Mori et al., 2014; Suzuki et al., 2009). Using CLIP-seq, we found that in addition to binding cellular RNAs, DDX17 also interacts with RVFV RNA, likely via structured viral RNA elements. We found striking similarities in the mode of recognition for host and viral RNA: DDX17 binds a subset of pri-miRNA hairpins along with a well-characterized hairpin on the RVFV genome. Cloning this hairpin into Sindbis virus (SINV) decreased its replication in a DDX17-dependent manner in human and insect cells, indicating a direct antiviral function for DDX17 binding to viral RNA. Taken together, these data expand our understanding of DDX17 recognition of cellular and viral RNAs as well as the scope of DEAD-box helicases in antiviral immunity, demonstrating that the immune functions of DEAD-box genes can be evolutionarily conserved from insects to humans.
RESULTS
DEAD-box helicase screen reveals that Rm62 restricts RVFV in Drosophila cells
Because DEAD-box helicases are widely conserved across species (Linder and Jankowsky, 2011), we performed a targeted RNAi screen in Drosophila cells to identify novel factors that control RVFV infection. Drosophila DL1 cells were treated with a panel of dsRNAs targeting 23 DEAD-box helicases that are conserved in humans (Table S1). The cells were then infected with the MP12 strain of RVFV, stained for RVFV nucleoprotein (N) to monitor infection and analyzed using automated microscopy (Hopkins et al., 2013).
Implementing a cut-off of a two-fold increase in percent infection and a p<0.05 across three independent experiments, we identified three DEAD-box helicase genes that restrict RVFV infection without impacting cell viability: me31B, CG10333 and Rm62 (Figure 1A and 1B). These results were independently corroborated by microscopy with depletion of Rm62, CG10333 or me31B resulting in increased infection (Figure 1C). The helicase me31B (DDX6) is a component of cytoplasmic granules known as processing bodies and activates mRNA decapping (Coller et al., 2001). Importantly, a recent genome-wide RNAi screen in Drosophila cells identified me31B as a RVFV restriction factor, validating our helicase screen (Hopkins et al., 2013). CG10333 is the homolog of human DDX23/PRP28, a component of the U5 small ribonucleoprotein that promotes spliceosome assembly during pre-mRNA splicing (Staley and Guthrie, 1999). Rm62, which has two human homologs (DDX5/p68 and DDX17/p72) (Figure S1A), has been studied in several contexts in flies and humans (Fuller-Pace, 2013). In Drosophila, Rm62 has been shown to promote dsRNA-mediated RNAi (Ishizuka et al., 2002), modulate chromatin insulation (Lei and Corces, 2006) and facilitate re-silencing of transcription loci after stimulation (Boeke et al., 2011; Buszczak and Spradling, 2006). In mammals, DDX5 and DDX17 have been associated with transcriptional coactivation and miRNA processing, among other functions (Fukuda et al., 2007; Fuller-Pace, 2013; Mori et al., 2014).
Figure 1. DEAD-box helicase screen identifies RVFV restriction factors.

A. Drosophila cells were treated with the indicated dsRNA, infected with RVFV (MOI=0.04) and processed for immunofluorescence (IF) and automated microscopy 30 hpi. Average fold increase in percent infection compared to control dsRNA-treated cells is shown. Mean±SEM. B. Average fold change in total nuclei from (A). C. Representative IF image of RVFV-infected Drosophila cells treated with the indicated dsRNA. D. Immunoblot for RVFV protein (Gn) from infected cells treated with the indicated dsRNA 30 hpi. E. Average fold increase in RVFV RNA in cells treated with the indicated dsRNA as quantified by Northern. Mean±SEM; *p<0.05, Student’s t-test. All data represent three independent experiments. See also Figure S1 and Table S1.
To further validate the antiviral activity of Rm62 and CG10333, we targeted these genes using independent dsRNAs, observing increased infection by immunofluorescence (Figure S1B), and verified efficient Rm62 silencing (Figure S1C). Rm62, CG10333 and me31B knockdown also increased RVFV glycoprotein Gn (Figure 1D) and RVFV RNA (Figure 1E), additional readouts of viral replication. Thus, these data identify Rm62 and CG10333 as new RVFV restriction factors.
Rm62 is antiviral against RVFV in adult flies
As Rm62 displayed the most potent antiviral effect against RVFV infection in Drosophila cells (Figure 1D and 1E), we next determined if Rm62 also controls viral replication at the organismal level. To silence Rm62 in adult flies, we performed in vivo RNAi by crossing transgenic flies expressing a UAS-controlled inverted repeat transgene directed against Rm62 (UAS-Rm62 IR) to heat shock-driven Gal4 (hs-Gal4). We validated Rm62 depletion by Northern blot (Figure S2A). Rm62-silenced flies (hs-Gal4> UAS-Rm62 IR) and their sibling controls (+> UAS-Rm62 IR) were challenged with RVFV and monitored for survival. Compared to control flies, Rm62-silenced flies exhibited increased mortality after infection (Figure 2A). This survival defect was associated with significantly elevated viral RNA replication, suggesting that Rm62 restricts RVFV infection in vivo (Figure 2B and 2C). Because the transgene is only expressed in adults, the increased susceptibility is not due to a developmental requirement for Rm62. Moreover, Rm62 depletion did not impact survival of uninfected flies (Figure S2B).
Figure 2. Rm62 restricts RVFV infection in adult flies.

A. Rm62-silenced flies (Hs-Gal4> UAS-Rm62 IR) or sibling controls were infected with RVFV and monitored for survival. Mean±SEM; p<0.05, Log-rank test. B. Northern for RVFV RNA from infected flies at d6 and d9 post-infection. C. Fold increase in RVFV S segment RNA from Rm62-silenced flies 6 dpi. Mean±SEM; *p<0.05, Student’s t-test. D. Survival of Rm62 mutant flies (Rm62CB02119/Rm6201086; MUT) or sibling control flies (Rm62CB02119/+; HET) infected with RVFV. Mean±SEM; p<0.001, Log-rank test. E. Representative RNA blot for RVFV RNA from RVFV-infected flies 6 dpi. F. Fold increase in RVFV S segment RNA in Rm62 mutant flies quantified by RNA blot. Mean±SEM; *p<0.05, Student’s t-test. All data represent at least three independent experiments. See also Figure S2.
To validate our in vivo RNAi results and control for potential off-target effects, we tested previously characterized trans-heterozygous Rm62 mutant flies that have substantially reduced Rm62 protein expression (Buszczak and Spradling, 2006). RVFV-infected Rm62 mutant flies (Rm62CB02119/Rm6201086), but not uninfected flies, showed a dramatic increase in mortality compared to sibling controls (Rm62CB02119/+) (Figure 2D and Figure S2C). Rm62 mutants also demonstrated significantly elevated RVFV RNA replication, similar to Rm62-silenced flies (Figure 2E and 2F). Taken together, these data reveal an in vivo requirement for Rm62 in controlling RVFV infection and protecting against lethality.
Rm62 specifically restricts bunyaviral infection in Drosophila
We next evaluated the specificity of Rm62 by testing its role during infection with additional viruses. To test if Rm62 restricts other bunyaviruses, we challenged Rm62-silenced Drosophila cells with LACV, a mosquito-transmitted bunyavirus that causes encephalitis in humans. Rm62 depletion dramatically increased the amount of recovered LACV RNA, indicating that Rm62 also controls LACV replication in vitro (Figure 3A and 3B). Furthermore, LACV-infected Rm62 mutant flies exhibited increased mortality and LACV RNA replication, with an approximately 35-fold elevation in LACV N expression (Figure 3C–E). These data indicate that Rm62 restricts both LACV and RVFV in cells and flies.
Figure 3. Rm62 specifically restricts bunyavirus infection in flies.

A. Drosophila cells were treated with the indicated dsRNA and infected with LACV (MOI=1). Viral RNA (LACV N) was monitored by RNA blot 36 hpi. B. Fold increase in LACV N RNA levels. Mean±SEM; *p<0.05, Student’s t-test. C. Survival of LACV- infected Rm62 mutant (Rm62CB02119/Rm6201086; MUT) or control flies (Rm62CB02119/+; HET). Mean±SEM; p<0.005, Log-rank test. D. Northern for LACV N mRNA from LACV-infected flies 6 dpi. E. Fold increase in LACV N in Rm62 mutant flies. Mean±SEM; *p<0.05, Student’s t-test. F. Survival of DCV-infected flies. G. Survival of VSV-GFP-infected flies. H. Survival of SINV-GFP-infected flies. I. Immunoblot for DCV capsid from DCV-infected flies 4 dpi. J. Fold increase in viral RNA (GFP) from VSV-GFP infected flies 6 dpi quantified by Northern. Mean±SEM. K. Fold increase in viral RNA (GFP) from SINV-GFP infected flies 6 dpi quantified by Northern. Mean±SEM. All data represent three independent experiments. See also Figure S3.
To examine if Rm62’s antiviral activity is limited to bunyaviruses or functions more broadly, we tested additional human arboviruses from distinct classes. Vesicular stomatitis virus (VSV) is a non-segmented negative-sense RNA virus in the Rhabdovirus family, while SINV is a positive-sense RNA alphavirus. Notably, we observed no increase in VSV or SINV protein expression in virus-challenged Drosophila cells treated with Rm62 dsRNA (Figure S3A and S3B). In addition, we infected Rm62 mutant flies with VSV, SINV and DCV, a picorna-like positive-sense RNA virus that is a natural Drosophila pathogen. Rm62 mutant flies showed no increase in mortality with DCV, VSV or SINV infection (Figure 3F–H). Furthermore, we observed no increase in DCV protein, VSV RNA or SINV RNA in Rm62 mutant flies (Figure 3I–K), demonstrating that Rm62 does not restrict these viruses in vivo. Therefore, Rm62 is a selective restriction factor for bunyaviruses in flies.
Human DDX17 controls RVFV and LACV infection
DEAD-box helicases are broadly conserved across species, with humans and mammals encoding two homologs of Rm62: DDX5 and DDX17 (Figure S1A). These proteins have both overlapping and unique functions, but their role in immunity is not defined. Thus, we evaluated whether DDX5 or DDX17 restrict bunyavirus infection in human cells.
To silence DDX5 and DDX17 expression, we transfected a human osteosarcoma cell line (U2OS cells) with gene-specific siRNAs or a non-targeting control siRNA (Figure S4A). Interestingly, while DDX17 silencing had no effect on DDX5 expression, DDX5 knockdown increased DDX17 protein levels, suggesting that DDX5 negatively regulates DDX17 expression, which has been observed in HeLa cells (Jalal et al., 2007). Compared to control cells, DDX17-depleted cells showed a significant increase in percent infection and viral RNA (Figure 4A–D). In contrast, we observed no difference in RVFV infection with DDX5 depletion, suggesting that DDX17 specifically limits RVFV replication in human cells. Independent siRNAs against DDX17 also augmented RVFV infection (Figure S4B and S4C). Furthermore, DDX17 but not DDX5 silencing increased LACV infection in U2OS cells (Figure 4E–G). Taken together, these data indicate that DDX17 restriction of bunyavirus infection is functionally conserved from flies to human cells.
Figure 4. DDX17 controls RVFV and LACV replication in human cells.

A. Representative IF image of RVFV-infected U2OS cells (MOI=0.3) transfected with the indicated siRNAs 16 hpi. B. Relative percent RVFV infection as quantified by automated image analysis. Mean±SEM; *p<0.05, Student’s t-test. C. Representative RNA blot from siRNA-transfected U2OS cells infected with RVFV 16 hpi. D. Fold increase in RVFV N mRNA compared to control siRNA-treated U2OS cells as quantified by Northern. Mean±SEM; *p<0.05, Student’s t-test. E. Representative IF image of LACV-infected U2OS cells (MOI=0.3) transfected with the indicated siRNAs 16 hpi. F. Relative percent LACV infection as quantified by automated image analysis. Mean±SEM; *p<0.05, Student’s t-test. G. Representative Northern for LACV N mRNA from siRNA-transfected U2OS cells infected with LACV 16 hpi. H. U2OS cells were transfected with the indicated siRNAs and infected with RVFV for 16h. Viral protein (Gn) was monitored by immunoblot. I. Immunoblot for viral protein (GFP) from U2OS cells treated with the indicated siRNAs and infected with VSV-GFP at 16 hpi (MOI=1). J. Ifit1 mRNA expression from uninfected or VSV-infected U2OS cells treated with the indicated siRNAs at 16 hpi by qRT-PCR, normalized to uninfected siCON-treated cells. Mean±SEM. K. Ifit1 mRNA expression from uninfected or RVFV-infected U2OS cells treated with the indicated siRNAs at 16 hpi by qRT-PCR, normalized to uninfected siCON-treated cells. Mean±SEM. All data represent three independent experiments. See also Figure S4.
Next we tested whether overexpression of DDX17 limits RVFV replication. Compared to control GFP overexpression, ectopic DDX17 expression reduced RVFV protein accumulation (Figure S4D) and the percentage of infected cells (Figure S4E). These results demonstrate that DDX17 may be limiting and that increased expression can modestly attenuate RVFV infection.
Finally, we tested the specificity of human DDX17 by challenging U2OS cells with VSV. While DDX17-depleted cells demonstrated increased RVFV Gn protein (Figure 4H), DDX17 silencing had no effect on VSV protein expression (Figure 4I). Therefore, as in Drosophila, DDX17 restricts the two bunyaviruses RVFV and LACV but not VSV, indicating selectivity for restricting bunyaviruses.
Some DEAD-box helicases such as RIG-I and MDA5 control virus-induced IFN-I expression. To determine if DDX17 similarly regulates IFN-I production, we tested if DDX17 silencing impacts upregulation of the interferon-stimulated gene (ISG) Ifit1 (Terenzi et al., 2006). Although VSV and RVFV infection induced Ifit1 RNA expression at 16 hours post-infection (hpi), DDX17 knockdown did not abrogate this response; in fact, Ifit1 levels were higher in DDX17-silenced cells with RVFV infection, most likely due to increased viral replication (Figure 4J and 4K). RVFV infection also did not induce IFNa or IFNb expression, likely because the virus encodes a potent interferon antagonist, NSs (Figure S4F and S4G). Notably, DDX17 depletion did not impact the expression of these genes, as well as IkBa, IRF7 and DDX58 (Figure S4H–J). Thus, our data suggest that DDX17 restricts RVFV infection in an interferon-independent manner.
Identification of DDX17-bound RNAs by CLIP-seq
Because DEAD-box helicases function as RNA-binding proteins, we hypothesized that DDX17 may directly bind RVFV RNAs to inhibit viral replication. To determine the specific RNAs bound to endogenous DDX17, we performed CLIP-seq, a method for purifying RNA-binding protein targets from cells under stringent conditions (Darnell, 2010). Briefly, uninfected or RVFV-infected U2OS cells were UV-irradiated, and endogenous DDX17-bound RNAs were digested to ~100 nt fragments, immunoprecipitated from cell lysates with anti-DDX17 or anti-FLAG as a control, and radiolabeled for visualization. We observed efficient depletion of DDX17 from the lysates with anti-DDX17 but not anti-FLAG (Figure 5A). Autoradiography of RNA-protein complexes revealed extensive signal for anti-DDX17 but not anti-FLAG immunoprecipitations, suggesting enrichment for DDX17-bound RNAs (Figure 5B). cDNA libraries were then generated from purified RNAs and submitted for Illumina deep-sequencing.
Figure 5. CLIP-seq analysis of DDX17-bound RNAs from uninfected and RVFV-infected U2OS cells.

A. Immunoblot from uninfected or RVFV-infected U2OS cells with immunoprecipitation (IP) using anti-DDX17 or anti-FLAG (control). Input, IP and unbound fractions are shown, with high efficiency of DDX17 IP. B. Autoradiograph of immunopurified and 32P-labeled DDX17-RNA complexes transferred to nitrocellulose membrane. Immunoprecipitation with anti-FLAG as a control shows high specificity of the DDX17-RNA signal. C. Flowchart of CLIP-seq alignment and processing pipeline, resulting in alignment-clusters. D. Alignment clusters overlapping annotated regions of the genome (refSeq) were further searched for significant peaks and the overlap between infected and uninfected DDX17 significant CLIP-seq peaks (FDR < 0.001) in protein-coding genes from refSeq at increasing peak height is plotted. R2= 0.88. E. Percentage of total nucleotides under significant CLIP-seq peaks within refSeq protein-coding genes broken down into transcript feature types extracted from refSeq. F. Composite motif logo of the multiple sequence alignment of the 20 most enriched hexamers under significant CLIP-seq peaks within protein-coding genes as identified by z-score, comparing hexamer frequencies to 100 permutations of binding site locations within bound transcripts for uninfected (top) or infected (bottom) cells. See also Figure S5 and Table S2.
From three pooled DDX17-CLIP experiments, we obtained ~80 million raw reads and ~90 million raw reads from uninfected and infected cells, respectively (Figure 5C). We generated a composite genome index incorporating the hg19 human genome and three genomic segments of RVFV (L, M, S), with over 55% of reads aligning unambiguously to the composite genome (unique alignments). Collapsed alignments were obtained by removing PCR duplicates and retaining only one alignment for each 5′ coordinate. Genomic intervals with at least 2 overlapping alignments were clustered together generating the alignment clusters. This yielded 733,542 clusters for uninfected cells and 426,135 clusters from RVFV-infected cells. Alignment clusters within human pre-mRNA loci were further searched for significant peaks (FDR<0.001) using an empirical algorithm (Shankarling et al., 2014). DDX17 pre-mRNA peaks showed strong overlap between uninfected and infected cells (Figure 5D), indicating that the overall profile of DDX17-bound cellular RNAs is similar during infection. Next, we determined the transcript features of DDX17 pre-mRNA peaks (Figure 5E). Interestingly, DDX17 peaks were enriched in coding exons, 5′UTRs and 3′UTRs, suggesting that DDX17 preferentially binds mature mRNA. Hexamer enrichment analysis of CLIP-seq peaks within protein-coding genes showed a bias for CT- and CA- repeat elements (Figure 5F and Table S2). Together, these data indicate both location and sequence preference for DDX17 binding to mRNAs.
To understand the functional targets of DDX17, we used DAVID to identify KEGG GO terms enriched among protein-coding genes associated with DDX17 CLIP-seq peaks. We observed enrichment for cell adhesion as well as several cellular signaling pathways (Figure S5A). Intriguingly, one of the most overrepresented KEGG pathways was mitogen-activated protein kinase (MAPK) signaling (Figure S5B). Previous data suggest that MAPK-activated protein kinase 2 (MK2) physically interacts with DDX5 to control its localization, and that DDX5/DDX17 regulate splicing of p38 MAPK (Hong et al., 2013; Samaan et al., 2013). Thus, DDX17-bound RNAs identified in our experiments overlap with known targets in MAPK signaling, suggesting that the CLIP-seq peaks reflect the biological activity of DDX17.
DDX17 directly binds to pri-miRNA stem loops
In addition to roles in transcriptional regulation and alternative splicing, DDX17 has been linked to miRNA biogenesis. DDX5 and DDX17 are components of the Microprocessor complex, which processes the pri-miRNA transcript into the 60–70-nt stem loop intermediate known as the pre-miRNA (Davis et al., 2008; Gregory et al., 2004; Mori et al., 2014; Suzuki et al., 2009). Loss of DDX17 results in decreased expression of a subset but not all miRNAs (Fukuda et al., 2007; Mori et al., 2014). Therefore, as further validation of our CLIP-seq data, we also analyzed the intersection of DDX17 CLIP signal with annotated miRNA stem loops.
We observed 160 pri-miRNA loci that were associated with DDX17 CLIP clusters (Table S3). There was strong correlation in normalized CLIP signal within pri-miRNAs from uninfected and RVFV-infected samples (Figure 6A), suggesting that similar pri-miRNAs are bound by DDX17 in uninfected and infected cells. In contrast, we found no correlation between CLIP-seq signal and level of miRNA expression reported in a previous study of small RNAs in U2OS cells, indicating that DDX17 clusters represent bias for certain miRNAs independent of expression level (Figure 6B). Among DDX17-bound miRNAs, miR-663a, miR99b and mir-6087 were some of the most highly represented miRNAs (Figure 6C). Analysis of DDX17 CLIP signal in relation to the predicted pri-miRNA stem-loop showed that DDX17 clusters were preferentially localized immediately 5′ and 3′ to the center of the loop (Figure 6D). These data suggest that DDX17 interactions are strongest with the stem region of the miRNA hairpin rather than the loop. Analysis of overrepresented hexamers in DDX17-associated miRNAs did not show any enrichment of the CA- or CT-repeat elements found with the DDX17 mRNA peaks (Table S4). Furthermore, de novo analysis of the bound pri-miRNAs identified no significantly enriched motifs compared to total pri-miRNA background. Thus, the interaction of DDX17 with pri-miRNAs is likely determined by RNA secondary structure.
Figure 6. DDX17 directly binds miRNA stem loops in human U2OS cells.

A. Normalized CLIP-seq signal (TPKM, tags per kilobase of pre-miRNA per million CLIP-seq reads) in pre-miRNA hairpin loci with CLIP signal extracted from miRBase. Linear regression of infected TPKM on uninfected TPKM is plotted, R2=0.79. B. Scatterplot of miRNAs that are bound; normalized pre-miRNA expression (RPKM) from small RNA-seq and the mean of normalized CLIP-seq signal (TPKM) between infected and uninfected U2OS cells are plotted, R2=0.001. C. Alignment clusters overlapping miRBase pre-miRNA hairpin loci on the UCSC genome browser with uninfected cells colored black, and infected cells colored red. D. RNA map of DDX17 CLIP signal in pre-miRNA hairpins. Fraction of 160 hairpins bound is plotted at single-nucleotide resolution relative to the center of the stem-loop. See also Tables S3 and S4.
DDX17 binds RVFV RNAs to restrict viral replication
To determine if DDX17 regulation of miRNA biogenesis is directly involved in antiviral defense, we silenced the Microprocessor component Drosha in U2OS cells (Figure S6A). Loss of Drosha had no impact on RVFV replication, suggesting that the antiviral mechanism of DDX17 is independent of Drosha and the canonical miRNA pathway (Figure S6B and S6C). Using luciferase reporter assays as previously described (Sabin et al., 2009), we also found that Rm62 is not required for siRNA- or miRNA-mediated silencing in Drosophila cells (Figure S6D). These data indicate that DDX17 does not act through RNAi to restrict RVFV infection.
Next, we tested if DDX17 directly interacts with viral RNA by analyzing the overlap of DDX17 CLIP clusters with the RVFV genome. We observed multiple DDX17 clusters, with the highest signal on the M and S segments (Figure 7A). These data suggest that DDX17 binds RVFV RNA in infected U2OS cells. In addition, DDX17 viral peaks did not overlap with CA- and CT-repeat motifs, suggesting that DDX17-viral interactions are not dependent on these elements (Figure S6E).
Figure 7. DDX17 binds RVFV RNA to restrict viral infection.
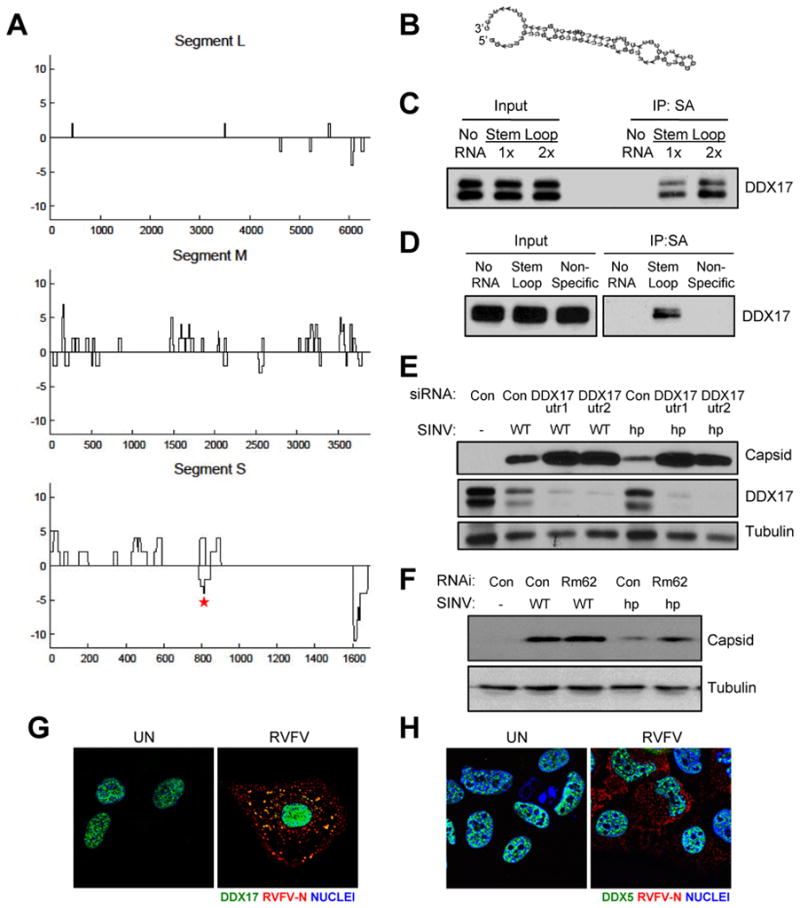
A. DDX17 CLIP-seq clusters aligned to the RVFV tripartite genome, plotted 3′ to 5′ (genome orientation) along the x-axis. Binding sites that map to the genome are below and to the antigenome are above the line. CLIP-seq signal intensity (black) is measured in total overlapping reads at each nucleotide position. B. Predicted secondary structure of a 75-nt RNA from DDX17 CLIP peak on the RVFV S segment between N and NSs as determined by RNAfold (asterisk in A). C. The 75-nt DDX17 CLIP peak RNA from (B) was synthesized by T7 in vitro transcription and biotinylated. Biotinylated RVFV RNA was incubated with U2OS cell protein lysates and immunoprecipitated, and DDX17-RVFV RNA complexes were analyzed by immunoblot. D. RNA-protein interaction assays were performed as in (C) using the biotinylated RVFV stem loop and non-specific control RNA from RVFV not bound in the DDX17 CLIP-seq dataset. E. Representative immunoblot of U2OS cells transfected with the indicated siRNAs and infected with wild type SINV (WT) or SINV encoding the RVFV hairpin (SINV-hp) 8 hpi. F. Representative immunoblot of Drosophila cells treated with control (β-gal) or Rm62 dsRNA and infected with SINV WT or SINV-hp 24 hpi (MOI=0.3). G. Representative IF images of DDX17 and RVFV N from uninfected or infected U2OS cells 12 hpi. H. Representative IF images of DDX5 and RVFV N from uninfected or infected U2OS cells 12 hpi. (Helicase, green; RVFV-N, red; Nuclei, blue). See also Figure S6.
Because viral RNAs are often highly structured and DDX17 was enriched at the stem region of pri-miRNA hairpins, we hypothesized that DDX17 may recognize structured elements in RVFV RNAs. Indeed, we observed a prominent CLIP cluster within the intergenic region (IGR) on the S segment (between N and NSs). The IGR on other ambisense bunyaviruses has been shown to form a highly complementary sequence that folds into a hairpin to control transcription termination (Emery and Bishop, 1987). This IGR in the RVFV antigenome similarly forms a hairpin that generates the majority of virus-derived siRNAs in infected Drosophila and mosquito cells (Sabin et al., 2013). We defined a 75-nt RNA that overlaps the largest S segment DDX17 CLIP cluster within the IGR on the genome strand, which is predicted to form a hairpin structure that resembles miRNA stem loops (Figure 7B). We synthesized this RNA in vitro using T7 RNA polymerase to test whether it is bound by DDX17. Biotinylated DDX17 peak RVFV RNA efficiently precipitated DDX17 from U2OS cell lysates in a dose-dependent manner, demonstrating that DDX17 physically interacts with RVFV RNA and validating our CLIP-seq results (Figure 7C). In contrast, a non-specific control from RVFV RNA not bound in our CLIP-seq dataset did not precipitate DDX17 (Figure 7D).
To determine if DDX17 binding on viral RNA can directly restrict viral infection, we cloned the RVFV DDX17 hairpin into the 3′ UTR of SINV under the control of a sub-genomic promoter (SINV-hp). This same strategy has been previously shown to tolerate the insertion of non-coding hairpin RNAs (Shapiro et al., 2010). We found that control cells supported substantially less infection of SINV-hp compared to wild type (WT) SINV (Figure 7E). Furthermore, while depletion of DDX17 led to modest increases in SINV capsid production of WT virus, loss of DDX17 led to large increases in capsid production from SINV-hp virus (Figure 7E). In addition, we tested if this RVFV hairpin also impacted SINV replication in Drosophila cells. WT SINV was unaffected by the loss of Rm62 (Figure 7F). Moreover, as we found in human cells, control RNAi-treated cells supported less infection of SINV-hp than WT SINV, and depletion of Rm62 led to a large increase in SINV capsid production from SINV-hp virus (Figure 7F). A second DDX17 peak at the 5′-end of the S genomic segment was also predicted to form a hairpin (Figure S6F), and cloning this hairpin into SINV (SINV-5′hp) also sensitized the virus to DDX17 restriction in Drosophila and human cells (Figure S6G and S6H). Together, these data demonstrate that the presence of a DDX17 binding site on viral RNA is restrictive and that this repression can be alleviated by loss of DDX17 across hosts.
We next assessed the localization of DDX17 and DDX5 during infection by immunofluorescence, as RVFV and SINV RNA replication occur exclusively in the cytoplasm. We validated the specificity of these antibodies for immunofluorescence using RNAi (Figure S6I and S6J). As previously reported, DDX17 was found in the nucleus in uninfected cells (Figure 7G) (Bortz et al., 2011). At 12 hpi, however, we observed some DDX17 staining in cytosolic puncta that colocalized with RVFV nucleocapsid protein N, which coats viral RNA and facilitates replication (Figure 7G). In contrast, DDX5 remained in the nucleus in the presence and absence of infection (Figure 7H), suggesting a distinct localization pattern for DDX17. Collectively, these data suggest that DDX17 may gain access to cytosolic RVFV replication complexes during infection and bind viral RNA to antagonize viral replication.
DISCUSSION
Emerging data have begun to uncover specialized functions for mammalian DEAD-box helicases in immunity, particularly in the sensing of viral nucleic acids to activate interferon induction (Fullam and Schroder, 2013). However, whether DEAD-box helicases play interferon-independent roles in antiviral immunity remains unclear. Moreover, whether antiviral DEAD-box helicases exist primarily in mammals or evolved immune functions in lower organisms has not been fully explored. We have discovered a specific and evolutionarily conserved role for the helicase DDX17 in restricting infection with RVFV, a major human arbovirus that lacks effective therapeutics.
Through an RNAi screen in Drosophila cells, we identified Rm62 as a novel anti-RVFV helicase gene. Rm62 is also essential for resistance to RVFV infection in vivo, as RVFV-challenged Rm62-deficient flies showed increased viral replication and mortality. Loss of Rm62 increased LACV infection but not SINV, VSV or DCV infection, and silencing closely related DEAD-box helicase genes had no effect on RVFV. Thus, Rm62 is as an essential and specific virus restriction factor in flies.
Mammals encode two orthologs of Rm62, DDX5 and DDX17, which have been widely studied in transcriptional coactivation, mRNA splicing and miRNA processing. In addition, previous studies have implicated DDX5 and DDX17 in promoting the replication of several viruses, such as hepatitis C virus and influenza virus (Bortz et al., 2011; Goh et al., 2004). We found that silencing DDX17 but not DDX5 increased RVFV replication in human U2OS cells, whereas DDX17 overexpression inhibited RVFV infection. While we cannot unequivocally rule out any antiviral function for DDX5, as DDX5 depletion up-regulated DDX17 expression and we could not efficiently knockdown both proteins simultaneously in U2OS cells, basal DDX5 levels were not able to compensate for DDX17 loss. Therefore, the antiviral activity of DDX17 is evolutionarily conserved from invertebrates to mammals, suggesting an ancient origin for DEAD-box helicases in innate immunity.
DDX17 joins a growing list of antimicrobial DEAD-box proteins that function as cytoplasmic sensors for viral nucleic acids. For example, DDX41 binds DNA to control IFN-I and pro-inflammatory cytokine induction (Zhang et al., 2011b). Additionally, DDX3 interacts with IKKε and TBK-1 to regulate IFN-I activation downstream of virus recognition (Soulat et al., 2008). In contrast, DDX17 is dispensable for antiviral gene expression, suggesting that DDX17 acts independently of IFN-I, which is distinct from previously defined antiviral DEAD-box genes.
Previous reports have proposed that Rm62 and DDX17 regulate RNAi, which is a well-characterized antiviral pathway in invertebrates. In Drosophila cells, Rm62 has been shown to bind Ago2 and control siRNA-mediated silencing (Ishizuka et al., 2002). Based on these findings, one study suggested that Rm62-deficient flies infected with Drosophila X virus (DXV) have increased mortality due to defective antiviral RNAi (Zambon et al., 2006); however, this study did not use sibling-matched or uninfected controls and did not monitor viral replication. In mammalian cells, DDX5 and DDX17 are found in the Microprocessor complex and regulate miRNA biogenesis. However, our data suggest that Rm62 and DDX17 restrict viral infection in an RNAi-independent manner. First, we found that Rm62 controls the replication of RVFV but not VSV, SINV or DCV, viruses that are restricted by antiviral RNAi in flies (Galiana-Arnoux et al., 2006; Mueller et al., 2010; Sabin et al., 2009; van Rij et al., 2006). Second, Rm62 has not been found to control siRNA or miRNA-mediated RNA silencing in more recent in vitro and in vivo screens, which we confirmed in our experiments (Cziko et al., 2009; Zhou et al., 2008). Third, RNAi is not generally thought to restrict viral infection in mammalian somatic cells, while its antiviral function in embryonic and undifferentiated mammalian cell types may be active (Cullen et al., 2013; Li et al., 2013; Maillard et al., 2013). Lastly, depletion of the Microprocessor component Drosha, which can act as an interferon-independent antiviral factor (Shapiro et al., 2014), did not impact RVFV infection. Although there may be additional complexity and interplay between DDX17, miRNA biology and antiviral defense, our data suggest that DDX17’s antiviral function is independent of its role in miRNA biogenesis.
CLIP-seq studies revealed that DDX17 physically associates with viral RNA to control viral infection. One of the DDX17 binding peaks, corresponding to the IGR between the N and NSs genes on the genomic S segment, efficiently precipitated DDX17 from cell lysates. Interestingly, this region forms an extensive hairpin (Sabin et al., 2013), suggesting that DDX17 may recognize highly structured stem loops on viral RNAs. Furthermore, in infected cells, DDX17 forms cytoplasmic puncta overlapping with RVFV N. This relocalization to viral replication complexes may allow DDX17 to access and bind structured viral RNA elements, and thereby limit replication.
Indeed, by expressing the RVFV hairpin from SINV (SINV-hp), we demonstrated that SINV becomes hypersensitive to DDX17 both in human and insect cells. This suggests that binding of DDX17 to viral RNA is sufficient to mediate its antiviral effect. How this binding limits viral replication remains to be clarified in future studies. DDX17 may associate with additional protein cofactors that mediate its antiviral function. For instance, DDX17 has been shown to bind Dcp2 and Dcp1a, which remove the 5′ cap from mRNAs, and the exonuclease Xrn1, which mediates 5′-to-3′ RNA degradation (Zhu et al., 2011). Interestingly, a recent study showed that Drosophila Dcp2 restricts RVFV infection, although this may be in an indirect manner by limiting the pool of cellular mRNA substrates that RVFV utilizes for its own replication (Hopkins et al., 2013). DDX5 and DDX17 also bind components of the RNA exosome, a complex that catalyzes 3′-to-5′ RNA degradation (Chen et al., 2008; Zonta et al., 2013). Consequently, DDX17 may act as the sensor that brings viral RNA targets to the RNA degradation machinery, or it may unwind viral RNAs to facilitate degradation. The decapping machinery, exosome and DDX17 have additionally been linked to the antiviral protein ZAP (Zhu et al., 2011). ZAP is known to restrict SINV replication in human cells (Bick et al., 2003), and we found a modest effect of DDX17 depletion on WT SINV, albeit this effect was not as strong as with SINV-hp. In contrast, flies do not encode a ZAP homolog and Rm62 silencing had no impact on WT SINV replication in insect cells. As maximal DDX17 restriction of SINV depended on the RVFV stem loop in both cells types, DDX17’s antiviral function in RVFV infection is likely independent of ZAP and dependent on direct viral RNA binding.
Why DDX17 specifically targets bunyaviral RNAs is also uncertain, as the rules that direct RNA binding activity of DDX17, as well as DEAD-box proteins in general, is difficult to decipher. We suspect that antiviral specificity derives from a combination of cellular localization and specific RNA structures. Our data suggest that SINV and VSV do not have the appropriate targeting signals. In addition, the correlation between DDX17 expression pattern and viral pathogenesis must be characterized. DDX17 is ubiquitously expressed and is not transcriptionally induced by IFN-I, and our data suggest that subcellular localization but not expression level is responsive to infection. Further studies will allow us to better define the regulation of DDX17 and explore the relationship between tissue type and antiviral activity.
Beyond elucidating new DDX17 functions in immunity, our data reveal important insights into the mechanism of DDX17 recognition for diverse RNAs. Our data suggest that DEAD-box proteins are highly amenable to CLIP-seq analysis. We found that DDX17 cellular mRNA targets are enriched for CT- and CA-repeat elements, suggesting that primary sequence contributes to mRNA recognition. In contrast, DDX17-bound pri-miRNAs were not enriched for this element or any other linear sequence (p> 0.05); instead, DDX17 was localized to the miRNA stem, suggesting that it recognizes pri-miRNAs via secondary structure. We compared our bound pri-miRNAs with two studies that monitored miRNAs upon loss of DDX17 (Fukuda et al., 2007; Mori et al., 2014). Fukuda et al. identified 94 miRNAs that were decreased upon loss of p72 in mouse embryonic fibroblasts that derive from 82 pre-miRNAs expressed in U2OS cells, of which 32% were directly bound by DDX17 in our studies. In another study, Mori et al. identified 317 miRNAs misregulated by DDX17 depletion in HaCaT cells; of the 160 DDX17-bound miRNAs from our study, 60 were analyzed in their cells, 30 of which they found were regulated by DDX17 (50%). Furthermore, Mori et al. identified a sequence motif in the 3′ flanking segment of a subset of pri-miRNAs that were impacted by DDX17 levels ([GTA]CATC[CTA]) and focused on miR-21, a miRNA we also identified as bound by DDX17. Mori et al. demonstrated by in vitro binding assays that both the motif, as well as a complete stem-loop, were required for full binding activity. Altogether, these data suggest that DDX17 recognizes the pri-miRNA stem in the context of a 3′ tail. This would bias DDX17 binding to pri-miRNAs over pre-miRNAs since some additional binding energy would be derived from the flanking regions and would facilitate DDX17 binding to stem loops within larger RNAs as is found in viral RNAs. In further support of this, the ([GTA]CATC[CTA]) sequence motif was over-represented in our mRNA peaks (p=3.7×10−17), suggesting that this sequence is indeed a preferred binding site for DDX17 in diverse RNAs.
In conclusion, our data reveal striking parallels between DDX17 recognition of pri-miRNAs and viral RNAs: in both cases, DDX17 targets a structured stem loop, either to facilitate miRNA processing or to mediate virus inhibition.
EXPERIMENTAL PROCEDURES
RNAi in Drosophila cells
dsRNAs were synthesized as described (Boutros et al., 2004). Drosophila cells were passaged into serum-free media and seeded onto 384-well or 6-well plates containing 250 ng or 4 μg of dsRNA/well, respectively. Complete media was added one hour later, and the cells were incubated for three days for knockdown.
CLIP-seq
U2OS cells were seeded onto 10-cm plates and infected with RVFV (MOI=3) for 16h. The cells were washed in ice-cold HBSS (Gibco) and irradiated at 254 nm (400 mJ/cm2) in a Stratalinker 1800 (Stratagene). The cells were pelleted, flash frozen in liquid nitrogen and stored at −80°C. Anti-DDX17 or anti-FLAG was bound to Dynabeads (Invitrogen) in binding buffer (0.1 M Na-phosphate pH 8 and 0.1% NP-40). Cell pellets were lysed in 1X PXL (1X PBS (no Mg2+ and no Ca2+), 0.1% SDS, 0.5% deoxycholate and 0.5% NP-40) with protease inhibitors and RNasin, and lysates were treated with RNase T1 (1:1000 dilution) and RQ1 DNase for 10 min at 37°C. Lysates were incubated with beads overnight at 4°C. RNA linkers (RL3 and RL5) and DNA primers (DP3 and DP5, DSFP3 and DSFP5) were prepared and used as described (Vourekas et al., 2012). Samples were resolved by NuPAGE and transferred to nitrocellulose membrane, which was exposed to film, and bands were excised above the expected MW of DDX17. RNA extraction, 5′-linker ligation, RT-PCR and re-amplification were performed as described (Vourekas et al., 2012). cDNA libraries were purified and submitted for Illumina deep sequencing.
CLIP-seq data analysis
Raw CLIP-seq reads were trimmed from the 3′ end to remove 2+ contiguous basecalls with Phred quality scores of 0. Sequencing adaptors were removed from the 3′ end with cutadapt version 0.9.4, and homopolymeric runs of 6+ basecalls of the same nucleotide were removed. Trimmed reads were aligned to a composite genome index containing hg19 and RVFV strain MP-12 genomes, allowing at most 2 mismatches and retaining only unambiguous alignments. PCR duplicates were removed by retaining only one alignment for each 5′ coordinate generating collapsed alignments. Genomic intervals with at least 2 overlapping alignments were clustered together generating the alignment clusters. Alignment clusters within annotated protein-coding genes (refSeq NM_*) and ncRNA genes (refSeq NR_*) were further searched for significant peaks with an empirical algorithm, using 100 iterations of permutations and a false discovery rate threshold of 0.001 (Xue et al., 2009).
DDX17-pre-miRNA interactions were identified by intersection of alignment clusters with pre-miRNA loci extracted from miRBase release 20. Mature miRNAs were associated with pre-miRNAs and 160 pre-miRNAs had at least one CLIP cluster in either uninfected or infected cells. For those pre-miRNAs, we included the total number of collapsed alignments for subsequent analysis. To analyze miRNA expression levels in U2OS cells, we aligned small RNA-seq reads from all three replicates of control-transfected U2OS cells (GEO submission GSM889286) (Wei et al., 2012) using the same alignment settings as above and computed mean RPKM values for each pre-miRNA hairpin from miRBase. To identify the relative location of DDX17 binding on pre-miRNA loci, center coordinates for each pre-miRNA locus from miRBase were computed, then intervals from −250nt to +250nt around the center were generated. Intervals that do not intersect any CLIP cluster were discarded. For each locus the clusters were compressed such that each nucleotide was bound or unbound (0 or 1). These were summed across the loci and divided by the total to present the fraction bound at each position.
Computation was executed on the Penn Genome Frontiers Institute High Performance Compute cluster, using Perl version 5.16.2, Python version 2.7.3, R version 3.0.2, bedtools version 2.16.2, and samtools version 0.1.18.
Motif enrichment analysis
Each hexamer occurring within DDX17-pre-mRNA interaction sites was assigned a z-score by comparing observed hexamer frequencies to the backgrounds computed by permutation of the coordinates of interaction sites 100 times within the pre-mRNA in which that site occurred. Multiple sequence alignments of the top 20 hexamers were generated with ClustalW2 and sequence logos were generated with WebLogo version 2.8.
Supplementary Material
Highlights.
dDDX17 (Rm62) restricts RVFV infection in Drosophila
DDX17 controls RVFV replication in human cells
DDX17 CLIP-seq demonstrates binding at pri-miRNA stem loops
DDX17 directly binds to a structured viral RNA element
Acknowledgments
We thank A. Spradling for Rm62 mutant flies; A. Bowie for DDX17 siRNA sequences; E. Dardenne for DDX17 plasmid; M. Tartell for technical help. This work was supported by grants from the National Institutes of Health to S.C. (R01AI074951, U54AI057168 and R01AI095500) and R.H.M. (T32-AI-007324). S.C. and B.T. are recipients of the Burroughs Wellcome Investigators in the Pathogenesis of Infectious Disease Award.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bick MJ, Carroll JW, Gao G, Goff SP, Rice CM, MacDonald MR. Expression of the zinc-finger antiviral protein inhibits alphavirus replication. Journal of virology. 2003;77:11555–11562. doi: 10.1128/JVI.77.21.11555-11562.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boeke J, Bag I, Ramaiah MJ, Vetter I, Kremmer E, Pal-Bhadra M, Bhadra U, Imhof A. The RNA helicase Rm62 cooperates with SU(VAR)3–9 to re-silence active transcription in Drosophila melanogaster. PloS one. 2011;6:e20761. doi: 10.1371/journal.pone.0020761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bortz E, Westera L, Maamary J, Steel J, Albrecht RA, Manicassamy B, Chase G, Martinez-Sobrido L, Schwemmle M, Garcia-Sastre A. Host- and strain-specific regulation of influenza virus polymerase activity by interacting cellular proteins. mBio. 2011;2 doi: 10.1128/mBio.00151-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boutros M, Kiger AA, Armknecht S, Kerr K, Hild M, Koch B, Haas SA, Paro R, Perrimon N. Genome-wide RNAi analysis of growth and viability in Drosophila cells. Science. 2004;303:832–835. doi: 10.1126/science.1091266. [DOI] [PubMed] [Google Scholar]
- Bowie AG, Unterholzner L. Viral evasion and subversion of pattern-recognition receptor signalling. Nature reviews Immunology. 2008;8:911–922. doi: 10.1038/nri2436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buszczak M, Spradling AC. The Drosophila P68 RNA helicase regulates transcriptional deactivation by promoting RNA release from chromatin. Genes & development. 2006;20:977–989. doi: 10.1101/gad.1396306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, Guo X, Lv F, Xu Y, Gao G. p72 DEAD box RNA helicase is required for optimal function of the zinc-finger antiviral protein. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:4352–4357. doi: 10.1073/pnas.0712276105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coller JM, Tucker M, Sheth U, Valencia-Sanchez MA, Parker R. The DEAD box helicase, Dhh1p, functions in mRNA decapping and interacts with both the decapping and deadenylase complexes. RNA. 2001;7:1717–1727. doi: 10.1017/s135583820101994x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cullen BR, Cherry S, tenOever BR. Is RNA interference a physiologically relevant innate antiviral immune response in mammals? Cell host & microbe. 2013;14:374–378. doi: 10.1016/j.chom.2013.09.011. [DOI] [PubMed] [Google Scholar]
- Cziko AM, McCann CT, Howlett IC, Barbee SA, Duncan RP, Luedemann R, Zarnescu D, Zinsmaier KE, Parker RR, Ramaswami M. Genetic modifiers of dFMR1 encode RNA granule components in Drosophila. Genetics. 2009;182:1051–1060. doi: 10.1534/genetics.109.103234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darnell RB. HITS-CLIP: panoramic views of protein-RNA regulation in living cells. Wiley interdisciplinary reviews. RNA. 2010;1:266–286. doi: 10.1002/wrna.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis BN, Hilyard AC, Lagna G, Hata A. SMAD proteins control DROSHA-mediated microRNA maturation. Nature. 2008;454:56–61. doi: 10.1038/nature07086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deddouche S, Matt N, Budd A, Mueller S, Kemp C, Galiana-Arnoux D, Dostert C, Antoniewski C, Hoffmann JA, Imler JL. The DExD/H-box helicase Dicer-2 mediates the induction of antiviral activity in drosophila. Nature immunology. 2008;9:1425–1432. doi: 10.1038/ni.1664. [DOI] [PubMed] [Google Scholar]
- Ding SW, Voinnet O. Antiviral immunity directed by small RNAs. Cell. 2007;130:413–426. doi: 10.1016/j.cell.2007.07.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emery VC, Bishop DH. Characterization of Punta Toro S mRNA species and identification of an inverted complementary sequence in the intergenic region of Punta Toro phlebovirus ambisense S RNA that is involved in mRNA transcription termination. Virology. 1987;156:1–11. doi: 10.1016/0042-6822(87)90430-2. [DOI] [PubMed] [Google Scholar]
- Fukuda T, Yamagata K, Fujiyama S, Matsumoto T, Koshida I, Yoshimura K, Mihara M, Naitou M, Endoh H, Nakamura T, et al. DEAD-box RNA helicase subunits of the Drosha complex are required for processing of rRNA and a subset of microRNAs. Nature cell biology. 2007;9:604–611. doi: 10.1038/ncb1577. [DOI] [PubMed] [Google Scholar]
- Fullam A, Schroder M. DExD/H-box RNA helicases as mediators of anti-viral innate immunity and essential host factors for viral replication. Biochimica et biophysica acta. 2013;1829:854–865. doi: 10.1016/j.bbagrm.2013.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuller-Pace FV. The DEAD box proteins DDX5 (p68) and DDX17 (p72): multitasking transcriptional regulators. Biochimica et biophysica acta. 2013;1829:756–763. doi: 10.1016/j.bbagrm.2013.03.004. [DOI] [PubMed] [Google Scholar]
- Galiana-Arnoux D, Dostert C, Schneemann A, Hoffmann JA, Imler JL. Essential function in vivo for Dicer-2 in host defense against RNA viruses in drosophila. Nature immunology. 2006;7:590–597. doi: 10.1038/ni1335. [DOI] [PubMed] [Google Scholar]
- Goh PY, Tan YJ, Lim SP, Tan YH, Lim SG, Fuller-Pace F, Hong W. Cellular RNA helicase p68 relocalization and interaction with the hepatitis C virus (HCV) NS5B protein and the potential role of p68 in HCV RNA replication. Journal of virology. 2004;78:5288–5298. doi: 10.1128/JVI.78.10.5288-5298.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory RI, Yan KP, Amuthan G, Chendrimada T, Doratotaj B, Cooch N, Shiekhattar R. The Microprocessor complex mediates the genesis of microRNAs. Nature. 2004;432:235–240. doi: 10.1038/nature03120. [DOI] [PubMed] [Google Scholar]
- Hong S, Noh H, Chen H, Padia R, Pan ZK, Su SB, Jing Q, Ding HF, Huang S. Signaling by p38 MAPK stimulates nuclear localization of the microprocessor component p68 for processing of selected primary microRNAs. Science signaling. 2013;6:ra16. doi: 10.1126/scisignal.2003706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopkins KC, McLane LM, Maqbool T, Panda D, Gordesky-Gold B, Cherry S. A genome-wide RNAi screen reveals that mRNA decapping restricts bunyaviral replication by limiting the pools of Dcp2-accessible targets for cap-snatching. Genes & development. 2013;27:1511–1525. doi: 10.1101/gad.215384.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikegami T, Makino S. The pathogenesis of Rift Valley fever. Viruses. 2011;3:493–519. doi: 10.3390/v3050493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishizuka A, Siomi MC, Siomi H. A Drosophila fragile X protein interacts with components of RNAi and ribosomal proteins. Genes & development. 2002;16:2497–2508. doi: 10.1101/gad.1022002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jalal C, Uhlmann-Schiffler H, Stahl H. Redundant role of DEAD box proteins p68 (Ddx5) and p72/p82 (Ddx17) in ribosome biogenesis and cell proliferation. Nucleic acids research. 2007;35:3590–3601. doi: 10.1093/nar/gkm058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim T, Pazhoor S, Bao M, Zhang Z, Hanabuchi S, Facchinetti V, Bover L, Plumas J, Chaperot L, Qin J, Liu YJ. Aspartate-glutamate-alanine-histidine box motif (DEAH)/RNA helicase A helicases sense microbial DNA in human plasmacytoid dendritic cells. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:15181–15186. doi: 10.1073/pnas.1006539107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei EP, Corces VG. RNA interference machinery influences the nuclear organization of a chromatin insulator. Nature genetics. 2006;38:936–941. doi: 10.1038/ng1850. [DOI] [PubMed] [Google Scholar]
- Li Y, Lu J, Han Y, Fan X, Ding SW. RNA interference functions as an antiviral immunity mechanism in mammals. Science. 2013;342:231–234. doi: 10.1126/science.1241911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linder P, Jankowsky E. From unwinding to clamping - the DEAD box RNA helicase family. Nature reviews Molecular cell biology. 2011;12:505–516. doi: 10.1038/nrm3154. [DOI] [PubMed] [Google Scholar]
- Loo YM, Gale M., Jr Immune signaling by RIG-I-like receptors. Immunity. 2011;34:680–692. doi: 10.1016/j.immuni.2011.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maillard PV, Ciaudo C, Marchais A, Li Y, Jay F, Ding SW, Voinnet O. Antiviral RNA interference in mammalian cells. Science. 2013;342:235–238. doi: 10.1126/science.1241930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyashita M, Oshiumi H, Matsumoto M, Seya T. DDX60, a DEXD/H box helicase, is a novel antiviral factor promoting RIG-I-like receptor-mediated signaling. Molecular and cellular biology. 2011;31:3802–3819. doi: 10.1128/MCB.01368-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori M, Triboulet R, Mohseni M, Schlegelmilch K, Shrestha K, Camargo FD, Gregory RI. Hippo Signaling Regulates Microprocessor and Links Cell-Density-Dependent miRNA Biogenesis to Cancer. Cell. 2014;156:893–906. doi: 10.1016/j.cell.2013.12.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moy RH, Gold B, Molleston JM, Schad V, Yanger K, Salzano MV, Yagi Y, Fitzgerald KA, Stanger BZ, Soldan SS, Cherry S. Antiviral autophagy restricts rift valley Fever virus infection and is conserved from flies to mammals. Immunity. 2014;40:51–65. doi: 10.1016/j.immuni.2013.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller S, Gausson V, Vodovar N, Deddouche S, Troxler L, Perot J, Pfeffer S, Hoffmann JA, Saleh MC, Imler JL. RNAi-mediated immunity provides strong protection against the negative-strand RNA vesicular stomatitis virus in Drosophila. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:19390–19395. doi: 10.1073/pnas.1014378107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parvatiyar K, Zhang Z, Teles RM, Ouyang S, Jiang Y, Iyer SS, Zaver SA, Schenk M, Zeng S, Zhong W, et al. The helicase DDX41 recognizes the bacterial secondary messengers cyclic di-GMP and cyclic di-AMP to activate a type I interferon immune response. Nature immunology. 2012;13:1155–1161. doi: 10.1038/ni.2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabin LR, Zheng Q, Thekkat P, Yang J, Hannon GJ, Gregory BD, Tudor M, Cherry S. Dicer-2 processes diverse viral RNA species. PloS one. 2013;8:e55458. doi: 10.1371/journal.pone.0055458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabin LR, Zhou R, Gruber JJ, Lukinova N, Bambina S, Berman A, Lau CK, Thompson CB, Cherry S. Ars2 regulates both miRNA- and siRNA- dependent silencing and suppresses RNA virus infection in Drosophila. Cell. 2009;138:340–351. doi: 10.1016/j.cell.2009.04.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samaan S, Tranchevent LC, Dardenne E, Polay Espinoza M, Zonta E, Germann S, Gratadou L, Dutertre M, Auboeuf D. The Ddx5 and Ddx17 RNA helicases are cornerstones in the complex regulatory array of steroid hormone-signaling pathways. Nucleic acids research. 2013 doi: 10.1093/nar/gkt1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankarling G, Cole BS, Mallory MJ, Lynch KW. Transcriptome-wide RNA interaction profiling reveals physical and functional targets of hnRNP L in human T cells. Molecular and cellular biology. 2014;34:71–83. doi: 10.1128/MCB.00740-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro JS, Schmid S, Aguado LC, Sabin LR, Yasunaga A, Shim JV, Sachs D, Cherry S, tenOever BR. Drosha as an interferon-independent antiviral factor. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:7108–7113. doi: 10.1073/pnas.1319635111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro JS, Varble A, Pham AM, Tenoever BR. Noncanonical cytoplasmic processing of viral microRNAs. RNA. 2010;16:2068–2074. doi: 10.1261/rna.2303610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soulat D, Burckstummer T, Westermayer S, Goncalves A, Bauch A, Stefanovic A, Hantschel O, Bennett KL, Decker T, Superti-Furga G. The DEAD-box helicase DDX3X is a critical component of the TANK-binding kinase 1-dependent innate immune response. The EMBO journal. 2008;27:2135–2146. doi: 10.1038/emboj.2008.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staley JP, Guthrie C. An RNA switch at the 5′ splice site requires ATP and the DEAD box protein Prp28p. Molecular cell. 1999;3:55–64. doi: 10.1016/s1097-2765(00)80174-4. [DOI] [PubMed] [Google Scholar]
- Suzuki HI, Yamagata K, Sugimoto K, Iwamoto T, Kato S, Miyazono K. Modulation of microRNA processing by p53. Nature. 2009;460:529–533. doi: 10.1038/nature08199. [DOI] [PubMed] [Google Scholar]
- Terenzi F, Hui DJ, Merrick WC, Sen GC. Distinct induction patterns and functions of two closely related interferon-inducible human genes, ISG54 and ISG56. The Journal of biological chemistry. 2006;281:34064–34071. doi: 10.1074/jbc.M605771200. [DOI] [PubMed] [Google Scholar]
- van Rij RP, Saleh MC, Berry B, Foo C, Houk A, Antoniewski C, Andino R. The RNA silencing endonuclease Argonaute 2 mediates specific antiviral immunity in Drosophila melanogaster. Genes & development. 2006;20:2985–2995. doi: 10.1101/gad.1482006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vourekas A, Zheng Q, Alexiou P, Maragkakis M, Kirino Y, Gregory BD, Mourelatos Z. Mili and Miwi target RNA repertoire reveals piRNA biogenesis and function of Miwi in spermiogenesis. Nature structural & molecular biology. 2012;19:773–781. doi: 10.1038/nsmb.2347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei W, Ba Z, Gao M, Wu Y, Ma Y, Amiard S, White CI, Rendtlew Danielsen JM, Yang YG, Qi Y. A role for small RNAs in DNA double-strand break repair. Cell. 2012;149:101–112. doi: 10.1016/j.cell.2012.03.002. [DOI] [PubMed] [Google Scholar]
- Xue Y, Zhou Y, Wu T, Zhu T, Ji X, Kwon YS, Zhang C, Yeo G, Black DL, Sun H, et al. Genome-wide analysis of PTB-RNA interactions reveals a strategy used by the general splicing repressor to modulate exon inclusion or skipping. Molecular cell. 2009;36:996–1006. doi: 10.1016/j.molcel.2009.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zambon RA, Vakharia VN, Wu LP. RNAi is an antiviral immune response against a dsRNA virus in Drosophila melanogaster. Cellular microbiology. 2006;8:880–889. doi: 10.1111/j.1462-5822.2006.00688.x. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Kim T, Bao M, Facchinetti V, Jung SY, Ghaffari AA, Qin J, Cheng G, Liu YJ. DDX1, DDX21, and DHX36 helicases form a complex with the adaptor molecule TRIF to sense dsRNA in dendritic cells. Immunity. 2011a;34:866–878. doi: 10.1016/j.immuni.2011.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Yuan B, Bao M, Lu N, Kim T, Liu YJ. The helicase DDX41 senses intracellular DNA mediated by the adaptor STING in dendritic cells. Nature immunology. 2011b;12:959–965. doi: 10.1038/ni.2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou R, Hotta I, Denli AM, Hong P, Perrimon N, Hannon GJ. Comparative analysis of argonaute-dependent small RNA pathways in Drosophila. Molecular cell. 2008;32:592–599. doi: 10.1016/j.molcel.2008.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Chen G, Lv F, Wang X, Ji X, Xu Y, Sun J, Wu L, Zheng YT, Gao G. Zinc-finger antiviral protein inhibits HIV-1 infection by selectively targeting multiply spliced viral mRNAs for degradation. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:15834–15839. doi: 10.1073/pnas.1101676108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zonta E, Bittencourt D, Samaan S, Germann S, Dutertre M, Auboeuf D. The RNA helicase DDX5/p68 is a key factor promoting c-fos expression at different levels from transcription to mRNA export. Nucleic acids research. 2013;41:554–564. doi: 10.1093/nar/gks1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.