

Abstract
Purpose
We analyzed inflamed mucosal/submucosal layers of ulcerative colitis (UC=63) and Crohn’s colitis (CC=50) and unexpectedly we unveiled a pool of free-hemoglobin-alpha (Hb-α) chain. Patients with colitides have increased ROS, DNA-oxidation products, free-iron in mucosa, in pre-neoplastic, and in colitis-cancers and increased risks of developing colorectal-cancer (CRC). All IBD-related-CRC lesions are found in segments with colitis. Linking this information we investigated whether free-Hb-α is key transformational stepping that increases colitis-related-CRC vulnerability.
Methods
UC/CC samples were profiled using MALDI-MS; protein identification was made by LCM. Diverticulitis (DV) was used as control (Ctrl). The presence of Hb(n) (n=α, β and hemin)/Hb was validated by Western blotting (WB) and immunohistochemistry (IHC). We tested for DNA-damage (DNAD) by exposing normal colonic-epithelial-cell-line, NCM460, to 10μM and 100μM of Hb(n)/Hb, individually for 2 h, 6 h, and 12 h. Quantification of Hb-α-staining was done by Nikon Elements Advance Research Analysis software. ROS was measured by the production of 8-OHdG. DNAD was assessed by Comet-assay. Colonic tissue homogenate antioxidants Nrf2-, CAT-, SOD- and GPx-expressions was analyzed densitometrically/ normalized by β-actin.
Results
IHC of CC/UC mucosal/submucosal-compartments stained strongly positive for Hb-α and significantly higher vs. Ctrl. NCM460 exposed to Hb(n)/Hb exhibited steadily-increasing ROS and subsequent DNAD. DNAD was higher in 10μM than 100μM in Hb-β/hemin the first 2 h then plateaued followed by DNAD-repair. This may be likely due to apoptosis in the later concentration. Nrf2 enzyme activities among UC, CC and UCAC were observed impaired in all IBD subjects. Decreased levels of Nrf2 among UC vs. CC patients with active disease was insignificant as well as vs. Ctrls but significantly lower in UCAC vs. Ctrl. SOD was decreased in UC and UCAC and GPx in CC but statistically not significant. Comparing CC vs. UC, SOD was significantly lower in CC (p< 0.05). CAT was observed increased among CC/UC/UCAC patients and GPx in UC and UCAC vs. Ctrl, respectively, and significantly increased in CC vs. Ctrl (p< 0.01)
Conclusion
In the colitides mucosal/submucosal tissue microenvironments demonstrated pool of free-Hb-α-chain. In vitro exposure of NCM460-cells to Hb(n)/Hb induced ROS and DNAD. Toxic effect of free-Hb-α, in colonic epithelial cells is therefore through production of ROS-formation modulated by impairment of antioxidant effects. Targeting reduction-oxidation-sensitive pathways and transcription factors may offer options for IBD-management and colitis-related-cancer prevention.
Keywords: IBD, Colon-tissue-profiling, Proteomics, Hb, Hb(n), ROS, DNA-damage, Nrf2, CAT, SOD, GPx, risks, Carcinogenesis
Introduction
Unambiguous diagnosis of inflammatory bowel disease (IBD) encompassing ulcerative colitis (UC) and Crohn’s colitis (CC), represent significant challenges in debuting stages of the disease. We previously analyzed human colonic mucosal and submucosal surgical pathology samples of 113 patients with IBD for proteomic patterns that delineated ulcerative UC and CC, using matrix-assisted laser desorption/ionization (MALDI) profiling.1,2 While looking for unique UC and CC phenotype specific biometric markers, we found a highly abundant mass-to-charge ratio spectral peak at m/z 5045 that was more intense in inflamed tissues of both colitides, than in adjacent non-inflamed tissues and in diverticulitis (DV) (control, Ctrl). Liquid chromatography-mass spectrometry (LC-MS/MS) analysis identified this signal as a triply charged hemoglobin alpha (Hb-α) chain. The presence of an abundant level of free Hb-α chain (a previously unrecognized tissue by-product of IBD), was intriguing, because it could have a biopathophysiological role in this tissue microenvironment. Organic iron is known to induce cellular DNA damage (DNAD) via the production of reactive oxygen species (ROS) and to be carcinogenic in IBD.3 There are also reports of significantly increased ROS, DNA oxidation products (8-hydroxy-deoxyguanosine, 8-OHdG) and free iron levels in inflamed and in pre-neoplastic mucosal IBD tissues compared to controls.4,5 In addition, immunoreactivity to major iron-binding proteins (lactoferrin, transferrin, and ferritin) was also increased.4 Such changes could trigger a self-perpetuating cycle, resulting in further tissue assault and damage and a trend toward neoplastic transformation.6,7 Free Hb is known to increase the production of tumor necrosis factor-α (TNF-α), interleukin-6 (IL-6) and interleukin-8 (IL-8), which are proinflammatory cytokines.8,9 Patients with IBD are known to be vulnerable and have an increased risk of developing colorectal cancer (CRC) than the general population.10,11 Our experience12,13 and that of others11–15 indicated that all CRC lesions in IBD subjects were found to co-occur in segments with evidence of colitis.11–15 Linking this information coupled with our evidence-based finding confirming the presence of abundant pool of free Hb-α chain in IBD surgical pathology samples, led us to investigate further, whether this accumulation of free Hb and Hb-subunits (Hb(n)) (n = α, β and hemin), in the cellular microenvironment is an initial key transformational step that increases the risk of CRC. Our hypothesis was that prolonged and repeated exposure of colon epithelial cells to free Hb and Hb(n), as a stepwise progression, induces cellular biochemical and morphological features that increases the vulnerability and eventual risk of CRC transformation through ROS overproduction, subsequent DNAD and carcinogenesis in susceptible patients with irreparable DNAD.
To understand the carcinogenesis induced by IBD, the emphasis has previously been rested on studies of epithelial cell behavior. However, over the last decade, the focus has shifted to studying the stromal and epithelial cells microenvironment.16,17 Nonetheless, there has been no report of inflammatory phenotype within pre-malignant human colonic carcinomas. Assessing the microenvironment of mucosal and submucosal layers is central of this laboratory to understand the mechanisms involved in the etiology of IBD and transition to pre-transformative to transformative disease.1,2,18–21
Given the fact that IBD-related ROS contributes to the propagation and exacerbation of disease activity,22 the contribution of enzymic antioxidants defense plays an important role in the disease biopathophysiology pathways. We therefore analyzed concentration levels of Nrf2, CAT, SOD and GPx, the major regulators of cytoprotective responses to oxidative stress and the primary cellular defense against the cytotoxic effects of oxidative stress.23–27 Representative immunoblot and densitometric analysis data for Nrf2 and SOD in inflamed UC and UCAC tissues compared to Ctrl showed impaired. The mean values for CAT and GPx was enhanced in all IBD conditions except for the GPx in CC patients was impaired. These observations corroborates with several other controversial studies previously reported about antioxidants in active IBD.24,26 As scientists we need to have a clear understanding that although the results in antioxidant levels among the studies is conflicting, the imperative point is the presence of an imbalance in antioxidant saturation, attesting the fact that vital organs/systems in IBD patients are oxidative stressed.
Methods
Clinical samples
This study was approved by the Meharry Medical College (IRB#: 100916AM206) and Vanderbilt University (IRB#s: 080898 and 100581) Institutional Ethical Review Board Committees and conducted in accordance with the Second International Helsinki Declaration.28 Informed consent was given, and participation in the study was voluntary. The meta-analysis of patients with IBD that were used in the study is depicted in Table 1. Mucosal and submucosal surgical samples of colon tissue compartments from 113 (n = 50, CC) and (n = 63, UC) patients were analyzed using histology-directed profiling for MALDI MS technology as previously described.1,2 All patients included in the study were adults with a definitive diagnosis of CC or UC; cases with any ambiguity were excluded. All samples were subjected to the same preprocessing and calibration parameters as previously described.1,2
Table 1.
Meta-analysis of the 113 patients diagnosed with IBD. The values are presented per disease and area of the colon affected. Moderate disease is categorized as patients with symptoms such as low grade fever, slight weight loss, mild abdominal pain and, tenderness and intermittent nausea or vomiting, or anemia. Severe disease is categorized as those with persistent symptoms despite conventional glucocorticoid or biologic agent therapy as outpatients, or individuals presenting with high fevers, persistent vomiting, intestinal obstruction, significant peritoneal signs, cachexia, or evidence of an abscess.
| Meta-analysis of subjects | Crohn’s colitis | Ulcerative colitis |
|---|---|---|
|
| ||
| Number of subjects | 50 | 63 |
|
| ||
| Age (mean ± SD) | 48.5 ± 13.2 | 47.8 ±17.8 |
| Range years | 18–78 | 18–84 |
|
| ||
| White | 42 (84%) | 54 (86%) |
| Black | 6 (12%) | 2 (3%) |
| Other | 2 (4%) | 7 (11%) |
|
| ||
| Male | 35 (70%) | 38 (60%) |
| Female | 25 (50%) | 25 (40%) |
|
| ||
| Moderate activity | 30(60%) | 34 (54%) |
| Severe activity | 20 (40%) | 29 (46%) |
|
| ||
| COLON (Sections Analyzed) | ||
|
| ||
| Ascending | 24 (48%) | 12 (19%) |
| Transverse | 4 (8%) | 19 (30%) |
| Descending | 16 (32%) | 29 (46%) |
| Hepatic flexure | 2 (4%) | 1 (2%) |
| Splenic flexure | 1 (2%) | 1 (2%) |
| Cecum | 3 (6%) | 1 (2%) |
Identification of hemoglobin-alpha chain by MALDI-MS
The basic steps of the histology-directed tissue profiling for MALDI MS are outlined in Fig. 1A–I. Samples were sectioned, stained, and regions of interest were identified by a pathologist. Protein identification was accomplished from extracted tissue samples and intact protein LC-MS/MS analysis.29 Data dependent MS/MS acquisition was used to select the top 5 most abundant MS signals for fragmentation by electron transfer dissociation (ETD) and collision induced dissociation (CID). MS/MS spectra (Fig. 1H) were manually interpreted and annotated with the assistance of ProteinProspector (http://prospector.ucsf.edu/prospector/mshome.htm). Among these 5 signals was triply charged Hb-α chain (Fig. 1I).
Fig. 1.

Workflow for histology-directed tissue profiling for MALDI MS. Digital photomicrographs acquired from histology and MALDI sections were used to identify and designate sites of interest for profiling. A) A 12 μm tissue section was placed on a gold-coated MALDI target. B) A serial section was mounted on a slide for H and E staining. C) A pathologist marked areas of interest on a picture of the H and E stained section. D) The H and E picture was overlaid on a picture of the MALDI plate and coordinates were generated. E) Tissue was spotted with matrix in the designated areas using a robotic spotter. F) Protein profiles were determined by MALDI MS. G) Mass spectra were generated from the matrix spots corresponding to different cell types. H) Spectral peak 5045 was generated from the matrix spots and classifiers were evaluated using LOOCV and LNOCV cross validation. I) LC-MS/MS analysis identified this signature as triply charged Hb-α chain.
Chemicals and cell lines
Human Hb and Hb alpha and beta chains and hemin were purchased from Sigma-Aldrich (St. Louis, MO, USA). The human NCM460 cell-line (normal colon epithleial cells) was obtained from INCELL Corporation Inc. (San Antonio, TX, USA). Some studies also used a human colon adenocarcinoma cell line (SW620) obtained from primary and secondary tumors from a 51 years old Caucasian patient.30 Cells were grown in proprietary M3TH Base complete medium supplemented with Fetal Bovine Serum (FBS) (Invitrogen, Grand Island, NY). They were plated at a density of 1 × 105 cells/ml in 6-well dishes and allowed to grow overnight. The media was then replaced with normal media (M3:BaseA (InCell Corp.) +10% FBS (Atlanta Biologocals) +1% PenStrep (Life Technologies)) or normal media containing different concentrations of test compounds for different period of times.
Western blotting
Full thickness frozen colon tissue samples were homogenized in ice-cold RIPA-buffer (Thermo Scientific, Rockford IL), containing protease inhibitors followed by sonification (500 watts using an Ultrasonic Processor – VCX Series) and centrifugation at 10,000 rpm at room temperature for 10 min. The protein concentration of the supernatant solution was determined by the Pierce® BCA method (Thermo Scientific, Rockford IL). Equal amounts of lysates were electrophoresed on a 4–20% precast gradient gel (Bio-Rad, Hercules, CA) and transferred overnight to low fluorescent polyvinylidene difluoride membranes (Immun-Blot® LF PVDF, Bio-Rad, Hercules, CA). Membranes were blocked with 5% milk, then incubated with primary antibody, goat polyclonal anti-human Hb-α or mouse polyclonal anti-human GAPDH (Santa Cruz Biotechnology, Inc.). For simultaneous two-color detection with the Odyssey® infrared imaging system, blots were incubated with a secondary antibody: donkey anti-mouse IRDye® 800CW or donkey anti-goat IRDye® 680 (all from Licor Biosciences, Lincoln NE). Membranes were scanned with an Odyssey® infrared imaging system. Odyssey Application software v3.0.16 (Licor Biosciences, Lincoln NE) was used to quantify bands. The results from three independent experiments per group (UC, CC and DV) were analyzed.
In addition, Nrf2 proteins (constitutively expressed nucleo-cytoplasmic) were quantified in colonic homogenates standard western blot (WB) analysis, as described in our previous studies.31,32 Proteins were separated by SDS polyacrylamide gel electrophoresis. The membrane was immunoblotted with polyclonal primary antibodies (Santacruz Biotechnology, CA) and their respective secondary antibodies. Nrf2 protein expression was normalized using β-actin.
Protein Lysate Collection for CAT, SOD and GPx proteins
Three samples from 4 different disease states (CC, UC, UC, UCAC), and DV were obtained for CAT, SOD and GPx proteins analyses. Protein was extracted using T-PER (Thermo Scientific). Before use, 100μL of Halt Protease Inhibitor EDTA-Free Cocktail (Thermo Scientific) was added to 10mLs T-PER reagent. Samples were weighed, and 20μL T-PER + Protease Inhibitors were added per milligram of tissue. Full thickness frozen colon tissue samples were homogenized using a rotor-stator homogenizer, and spun at 10,000 × g for 5 minutes to pellet debris. Cleared supernatant was removed to a new 1.5mL tube, and samples frozen at −80°C for later use. All steps above were performed on ice.
Bradford Assay
In order to determine protein concentration, a Bradford Assay was performed. 10μL of sample was diluted two-fold in T-PER + protease inhibitors, then 5μL of each sample were added to a 96 well plate, and 250μL of Quick Start Bradford Dye reagent (Bio-Rad) was added. Samples were incubated at room temperature for 10 minutes, and then read using Tecan plate reader Bradford Assay Protocol. In addition to samples, a Bovine Serum Albumin (BSA) standard curve was also utilized, using concentrations of 0, 0.125mg/mL, 0.25mg/mL, 0.5mg/mL, 0.75mg/mL, 1mg/mL, 1.5mg/mL, and 2mg/mL.
SDS-PAGE
25μL of β-mercaptoethanol (BME) was added to 425μL Laemmli Buffer (Bio-Rad) to make loading buffer for reducing conditions. 8μg of sample was added to equal amounts of loading buffer and boiled at 100°C for 10 m to further reduce the samples. Samples were loaded onto a 4–20% tris-glycine gel (Bio-Rad) and ran on ice at 50 volts for 1 h. Voltage was increased to 130 until dye front ran off gel.
Protein Transfers
PVDF membrane (Bio-Rad) was equilibrated in methanol and transfer buffer prior to use. Gels were transferred to PVDF at 100 volts for 60 m on ice.
Western Blots – Catalase, GPx, SOD-1, and β-actin
Note: All incubations were performed at room temperature on a shaker.
Membranes were blocked in 3% milk for 1 h, and then washed twice in PBS-T for 5 m each time. Primary antibody was incubated on the blots for 1 hour. Blots were washed twice in PBS-T for 5 m each time, and then incubated with secondary antibody for 1 h. Finally, the blots were washed three times in PBS-T for 5 m each time. Blots were developed with Opti-4CN detection kit (Bio-Rad) according to manufacturer’s protocol and band intensity analyzed using Bio-Rad ChemiDoc XRS+ imaging system and Image Lab software.
Assessment of ROS
ROS was detected using a 2′-7′-dichlorodihydrofluorescein diacetate (DCFDA) - cellular ROS detection assay kit from Abcam (Cambridge, MA). Briefly, NCM460 and SW620 cells (2.5 × 104/100 μl) were plated in Costar 96-well black solid-bottom culture plates (Corning, Corning, NY) and incubated overnight at 37°C. Complete media was replaced with starvation media (M3:BaseA (InCell Corp.) +FBS (0.5%), +PenSptrep (0.5%)) then the cells were incubated overnight at 37°C. Cell were washed with 1 x PBS and incubated with 25 μM DCFDA for 45 m at 37°C. After incubation, 100 μl/well of the various test compounds (Hb(n) or Hb) at various concentrations were added to the wells. Appropriate blanks (with, without DCFDA) were used to correct for background autofluorescence of controls, and cells treated with 50 μM tert-butyl hydroperoxide and DCFDA were used as positive controls. Fluorescence was measured at 1 h intervals for up to 24 h (data shown represents 0 h, 2 h, 6 h, 12 h, and 24 h only) in a BioTek Synergy 2 plate reader (BioTek Instruments) using excitation and emission wavelengths of 485 nm and 535 nm. Fluorescence is presented as a percent of control values by the formula [Ftexp/Ftcontrol], where Ftexp = fluorescence at any time after treatment in a given well and Ftcontrol = mean fluorescence of the respective untreated control replicates.
Co-localization studies
We used double immunofluorescence staining for Hb-α and the macrophage specific marker CD163 in paraffin-embedded sections from patients with UC and CC. Patients in both of these groups included moderate to severe inflammatory disease activity (according to the final surgical pathology report). The primary antibodies used were: anti-human hemoglobin-α (Abcam, Cambridge) rabbit monoclonal antibody and anti-human CD163 mouse monoclonal antibody (AbD Serotec, Raleigh NC). The secondary antibodies used were: Alexa Fluor 488 goat anti-mouse IgG (H+L), and Alexa Fluor 549 goat anti-rabbit IgG (H+L), (Invitrogen, Carlsbad, CA). Nuclei were counterstained with ProLong Gold anti-fade reagent containing DAPI (Invitrogen).
Comet and cell viability assays
We used the OxiSelect Comet™ Assay kit (Cell Biolabs, Inc.) to evaluate cellular DNA damage. The resulting image that is obtained resembles a “comet” with a distinct head and tail. The head is composed of intact DNA, while the tail consists of damaged (single-strand or double-strand breaks) or broken pieces of DNA. The MTT (3-[4,5-dimethylthiazol-2-yl]-2,5 diphenyl tetrazolium bromide) cell viability assay is a colorimetric assay based on the conversion of MTT into formazan crystals by living cells, which determines mitochondrial activity.33 Since for most cell populations, the total mitochondrial activity is related to the number of viable cells, this assay is broadly used to measure the in vitro cytotoxic effects of drugs. It is a yellow substrate that is cleaved by living cells to yield a dark blue formazan product. The assay began by plating 20,000 cells/well into 96 well plates. After an overnight incubation at 37°C, the cells were treated with Hb or Hb(n) or media only. Cells were then incubated for 24 h. After the duration of exposure was complete the media were aspirated and the cells washed twice with PBS. At this point 10 μl MTT was added to each well followed by 4 h incubation at 37°C. At the end of this time, 100 μl of isopropanol containing 0.04 N HCl was added to each well. Within an h, the absorbance was measured on a plate reader with a test wavelength of 570 nm and a reference wavelength of 630 nm.
Statistical analysis
For MALDI-MS findings, a whole spectrum screen was performed to identify candidate m/z peaks. We used the weighted flexible compound covariate method (WFCCM) of statistical analysis that collectively includes the mutual information scoring (Info Score), Kolmogorov-Smirnov test, weighted gene analysis (WGA), significance analysis of microarray (SAM), and permutation t-test.34,35 Misclassification rate is assessed using Leave One-Out Cross-Validated (LOOCV) class prediction. Floating search algorithm was applied to select the optimal feature combination during cross-validation of a classifier development. LOOCV and leave-N-out cross-validation (LNOCV) tests were performed. The k parameter was assessed for optimal feature performance. Both the k parameter and the consistency of the k parameter through the KNN tables were considered in designing the classifier. Using this method, hemoglobin alpha chain was identified by its known m/z ratio of 5045.
For the remainder of the data, values are shown as mean ± S.E. Statistical significance was determined by Turkey test after one-way ANOVA. Each maneuver was done three times, each in replicate. Unless stated otherwise, calculated differences were regarded as significant for a probability value of p< 0.05. Correlation of test results was accomplished with linear regression calculations.
Results
Using MALDI MS technology, we analyzed surgical pathology biospecimens from 113 patients with IBD (CC, n = 50 and UC, n = 63). The meta-data information about these subjects is depicted in Table 1. The age of patients with CC at the time of surgery was 48.5 ± 13.2 and 47.8 ± 17.8 years for the UC group. A MALDI-TOF-MS signal at m/z 5045 (Fig. 1H) was abundantly more intense in inflamed areas of both inflammatory colitides tissues. This fingerprint was identified as triply charged Hb-α chain (Fig. 1I). Validation of this signal using protein extracts from full thickness colon samples and a commercially available antibody for Hb-α protein confirmed the presence of Hb-α (Fig. 2A). Quantification of Hb-α in UC vs. CC was higher in CC but showed no statistically difference. However, the level of Hb-α chain was significantly higher in the colitides tissues than in samples from patients with Ctrl; UC and CC vs. Ctrl (24.47 ± 5.03 and 31.88 ± 3.56 vs. 1.28 ± 0.33 (p< 0.006 and p<0.0001, respectively) (Fig. 2B). Immunohistochemistry staining for Hb-α was greater in the CC than in the UC samples but statistically not significant from each other in both the mucosa and submucosa areas (Fig. 2C and 2D). In DV the staining was restricted to the erythrocytes found only in the capillaries of the submucosa (Fig. 2C, lower panel). Staining for Hb-β was negative in UC, CC and in Ctrl in both the mucosal and submucosal layers (Fig. 2E). Antibodies were also used against spleen and liver controls.
Fig. 2.

Western blots were used to determine the molecular weights of proteins and to measure relative amounts of the protein present in tissue samples. A) Quantification of Hb-α staining of UC, CC and. DV using Nikon Elements Advance Research Analysis software; a threshold was established using the red channel for the Hb-α staining and the area fraction of the staining was determined. B) Staining of Hb-α by IHC in colonic mucosal (left) and mucosa-submucosal (right) layers at a 30× and 10 × magnifications, respectively. DV staining was restricted to RBC found in submucosal capillaries. C) Quantification of Hb-α staining of UC, CC and DV using Nikon Elements Advance Research Analysis software. D) Staining of Hb-β by IHC in colonic mucosal (left) and mucosa-submucosal (right) layers at a 30 × and 10 × magnification, respectively. Colitides and DV staining of Hb-β were restricted to RBC found in submucosal capillaries. E) Mean intensity of Hb-α chain was higher in the mucosa (UC = 90AU, CC = 102AU and DV = 38AU) than submucosa (UC = 27AU, CC = 17AU and DV = 4AU), respectively.
DNA damage
We treated NCM460 cells with 150 μM of Hb for 4 h and used the comet assay to assess DNAD. Comet slides were manually scored. In this assay, damaged DNA (containing cleaved DNA and DNA with strand breaks) migrates further than intact DNA and produces a “comet tail”. As shown in Fig. 3A, control cells show no tail fragments indicating intact DNA, while cells exposed to Hb, showed tail fragments (Fig. 3B) indicating DNAD. Insets in Fig. 3C show a representation of: a) undamaged cell, b) damaged cell, and c) severely damaged cell. We found that the DNAD cell count was significantly higher in Hb-treated cells (p< 0.00001) compared to controls when counted by a Nikon DS-Fi1 color CCD camera on a TE2000E microscope. Quantification of the extent of DNAD between the two groups is depicted in Fig. 3D. The mechanism leading to DNAD is thought to be through the overproduction of ROS in the presence of Hb. To validate this assumption, we present the experimental results of the effects of Hb(n) and Hb on ROS production in Table 2.
Fig. 3.
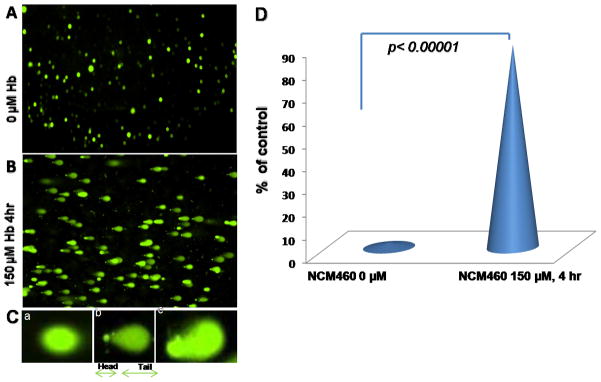
Comet assays. A) Normal human colon cells (NCM 460) were fed with fresh complete media alone, as a control or B) with 150 μM Hb for 4 h followed by a comet assay to assess DNA damage (DNAD) using a denaturing electrophoresis. C (a) (b) and (c), show an interpretation of the intensity of DNAD: a = undamaged cells, b = damaged cells and, c = severely damaged cells. D) Quantification of DNAD.
Table 2.
Depicts data results of NCM 460 cells treated with Hb(n) and Hb, individually, are mediators for ROS-dependent cell formation. Effects of Hb(n) and Hb, individually, on ROS with concentration-time dependent manner of 10 μM and 100 μM at 2 h, 6 h, 12 h and 24 h analytical points. The generation of ROS was measured and quantified by the production of 8-OHdG. Data are given as mean ± S.D and S.E. Statistical significance was determined by Turkey test after one-way ANOVA. Unless stated otherwise calculated differences were regarded as significant for a probability value of less than 0.05.
| Hemoglobin alpha (Hb-α) | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Hb-α 10 | Hb-α 100 | p-value | |||||||
| Time | Mean | Standard Deviation | Standard Error | Mean | Standard Deviation | Standard Error | Hb-α, 10 μM vs. 100 μM | TbHP vs. Hb-α 10 μM | TbHP vs. Hb-α 100 μM |
| 0 h | 2.25 | 0.43 | 0.22 | 1.75 | 0.43 | 0.22 | 0.1817 | 1.0000 | 0.0032 |
| 2 h | 5.25 | 0.43 | 0.22 | 5.00 | 0.71 | 0.35 | 0.3910 | 0.005 | 0.0042 |
| 6 h | 12.50 | 1.12 | 0.56 | 8.00 | 0.71 | 0.35 | 0.0006 | 0.04 | 0.0022 |
| 12 h | 25.50 | 2.06 | 1.03 | 15.75 | 1.92 | 0.96 | 0.001 | 0.0003 | 1.0000 |
| 24 h | 46.75 | 5.36 | 2.68 | 31.25 | 7.76 | 3.88 | 0.0114 | 0.0005 | 0.0234 |
| Hemoglobin Beta (Hb-β) | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Hb-β 10 μM | Hb-β 100 μM | p-value | |||||||
| Time | Mean | Standard Deviation | Standard Error | Mean | Standard Deviation | Standard Error | Hb-β, 10 μM vs. 100 μM | TbHP vs. Hb-β 10 μM | TbHP vs. Hb-β 100 μM |
| 0 h | 2.50 | 0.50 | 0.25 | 1.75 | 0.83 | 0.41 | 0.06 | 0.006 | 0.003 |
| 2 h | 6.25 | 0.43 | 0.22 | 4.50 | 0.50 | 0.25 | 0.006 | 0.008 | 0.005 |
| 6 h | 15.00 | 2.45 | 1.22 | 7.50 | 1.50 | 0.75 | 0.0014 | 0.2152 | 0.0001 |
| 12 h | 33.50 | 4.97 | 2.49 | 14.25 | 1.30 | 0.65 | 0.0029 | 0.002 | 0.0577 |
| 24 h | 62.00 | 9.41 | 4.70 | 22.00 | 2.55 | 1.27 | 0.0022 | 0.001 | 0.0663 |
| Hemin | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Hemin 10 μM | Hemin 100 μM | p-value | |||||||
| Time | Mean | Standard Deviation | Standard Error | Mean | Standard Deviation | Standard Error | Hm, 10 μM vs. 100 μM | TbHP vs. Hm 10 μM | TbHP vs. Hm 100 μM |
| 0 h | 2.75 | 0.83 | 0.41 | 2.50 | 0.50 | 0.25 | 0.3910 | 0.01 | 0.006 |
| 2 h | 4.25 | 0.43 | 0.22 | 3.75 | 0.83 | 0.41 | 0.1817 | 0.004 | 0.002 |
| 6 h | 5.00 | 0.71 | 0.35 | 5.25 | 0.83 | 0.41 | 0.3910 | 0.0007 | 0.001 |
| 12 h | 8.75 | 1.48 | 0.74 | 11.25 | 1.79 | 0.89 | 0.02 | 0.0004 | 0.0061 |
| 24 h | 7.50 | 1.66 | 0.83 | 13.25 | 1.30 | 0.65 | 0.0002 | 0.001 | 0.01 |
| Hemoglobin | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Hb 10 μM | Hb 100 μM | p-value | |||||||
| Time | Mean | Standard Deviation | Standard Error | Mean | Standard Deviation | Standard Error | Hb, 10 μM vs. 100 μM | TbHP vs. Hb 10 μM | TbHP vs. Hb 100 μM |
| 0 h | 2.25 | 0.43 | 0.22 | 2.50 | 0.50 | 0.25 | 0.3910 | 1.0000 | 0.0060 |
| 2 h | 9.25 | 2.17 | 1.08 | 7.50 | 0.50 | 0.25 | 0.2126 | 0.0006 | 0.0154 |
| 6 h | 17.75 | 2.49 | 1.24 | 6.75 | 0.83 | 0.41 | 0.002 | 0.006 | 0.0006 |
| 12 h | 32.50 | 3.04 | 1.52 | 14.75 | 0.43 | 0.22 | 0.002 | 0.0003 | 0.4228 |
| 24 h | 57.00 | 4.90 | 2.45 | 25.50 | 2.06 | 1.03 | 0.0009 | 1.0000 | 0.0318 |
ROS-dependent cell formation, cell-induced DNA damage and cell viability
In the case of free Hb and Hb(n) mediated ROS-dependent cell formation we anticipated that DNA may not be protected from oxidative assault and could be damaged. ROS production was measured in NCM460 cells during incubations with various concentrations (10 μM and 100 μM) of Hb(n) or Hb, and assayed at 2 h, 6 h, 12 h and 24 h compared to 0 h. Cells treated with 10 μM, and 100 μM of Hb-α, hemin and Hb, showed increases of ROS production at 2 h but those values were not significantly different from each other. Only Hb-β demonstrated a significantly higher level of ROS at 100 μM compared to 10 μM concentrations (p< 0.006). At analytical time point of 6 h, 12 h and 24 h, Hb(n) and Hb, individually, induced a significant increase of ROS, (p< 0.0001, p< 0.0006, p< 0.001 and p< 0.01), (except for hemin at 6 hour, p= 0.3910) respectively at the higher compared to the lower concentration (Table 2). There were no ROS observed in cells treated with 0.001 μM, 0.01 μM and 0.1 μM of Hb(n) and Hb (data not shown) possibly because the concentrations were too low. The rank order of ROS generation at 10 μM concentrations at 12 h and 24 h was Hb-β ≈ Hb>Hb-α≫hemin. However, during these extended time periods, cells treated with 100 μM compared to 10 μM concentrations demonstrated less ROS (Table 2, Fig. 4). This may be likely due to an effect on cell integrity (Fig. 5). Hemin was particularly destructive at both concentrations. While low concentrations of Hb-α chain, Hb-β chain and Hb significantly stimulated mitochondrial activity compared to controls, the higher concentrations (100 μM) significantly blunted these effects. Thus it appears that the lower production of ROS in Table 2 and Fig. 4 at the 100 μM concentrations, negatively altered cell viability, and possibly led to apoptosis (Fig. 5).
Fig. 4.

Comet assay of the effect of the dose and term of incubation with Hb-α, Hb-β, hemin and Hb on DNAD and ROS generation in NCM460 cells. A) Two thousand NCM460 cells/well were treated with 10 μM or 100 μM of Hb-α or hemim or Hb for 2h, 6h and 12h. DNA was assessed by the comet assay. Damage in these experiments is expressed as the olive tail moment (OTM). Values shown are the mean ± SEM of 2000 cells/ well (25 cells per slide) p< 0.001 (One Way ANOVA) compared to controls. B) DNA damage in colonocytes treated with 10 μM or 100 μM of Hb-α, Hb-β, hemin and Hb, as a function of time. C) Time and dose dependent changes in ROS. Tertiary butyl hydroperoxide (TBHP was set as control. Colonocytes were isolated, washed and treated with 50 μM concentrations TBHP or 10 μM or 100 μM of Hb-α, Hb-β, hemin and Hb and incubated at 37°C for the times shown. Intracellular ROS was estimated spectrofluometrically by incubating washed colonocytes with 10 μM of the fluorescent dye DCFDA (Cellular Reactive Oxygen Species Detection Assay) for 30 min at 37°C in the dark. ROS production in colonocytes was assessed with increase in time (0 hour, 2 hours, 6 hours, 12 hours and 24 hours). Each point indicates mean ± S.E. Data were analyzed by one-way ANOVA, with Student-Newman-Keuls post hoc tests. Significant differences were attributed at p< 0.05.
Fig. 5.
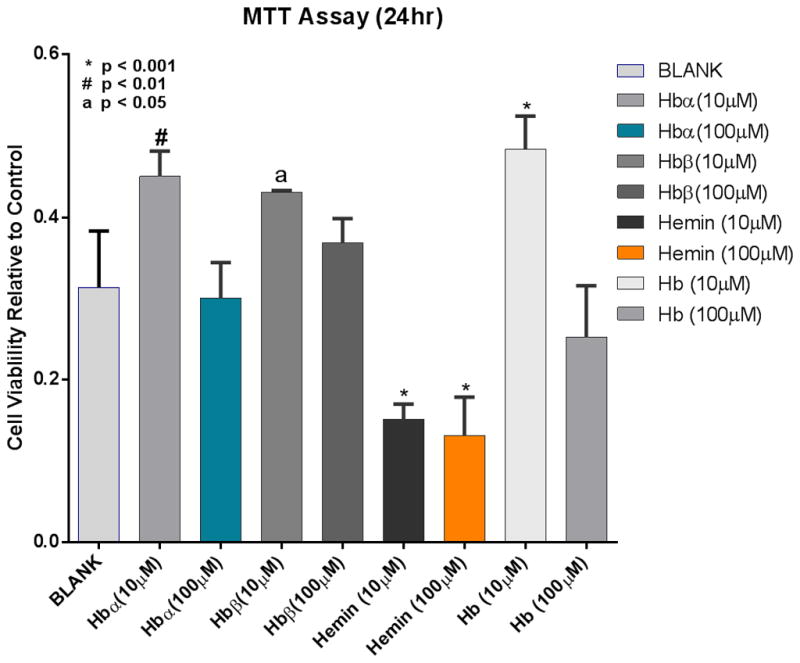
Cell viability, measured by MTT assays, relative to controls in NCM460 cells exposed to Hb(n) or Hb (10 μM and 100 μM) for 24 h.
A significant correlation was noted between ROS production and DNAD, Table 3 and Fig. 4. Cells that were incubated for 12 h appeared to have their DNAD partially recovered despite the fact that ROS was steadily increasing with time up to 24 h, the period of follow-up. This is likely due to metabolism of H2O2 to H2O and O2 and the effects of DNA repair mechanisms and the activation of antioxidant enzymes. These observations confirm the toxic effects of Hb(n) and Hb, based on the production of ROS, DNAD and even cell viability.
Table 3.
Pearson’s correlation coefficient: DNA damage vs. ROS at 2 h time points. A significant correlation between ROS vs. DNAD was noted.
| Hb-α 10 μM | Hb-α 100 μM | Hb-β 10 μM | Hb-β 100 μM | Hemin 10 μM | Hemin 100 μM | Hb 10 μM | Hb 100 μM | |
|---|---|---|---|---|---|---|---|---|
| r2 value | 0.9031 | 0.9527 | 0.9329 | 0.9533 | 0.9555 | 0.9759 | 0.9329 | 0.7670 |
| p-value | 0.002 | 0.0003 | 0.0007 | 0.0002 | 0.0002 | 0.00003 | 0.0007 | 0.03 |
Hb permeability of cultured colonic epithelial cells
To test whether hemoglobin could penetrate colonic normal epithelial (NCM460) and/ or colon cancer (SW620) cell-lines, we incubated cells with Hb for up to 1 h. Cytosolic and nuclear fractions were then blotted for Hb. The content of Hb in both fractions was identical. In order to visualize the uptake of Hb, cells were grown on cover slips mounted onto a slide cover incubated with 100 μM Hb for 1 h and then fixed. Nuclei were stained with DAPI and the autofluorescence of Hb was used to detect its presence. Both cytosolic and nuclear cell extracts expressed GAPDH at similar levels (Fig. 6A). The cytosolic protein extracts showed presence of Hb-α at similar levels. Presence of Hb-α level in the nuclear extracts increased over time. Longer exposure to Hb caused increased Hb-α expression. Fig. 6B, experiment validates that the Hb-α, which is a part of heterotetrameric Hb complexes that penetrated the cell, is likely could be the causal agent of ROS and subsequent DNAD.
Fig. 6.

Western blot and immunofluorescent detection of hemoglobin. A) Cytosolic and nuclear extracts were prepared from normal NCM460 and cancerous (SW620) colon cells which had been exposed to 150 μM hemoglobin for the times shown. The samples were subjected to SDS-PAGE followed by immunoblotting. The membranes were probed using antibodies against hemoglobin-α (red, 14kDa), GAPDH (green, 37 kDa) and simultaneous two-color fluorescent secondary antibodies and analyzed on an Odyssey-infrared imaging system (LI-COR®). Both cytosolic and nuclear cell extracts expressed GAPDH at similar levels. The cytosolic protein extracts expressed Hb-α at similar levels while the nuclear extracts expressed hemoglobin-α in a more time-dependent manner. B) Immunofluorescent staining of normal cells incubated for 4 h in complete media without or with 100 μM Hb. The cells were treated with Prolong® Gold antifade reagent with DAPI for visualization of the nucleus using a blue filter while the autofluorescent properties of Hb were visualized using a red filter. Both frames show nuclear and cytosolic images superimposed at a 30 × magnification; Hb uptake into both the cytosol and nucleus is shown.
Co-localization studies
To determine the source of free Hb-α as a product of macrophage erythrophagocytosis by co-localizing macrophages, erythrocytes and free Hb-α, we used double immunofluorescence staining for α-Hb and the macrophage marker CD163 in paraffin-embedded sections from patients with UC and CC who had moderate disease activity. The study validated macrophage phagocytosis of erythrocytes that were outside of the vascular bed (Fig. 7). The analysis also show macrophages (Fig. 7, yellow arrow) actively engulfing/phagocytizing the extravasated erythrocytes. The presence of extravasated macrophage erythrophagocytosis in hemorrhagic tissues of UC and CC samples (Fig. 7A, red arrows show erythrocytes; white arrows show macrophages) illustrates that the phagocytized digested erythrocyte by macrophages (releases free Hb and Hb(n) into cellular microenvironment). This provides evidence that the source of free Hb and Hb(n) is partly from extravasated macrophage erythrophagocytosis. Fig. 7B shows colonic mucosa with extravasated erythrocytes and macrophage in close vicinity to crypts.
Fig. 7.
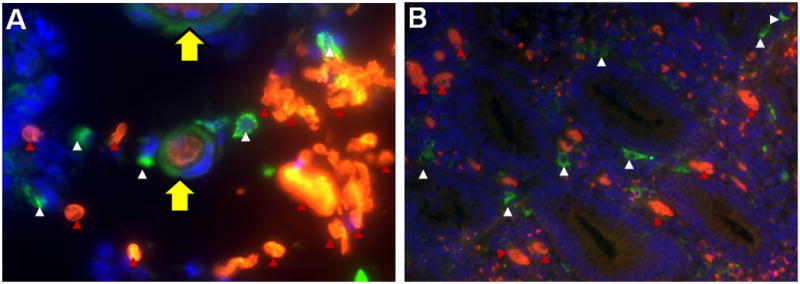
Double immunofluorescent staining for hemoglobin-α (red) and the macrophage marker CD163 (green) in paraffin-embedded sections from a patient with UC. Nuclei were counterstained with DAPI (blue): red arrows indicate extravasated erythrocytes and white arrows indicate macrophages. Pictures were taken at 60× (A) and 20× (B) magnification. A) Depicts macrophage erythrophagocytosis: a macrophage (yellow arrow) engulfed three red blood cells from extravasated erythrocytes (red arrow). B) Colonic mucosa with extravasated erythrocytes and macrophages in close vicinity to crypts.
Potential role of free Hb induced ROS production on antioxidants
To explain the differences in the amounts of ROS detected in the tissues, we speculate that the colitis-tissues express lower levels of both the nuclear and cytosolic antioxidants. We therefore analyzed the nuceo-cytoplasmic Nrf2, and cytosolic-based CAT, SOD and GPx enzyme/protein levels.
Nuclear factor erythroid-2-related factor 2 (Nrf2)
We tested Nrf2 by performed western blotting using colonic tissue cell homogenates from UC, CC, and UCAC. This analysis showed that Nrf2 levels were lower in the inflamed tissue in both UC/CC samples compared to Ctrls, although the effects were not significantly different (Fig. 8A). There was however a significant difference between DV values and those in UCAC samples. Nrf2 protein levels were twice as much in DV versus UCAC samples (15 ± 7 vs. 32 ± 8, p< 0.043394), (Fig. 8B). Although western blots showed no significant difference in Nrf2 expression levels between the colitides and colitides vs. diverticulitis, notably because Nrf2 is essentially a constitutively expressed nucleo-cytoplasmic protein that the T-PER protein extraction protocol used is not efficient at extracting nuclear protein from cells. There was no correlation found between Nrf2 and 8-OHdG. Further, in inflamed colonic tissue of UC and CC patient samples, representative immunoblot and densitometric analysis data for the antioxidant protein Nrf2 showed a significant decrease in Nrf2 compared to adjacent uninflamed tissue (control, NL) from the same patients. The mean ± S.E for UC vs. NL-UC analyses was observed to be 1.48 ± 0.10 vs. 2.09 ± 0.17 (p< 0.05) and for CC vs. NL-CC was 1.43 ± 0.03 vs. 1.99 ± 0.08 (p< 0.05), respectively, Fig. 9.
Fig. 8.

Western blots for Nrf2. A) Shows Nrf2 (57kDa) and β-actin (43kDa); levels are graphed as a percentage of β-actin. All blots were performed on the same PVDF membranes, and all incubations were performed at room temperature on a shaker. Membranes were blocked in 3% milk for 1 h, and then washed twice in PBS-T for 5 min each time. The primary antibody was incubated on the blots for 1 h. Blots were washed twice in PBS-T for 5 min each time, and then incubated with secondary antibody for 1 h. Finally, the blots were washed three times in PBS-T for 5 min each time. B) Quantification of Nrf2 was calculated based on levels graphed as a percentage of β-actin. Abbreviations: DV = Diverticulosis; CC = Crohn’s colitis, UC = ulcerative colitis, and UCAC = ulcerative colitis associated colon cancer.
Fig. 9.
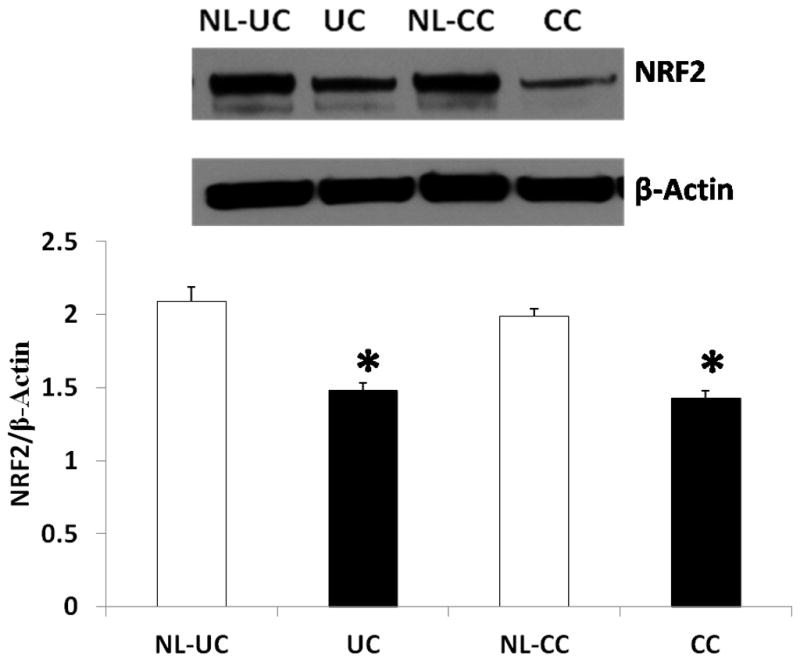
Expression of Nrf2 protein, in ulcerative colitis and Crohn’s colitis (CC), inflamed vs. uninflamed colonic muscular tissues from same patients. A) Representative immunoblot and densitometric analysis data for colonic Nrf2 proteins in UC and CC subjects as well as values in adjacent normal tissue from the same two patients (NL-UC; NL-CC). The bands show a significant decrease in Nrf2 saturation in both inflamed UC and CC colonic specimens compared to adjacent uninflamed tissues specimens. B) The lower panel (histogram) represents the data for n= 4 UC and N=4 CC, plus their adjacent normal (NL) tissue controls. Statistical significance was determined by Turkey test after one way ANOVA. *p < 0.05 compared with respective control (NL) group.
Further, we analyzed the cytoplasmic/cytosolic antioxidant enzymes i.e. Catalase (CAT), superoxide dismutase (SOD) and Glutathione peroxidase (GPx). The results indicate that the median levels in active IBD CAT was enhanced in all active disease colonic tissues and significantly increase in CC compared to Ctrl (mean ± STDev, 2.61 ± 0.57 vs. 0.01 ± 0.12, p< 0.01). Concentrations of GPx in UC was significantly enhanced compared to Ctrl (mean ± STDev, 2.74 ± 1.83 vs. 1.00 ± 0.23, p< 0.5). When comparing these antioxidants between CC vs. UC, SOD was the only enzyme observed significantly depleted in UC than CC (mean ± STDev, 0.47 ± 0.27 vs. 0.97 ± 0.13, p< 0.05). Respective WB and quantification of CAT, SOD and GPx calculated based on levels graphed as a percentage of β-actin are depicted in Fig. 10A–B.
Fig. 10.

Cytosolic antioxidant CAT, SOD and GPx levels in the tissue of patients with Diverticulitis (DV) Crohn’s colitis (CC), ulcerative colitis (UC) and ulcerative colitis associated colon cancer (UCAC). A) Show CAT, SOD, GPx and β-actin; levels are graphed as a percentage of β-actin. Note: All incubations were performed at room temperature on a shaker. Membranes were blocked in 3% milk for 1 hour, and then washed twice in PBS-T for 5 minutes each time. Primary antibody was incubated on the blots for 1 hour. Blots were washed twice in PBS-T for 5 minutes each time, and then incubated with secondary antibody for 1 hour. Finally, the blots were washed three times in PBS-T for 5 minutes each time. Blots were developed with Opti-4CN detection kit (Bio-Rad) according to manufacturer’s protocol and band intensity analyzed using Bio-Rad ChemiDoc XRS+ imaging system and Image Lab software. B) Quantification of CAT, SOD and GPx was calculated based on levels graphed as a percentage of β-actin.
Discussion
The molecular mechanisms of colitis-associated carcinogenesis has been described as a consequence of inflammatory infiltration (herein free Hb(n), not reported earlier) resulting to increased production of ROS, impairment of antioxidant defenses leading in oxidative stress (OS) following lipid peroxidation, DNA damage and genetic and epigenetic alterations in transforming epithelial cells, eventually resulting in colorectal cancer. The potential role of antioxidant defense is that oxidative damage occurs continually in vivo and organism/cells have to constantly repair or replace the damaged biomolecules.36 Damaged DNA is repaired and unrepaired DNA and damaged proteins are removed. Failures in these repair and removal systems can contribute to pathology (e.g. cancer) and perhaps even to process of aging.37–39
The primary aim of this study was to assess the possible implications of the deposition of free Hb-α chain pool abundantly found in inflamed colonic mucosal and submucosal microenvironment of IBD surgical pathology samples compared to that seen in diverticulitis. This coincidental finding co-localizes IBD-related CRC which also is found to co-occur in segments with colitis.11,40–44 We observed levels of free Hb-α, ROS and Nrf2 to be indifferent between the active colitides, as is the reported incidence and prevalence of CRC in these patients. 13,37–41,45, The development of CRC in IBD patients therefore, may not be influenced by disease activity and the extent or duration of disease alone, but by hypersensitivity to ROS, which may partly explain the identical incidence and prevalence of CRC complicating the two pathologies. 12,44,46,47 In reference to the published cumulative risk data reported that patients with IBD who were followed and treated for a long time period who developed CRC before or after colectomy surgery had the prior disease for not less than 8–10 years.12,13,44–48 For that reason, even after prophylactic surgery “proctocolectomy with mucosectomy”, the microscopic intact remnant anorectal mucosa island residues, which are potential for cancer transformation, may be retained.12,13,49
In in-vitro analysis we observed an apparent Hb-ROS-DNAD sequence. In most cases damaged DNA is repairable,50,51 however in IBD a damaged DNA base repair mechanism has been reported in UC patients.52 In certain cases the repair of damaged DNA may fail because the maintenance of genome integrity and fidelity, which is essential for the proper function and survival of all organisms, is compromised.53,54 This task is particularly daunting due to constant assault on the DNA by genotoxic agents (e.g. free Hb/Hb(n)), nucleotide disincorporation during DNA replication, and the intrinsic biochemical instability of the DNA itself.50–56 Failure to repair DNA lesions may result in blockages of transcription and replication, mutagenesis, and/or cellular cytotoxicity.57,58 In humans, DNAD has been shown to be involved in a variety of genetically inherited disorders and in carcinogenesis.58–62
It is recognized that overproduction of ROS, which is evident in the mucosa of IBD patients4,5 is a common feature affecting cell growth and apoptosis.63,64 The analysis in this study showed that hemin significantly impacted cell viability after a 24 h incubation of cells treated with either 10 μM and 100 μM concentrations. Hb at a 100 μM concentration also did affect viability negatively but the effect was not significant.
Cumulative Hb-catalyzed oxidant damage to DNA damage might occur with Hb-α and hemin overload, perhaps explaining the often lethal evolution of IBD associated conversion to carcinogenesis. We now realize that, in IBD patient surgical pathology samples, free Hb-α, ROS, DNAD (analyzed by a H2AX assay, a DNA double-strand breaks biomarker, not presented here) and colitis-associated CRC are all co-found to co-localize/ co-occur in segments with colitis.11–15,45,46,65–69 To the best of our knowledge, these studies upon which this application is based represent the first reports using histology-directed, MALDI MS, a cutting-edge technology, to investigate and determine disease-related proteomic patterns that may improve the diagnostic accuracy in IBD patients. These studies could lead to the early detection of CRC through a relatively non-invasive, easier, accurate, affordable, and fast screening process to promote therapy and prevention.
Further, we investigated if the stress response proteins Nrf2, CAT, SOD and GPx was altered in IBD surgical pathology specimens compared to Ctrls. Systemic circulating Plasma/serum levels of CAT, GPx and SOD have been investigated in IBD patients with controversial results reported either identical, down- or upregulation of these enzymic activities.70–73 To datethere is no agreement as to whether a drop or a rise of these feature activities is linked to IBD.70 These controversies may as well depend on the sensitivity of numerous techniques available, with varying precision to measure ROS or the reduction of antioxidants. Because ROS have short biological half-lives, attempts to directly quantify their levels are limited to electron spin resonance spectroscopy74,75 and chemiluminescence,76 expensive techniques that are restricted in their application. Oxidative stress is a potential etiological and/or triggering factors for IBD,4,73,77–82 The detrimental effects of reactive oxygen molecules (ROM) have been well studied and established in the inflammation sequence.83 However, the specific pathways leading to cellular damage are not completely elucidated and remain poorly defined, noncomprehensive, and impractical in clinical medicine and in therapeutic advancements.84
Nuclear factor erythroid-2-related factor 2 (Nrf2)
Nrfs is a basic leucine zipper transcription factor that is essential for cellular response to oxidative stress.85–87 Under normal and unstressed conditions, Nrf2 is a highly unstable protein with a very short half-life (t1/2 ~ 15–25 min)88,89 and is kept in the cytoplasm by a cluster of proteins that degrade it quickly.88,93 However, under oxidative stress, such as in active IBD, Nrf2 is not degraded, but instead travels to the nucleus where it binds to a DNA promoter and initiates transcription of antioxidative genes and their proteins.90 Herein data show that the expression of Nrf2 was identically reduced in both UC and CC samples while in UCAC tissue it was two-fold lower compared to Ctrl. The results of Kalthoff et al. suggest that Nrf2 is critical for protecting the GI tract against disease by regulating a multifaceted cellular antioxidant defense.91 Other studies have also shown that Nrf2 regulated antioxidant enzymes SOD and GPx are reduced in IBD patients.16,41 Colonic inflammation observed in UC and CC patients are known to impair enteric neurons and smooth muscle mediated motility function.92–96 In addition, Winston et al. studies have shown that a decrease in two myenteric neuron proteins, neuronal nitric oxide synthase (nNOS) and ChAT, and an increase in H202 (a component of oxidative stress) in animal models of colitis.93 Our recent studies97 have shown that the nitrergic (nNOS) mediated gastric motility is impaired in Nrf2 knock-out mice known to show oxidant burden.97 The above studies suggest that elevated inflammation and oxidative stress may impair nitrergic mediated motility function and Nrf2-Phase II system may be critically involved in this path. An in-depth mechanistic experiment is warranted in future to investigate the motility abnormalities associated with colonic inflammation in IBD patients.
A study by D’Odorico et al.24 analysed the blood circulating antioxidant concentrations in IBD patients and in Ctrl subjects. They found significantly reduced antioxidant saturation in IBD patients, especially in those with active disease, compared to Ctrls (p< 0.0001). They also found 8-OHdG concentrations (a measurement for ROS generation) were significantly increased in IBD patients compared to controls, independent of disease activity (p< 0.05). They also found no differences were detected between the colitides. No correlation was found between antioxidants and 8-OHdG concentrations. These observed are consistent with our observations with regard to Nrf2 protein concentrations in IBD patients with inflamed compared to adjacent uninflamed colon tissues and Ctrls. In inflamed colonic tissue of UC and CC patient samples, representative immunoblot and densitometric analysis data for the antioxidant protein Nrf2 show significant decrease compared to adjacent uninflamed tissue (normal, NL). We should keep in mind that the depletion of systemic antioxidant levels found in IBD patients,24 can depend on several other factors than different technology technique mentioned earlier herein. Dietary intake may be one of the major causes. A combination of other mechanisms could also be involved, e.g. malabsorption, insufficient vitamin intake, impaired absorption, increased vitamin requirements and increased losses, especially during active disease.24,98 In many instances, more than one of these factors could be responsible for the resulting systemic depletion. Several agents that can interfere with redox cell signaling pathways could collectively alter the cellular microenvironment and be therapeutically leveraged. 99–101 Microenvironment is capable of normalizing pathological cells, suggesting that re-education of cells may be effective strategy for treatment and prevention of colitis related neoplasia. Nutraceuticals derived from fruits, vegetables, spices, grains and cereals may play an important role.
Catalase (CAT)
CAT is a tetramer of four polypeptide heme (iron) chains common enzyme found in living organisms and a very important enzyme in protecting the cell from oxidative damage by ROS.102 It catalyzes the decomposition of hydrogen peroxide to water and oxygen.103,104 Likewise, CAT has one of the highest turnover numbers of all enzymes; one CAT molecule can convert millions of molecules of hydrogen peroxide to water and oxygen each second.102
Previous studies,22,70 on antioxidant CAT capacity in IBD focused on circulating concentrations70 reported to increase70 or decreased22 in fulminant and/or active IBD. CAT is a key antioxidant enzyme that is speculated to be actively released into the circulation as part of inflammatory response 105 and is present in the peroxisome defense against oxidative stress.105 Polymorphisms in this gene have been associated with decreases in catalase activity. In our IBD-patient tissue studies we found out that the activities of antioxidant CAT which is predominantly intracellular/cytosolic enzymes was enhanced in all active CC and UC and significantly increased in CC compared to Ctrl. Despite the statistical indifference shown between the inflammatory colitides, polymorphism within the human population as well as the sample size (cohort, n=3 per group) must be considered in evaluating the data. Discrepancies concerning results from a small number of enrolled patients in the majority of studies, could lead to type II error.106,107 Since there was no difference in their results in the enzyme activities between CAT and GPx Krzystek-Korpacka et al22 evaluated these enzymes whether could serve as indicators of active disease. Patients from both cohort were combined and the results of GPx level was comparable to that of C-reactive protein (CRP) and erythrocyte sedimentation rate (ESR), whereas that of CAT was poor. GPx showed a diagnostic accuracy of 73%, whereas CAT showed 63% as compared to 74% of CRP and ESR.
Glutathione (GPx)
GPx are important antioxidant enzymes that reduce H2O2 and other ROS by oxidizing glutathione.3 In this study, GPx levels was not changed as compared to Ctrl except in patients with active UC was significantly increased compared to Ctrl. In animal experiments when members of the GPx-gene family were disrupted, they developed severe ileocolitis and later, about 30% developed adenocarcinoma.108 It has been observed that the fall in GPx activity is associated with the coexistence of the severity of IBD, reflected by the closest of all enzyme relationships with inflammatory indices both on univeriate and multivariate analyses.22 There is a speculation that the impact of the disease and/or inflammation severity on GPx may be related to reduce glutathione, a vital intracellular antioxidant and GPx substrate, depletion of which is considered a crucial event in colonic tissue damage during IBD etiopathogenesis.109 Our previous experience has shown that most patients with active IBD are homeostatic depleted110–112 because of loses with subsequent adaptive returns following pouch surgery (restorative proctocolectomy and ileal pouch-anal anastomosis)110–112 and a significant and stable composition was achieved after closure of a diverting loop ileostomy.111–114 Accordingly, the direct relationship between GPx and cholesterol, iron, transferrin, and albumin, may reflect a more general association with nutritional status of IBD patients, impaired already at the time of disease diagnosis.22,115 GPx seems to be of special interest in the context of IBD etiopthogenesis. Studies on animals showed that mice with concomitant disruption of genes coding for GPx1 and GPx2 enzymes developed UC and that their colonic inner lining (the mucosa) had elevated activities of myeloperoxidase and the levels of lipid hydroperoxides, stressing the importance of both isozymes for the prevention of intestinal inflammatory response.116
Superoxide dismutase (SOD)
Herein observations show statistically insignificant impairments’ of SOD in UC and in UCAC subjects vs. Ctrls. When CC vs. UC was compared a significant depletion was noted in UC than in CC. SOD is an unusually ubiquitous stable cytoplasmic enzyme although its apoenzyme is very unstable117 that function to effectively catalyze the dismutation of superoxide (O2 ) inions into oxygen and hydrogen peroxide. Hence, they are an important antioxidant defense in nearly all cells exposed to oxygen. What role SOD play in both normal and disease states is only slowly beginning to be understood. In case of ongoing intestinal inflammation, like in active IBD, SOD leads to depletion of circulating antioxidant, but the blood activity of SOD was reported to have remained either unaltered or increased in the majority of studies.70,118
The Colitides
The IBD encompasses of UC and Crohn’s disease (CD),119 (and an indeterminate colitis (IC) where a non-definitive evaluation cannot be made).120 CD of the colon is termed Crohn’s colitis (CC). UC and CD are two chronic, relapsing and remitting diseases that (when predominately colonic) remain as major diagnostic, therapeutic, and surgical challenges, have no permanent drug cure and have the potential to causing significant long-term morbidity.119,121 Despite extensive research work, to date, an integrated, pathophysiologic model of the etiology and/or propagation of IBD have not emerged and is still obscure but considered to be multifactorial, including a genetic predisposition, the gut flora and environmental agents.122–124 As a result of these multifactorial theories it is generally hypothesized that IBD is caused by gastrointestinal tract immune dysregulation, because the disease is accompanied by a considerable infiltration of inflammatory cells in gut mucosa.125 There is no difference in IBD incidences by gender,126,127 although in up to 20% of patients the disease presents before the age of 18 years.128–131 UC affects only the colon and is confined mostly to the mucosal and to a lesser degree, the submucosal layers. In contrast, Crohn’s disease (CD) may involve any part in the GI tract from the mouth to the anus and all layers (transmural) of the GI system;121 it may also involve other organs outside the GI system through fistulization.132–136 Despite data supporting primary chemoprevention with pharmacotherapy and secondary prevention with surveillance endoscopy, CRC continues to constitute 10–15% of IBD-associated mortality.137,138 Within this population however, there is a considerable variability in the cancer risk possibly due to co-inheriting a risk of developing both IBD and CRC.40 This secondarily has been attributed to a complex sequence of molecular and histologic derangements of colonocytes that are initially and at least partly sustained by a long-standing, extensive, chronic, relapsing and remitting inflammation.41,42,46,67
Conclusion
We found pool of Hb-α chain in the inflamed colonic mucosal and submucosal tissues of IBD patients. The pool accumulated overtime as a result of Hb degradation and extravasated erythrocyte macrophage erythrophagocytosis. More importantly, we observed and demonstrated that free Hb-α is a mediator for ROS-dependent DNAD. DNAD may potentially and independently increase risk vulnerability of transformation to CRC. The fact that a level of Hb-α is identical between UC and CC may partially explain the indifference of CRC incidence between the two pathologies since both are co-localized in segments with evidence of colitis. Depletion of antioxidants is likely to be important in the biopathophysiology of IBD and may partly be a primary phenomenon that severely compromises the mucosa and therefore increases the susceptibility to oxidative tissue damage. Despite the fact that a rigid basis for causality of IBD, it is likely that OS has an etiologic triggering potential. However, whether the OS biometrics are vital to be useful in monitoring disease activity or determining prognosis remains uncertain to date. This may however, represent a risk factor for the disease and can partly be an additional argument for the direct implication of free-Hb(n) induced OS in the pathogenesis of IBD and the subsequent colitis related CRC. Therapeutic applicability of antioxidants must be elucidated, randomized, clinical trials in future. Targeting Redox (reduction-oxidation)-sensitive pathways and transcription factors as well as the use of high-dose potential antioxidants, along follow-up period, and emphasis on active cases of the colitides may therefore offer great promise options in the treatment and prevention of IBD and colitis associated CRC.
Acknowledgments
Funding: NIH R21DK095186; 1UL1 RR024975-01; 2UL1 TR000445-06; 3U54 CA091408–09S1; 5U54RR026140-03; S10RR0254970; R21DKO76704; G12MD007586; U54MD007593; U54CA163069.
The authors thank Kay M. Washington, M.D., Ph.D and Billy R. Ballard, DDS, M.D., for guidance in surgical pathology reports of the biospecimens. We are grateful to Ifeanyi J. Arinze, Ph.D and Amos Sakwe, Ph.D., MSCI for intellectual critics, comments and suggestions. We are thankful to Kerry R. Wiles and Anthony L. Frazier for the coordination of biospecimens collection. We are grateful to Marry P. Moyer, Ph.D for providing NCM460 colonocytes. We are grateful to Melissa B. Downing, Kieosha D. Williams and Tanusri Ghosh for assistance with sample preparations, Western blot and for immunohistochemistry analyses. We are thankful to Naji N. Abumrad, M.D. and Alan Herline, M.D. for assistance with patient clinical guidelines and J. Shawn Goodwin, Ph.D for protein imaging and quantifications. We are grateful to Krista Meyer, Ph.D and Julia Grigorieva, Ph.D for guidance in statistical data analyses.
Abbreviations
- IBD
inflammatory bowel disease
- UC
ulcerative colitis
- CC
Crohn’s colitis
- DV
diverticulosis
- UCAC
ulcerative colitis-associated colon cancer
- DNAD
Deoxyribonucleic acid damage
- Hb
hemoglobin
- Hb(n)
(n, α, β, and hemin)
- IHC
immunohistochemistry
- CRC
colorectal cancer
- ROS
reactive oxidative stress
- MALDI MS
matrix-assisted laser desorption/ionization mass spectrometry
- m/z
mass-to-charge ratio
- LC-MS/MS
liquid chromatography-mass spectrometry
- ETD
electron transfer dissociation
- CID
collision induced dissociation
- Nrf2
Nuclear factor E2-related factor 2
- CAT
Catalase
- GPx
Glutathione peroxidase
- SOD
superoxide dismutase
- CPR
C-reactive protein
- ESR
erythrocyte sedimentation rate
Footnotes
Presented
in parts, at The 103nd Annual Congress, the American Association for Cancer Research, Chicago, IL, March 31-April 4, 2012; at AACR Special Conference on Molecular Targeted Therapies: Mechanisms of Resistance, San Diego, CA, May 9–12, 2012; at the Fifth AACR Conference on The Science of Cancer Health Disparities, San Diego, CA, October 27–30, 2012; at 2nd International Conference on Proteomics & Bioinformatics, Las Vegas, NA July 2–4, 2012; and at 3rd International Conference on Proteomics & Bioinformatics, Philadelphia, PA July 15–17, 2013.
Conflict of interest
The authors confirms of compliance with ethical and legal obligations including but not limited to compliance with ICMJE authorship and competing interests guidelines, that the article is neither under consideration for publication nor published elsewhere. This article is subject to blind, independent, expert peer review.
Writing Assistance
None
Author contributions
Jeremy N. Myers: study concept and design, acquisition of data, analysis and interpretation of data, statistical analysis
Michael W. Schäffer: study concept, acquisition of data; data analysis, interpretation of data, statistical analysis
Olga Y. Korolkova: acquisition of data, analysis and interpretation of data, material and technical support
Amanda D. Williams: data analysis, interpretation of data, material support
Pandu R. Gangula: expertise in antioxidants and critical reading of manuscript for intellectual content
Amosy E. M’Koma: study concept and design, analysis and interpretation of data, drafting and revision of the manuscript, critical writing of manuscript for important intellectual content, statistical analysis, obtained funding, study architect and supervision
References
- 1.M’Koma AE, Seeley EH, Washington MK, et al. Proteomic profiling of mucosal and submucosal colonic tissues yields protein signatures that differentiate the inflammatory colitides. Inflamm Bowel Dis. 2011;17:875–83. doi: 10.1002/ibd.21442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Seeley EH, Washington MK, Caprioli RM, M’Koma AE. Proteomic patterns of colonic mucosal tissues delineate Crohn’s colitis and ulcerative colitis. Proteomics Clin Appl. 2013;7:541–9. doi: 10.1002/prca.201200107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Roessner A, Kuester D, Malfertheiner P, Schneider-Stock R. Oxidative stress in ulcerative colitis-associated carcinogenesis. Pathol Res Pract. 2008;204:511–24. doi: 10.1016/j.prp.2008.04.011. [DOI] [PubMed] [Google Scholar]
- 4.Lih-Brody L, Powell SR, Collier KP, et al. Increased oxidative stress and decreased antioxidant defenses in mucosa of inflammatory bowel disease. Dig Dis Sci. 1996;41:2078–86. doi: 10.1007/BF02093613. [DOI] [PubMed] [Google Scholar]
- 5.Grisham MB. Oxidants and free radicals in inflammatory bowel disease. Lancet. 1994;344:859–61. doi: 10.1016/s0140-6736(94)92831-2. [DOI] [PubMed] [Google Scholar]
- 6.Halliwell B, Gutteridge JM. Oxygen free radicals and iron in relation to biology and medicine: some problems and concepts. Arch Biochem Biophys. 1986;246:501–14. doi: 10.1016/0003-9861(86)90305-x. [DOI] [PubMed] [Google Scholar]
- 7.Sturniolo GCTG, D’Inca R. Mucosal iron concentration and immunohistochemical evidence of iron-binding proteins in long-standing ulcerative colitis. Intal J Gastroenterol. 1993;25:A68. [Google Scholar]
- 8.McFaul SJ, Bowman PD, Villa VM. Hemoglobin stimulates the release of proinflammatory cytokines from leukocytes in whole blood. J Lab Clin Med. 2000;135:263–9. doi: 10.1067/mlc.2000.105180. [DOI] [PubMed] [Google Scholar]
- 9.McFaul SJ, Bowman PD, Villa VM, Gutierrez-Ibanez MJ, Johnson M, Smith D. Hemoglobin stimulates mononuclear leukocytes to release interleukin-8 and tumor necrosis factor alpha. Blood. 1994;84:3175–81. [PubMed] [Google Scholar]
- 10.Connelly TM, Koltun WA. The Cancer “Fear” in IBD Patients: Is It Still REAL? J Gastrointest Surg. 2014;18:213–8. doi: 10.1007/s11605-013-2317-z. [DOI] [PubMed] [Google Scholar]
- 11.Averboukh F, Ziv Y, Kariv Y, et al. Colorectal carcinoma in inflammatory bowel disease: a comparison between Crohn’s and ulcerative colitis. Colorectal Dis. 2011;13:1230–5. doi: 10.1111/j.1463-1318.2011.02639.x. [DOI] [PubMed] [Google Scholar]
- 12.Um JW, M’Koma AE. Pouch-related dysplasia and adenocarcinoma following restorative proctocolectomy for ulcerative colitis. Tech Coloproctol. 2011;15:7–16. doi: 10.1007/s10151-010-0664-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.M’Koma AE, Moses HL, Adunyah SE. Inflammatory bowel disease-associated colorectal cancer: proctocolectomy andmucosectomy does not necessarily eliminate pouch related cancer incidences. Int J colorect Dis. 2011;26:533–52. doi: 10.1007/s00384-011-1137-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ekbom A, Helmick C, Zack M, Adami HO. Ulcerative colitis and colorectal cancer. A population-based study. N Engl J Med. 1990;323:1228–33. doi: 10.1056/NEJM199011013231802. [DOI] [PubMed] [Google Scholar]
- 15.Ekbom A, Helmick C, Zack M, Adami HO. Increased risk of large-bowel cancer in Crohn’s disease with colonic involvement. Lancet. 1990;336:357–9. doi: 10.1016/0140-6736(90)91889-i. [DOI] [PubMed] [Google Scholar]
- 16.Mantovani A, Romero P, Palucka AK, Marincola FM. Tumour immunity: effector response to tumour and role of the microenvironment. Lancet. 2008;371:771–83. doi: 10.1016/S0140-6736(08)60241-X. [DOI] [PubMed] [Google Scholar]
- 17.Lorusso G, Ruegg C. The tumor microenvironment and its contribution to tumor evolution toward metastasis. Histochem Cell Biol. 2008;130:1091–103. doi: 10.1007/s00418-008-0530-8. [DOI] [PubMed] [Google Scholar]
- 18.M’Koma AE, Seeley EH, Wise PE, et al. Proteomic analysis of colonic submucosa differentiates Crohn’s and ulcerative colitis. Annual Congress - Digestive Disease Week; Chicago, IL. 2009. p. 600. [Google Scholar]
- 19.M’Koma AE, Seeley EH, Wise PE, et al. Proteomic Patterns of Colonic Submucosa Discriminates Inflammatory Colitides. Annual Congress - The American Society of Colon and Rectal Surgeons; Hollywood, Florida. 2009. p. 47.p. 166. [Google Scholar]
- 20.M’Koma A, Wise PE, Seeley EH, et al. Human Alpha Defensins are Differentially Expressed Between the Inflammatory Colitides. Annual Congress - Digestive Disease Week; New Orleans, LA. 2010. p. 720. [Google Scholar]
- 21.M’Koma A, Wise PE, Schwartz DA, et al. Gene Expression of Colonic Submucosa Differs Between the Inflammatory Colitides. Annual Congress - The American Society of Colon and Rectal Surgeons; Minneapolis, MN. 2010. p. 117. [Google Scholar]
- 22.Krzystek-Korpacka M, Neubauer K, Berdowska I, et al. Impaired erythrocyte antioxidant defense in active inflammatory bowel disease: impact of anemia and treatment. Inflamm Bowel Dis. 2010;16:1467–75. doi: 10.1002/ibd.21234. [DOI] [PubMed] [Google Scholar]
- 23.Kruidenier L, Verspaget HW. Review article: oxidative stress as a pathogenic factor in inflammatory bowel disease--radicals or ridiculous? Aliment Pharmacol Ther. 2002;16:1997–2015. doi: 10.1046/j.1365-2036.2002.01378.x. [DOI] [PubMed] [Google Scholar]
- 24.D’Odorico A, Bortolan S, Cardin R, et al. Reduced plasma antioxidant concentrations and increased oxidative DNA damage in inflammatory bowel disease. Scand J Gastroenterol. 2001;36:1289–94. doi: 10.1080/003655201317097146. [DOI] [PubMed] [Google Scholar]
- 25.Krzystek-Korpacka M, Neubauer K, Berdowska I, et al. Enhanced formation of advanced oxidation protein products in IBD. Inflamm Bowel Dis. 2008;14:794–802. doi: 10.1002/ibd.20383. [DOI] [PubMed] [Google Scholar]
- 26.Boehm D, Krzystek-Korpacka M, Neubauer K, et al. Paraoxonase-1 status in Crohn’s disease and ulcerative colitis. Inflamm Bowel Dis. 2009;15:93–9. doi: 10.1002/ibd.20582. [DOI] [PubMed] [Google Scholar]
- 27.Koutroubakis IE, Malliaraki N, Dimoulios PD, et al. Decreased total and corrected antioxidant capacity in patients with inflammatory bowel disease. Dig Dis Sci. 2004;49:1433–7. doi: 10.1023/b:ddas.0000042242.22898.d9. [DOI] [PubMed] [Google Scholar]
- 28.Puri KS, Suresh KR, Gogtay NJ, Thatte UM. Declaration of Helsinki, 2008: implications for stakeholders in research. J Postgrad Med. 2009;55:131–4. doi: 10.4103/0022-3859.52846. [DOI] [PubMed] [Google Scholar]
- 29.Schey KL, Anderson DM, Rose KL. Spatially-directed protein identification from tissue sections by top-down LC-MS/MS with electron transfer dissociation. Anal Chem. 2013;85:6767–74. doi: 10.1021/ac400832w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Maamer-Azzabi A, Ndozangue-Touriguine O, Breard J. Metastatic SW620 colon cancer cells are primed for death when detached and can be sensitized to anoikis by the BH3-mimetic ABT-737. Cell Death Dis. 2013;4:e801. doi: 10.1038/cddis.2013.328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gangula PR, Chinnathambi V, Hale AB, et al. Impairment of nitrergic system and delayed gastric emptying in low density lipoprotein receptor deficient female mice. Neurogastroenterol Motil. 2011;23:773–e335. doi: 10.1111/j.1365-2982.2011.01695.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ravella K, Yang H, Gangula PR. Impairment of gastric nitrergic and NRF2 system in apolipoprotein E knockout mice. Dig Dis Sci. 2012;57:1504–9. doi: 10.1007/s10620-012-2070-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.van Meerloo J, Kaspers GJ, Cloos J. Cell sensitivity assays: the MTT assay. Methods Mol Biol. 2011;731:237–45. doi: 10.1007/978-1-61779-080-5_20. [DOI] [PubMed] [Google Scholar]
- 34.Megalopoulou TM, Koutroumbas K, Pouliakis A, et al. The potential of feature selection by statistical techniques and the use of statistical classifiers in the discrimination of benign from malignant gastric lesions. Oncol Rep. 2006;15(Spec no):1033–6. doi: 10.3892/or.15.4.1033. [DOI] [PubMed] [Google Scholar]
- 35.Webb A. Statistical Pattern Recognition. Chichester, UK: John Wiley & Sons; 2000. [Google Scholar]
- 36.Halliwell B. Free radicals and antioxidants: updating a personal view. Nutrition reviews. 2012;70:257–65. doi: 10.1111/j.1753-4887.2012.00476.x. [DOI] [PubMed] [Google Scholar]
- 37.Mason PA, Cox LS. The role of DNA exonucleases in protecting genome stability and their impact on ageing. Age. 2012;34:1317–40. doi: 10.1007/s11357-011-9306-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pickering AM, Koop AL, Teoh CY, et al. The immunoproteasome, the 20S proteasome and the PA28alphabeta proteasome regulator are oxidative-stress-adaptive proteolytic complexes. Biochem J. 2010;432:585–94. doi: 10.1042/BJ20100878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dasuri K, Zhang L, Ebenezer P, et al. Proteasome alterations during adipose differentiation and aging: links to impaired adipocyte differentiation and development of oxidative stress. Free Radic Biol Med. 2011;51:1727–35. doi: 10.1016/j.freeradbiomed.2011.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rhodes JM. Unifying hypothesis for inflammatory bowel disease and associated colon cancer: sticking the pieces together with sugar. Lancet. 1996;347:40–4. doi: 10.1016/s0140-6736(96)91563-9. [DOI] [PubMed] [Google Scholar]
- 41.Rubin DT, Parekh N. Colorectal cancer in inflammatory bowel disease: molecular and clinical considerations. Curr Treat Options Gastroenterol. 2006;9:211–20. doi: 10.1007/s11938-006-0040-5. [DOI] [PubMed] [Google Scholar]
- 42.Harpaz N, Talbot IC. Colorectal cancer in idiopathic inflammatory bowel disease. Semin Diagn Pathol. 1996;13:339–57. [PubMed] [Google Scholar]
- 43.Loftus EV., Jr Epidemiology and risk factors for colorectal dysplasia and cancer in ulcerative colitis. Gastroenterol Clin North Am. 2006;35:517–31. doi: 10.1016/j.gtc.2006.07.005. [DOI] [PubMed] [Google Scholar]
- 44.Vleggaar FP, Lutgens MW, Oldenburg B, et al. British and American screening guidelines inadequate for prevention of colorectal carcinoma in patients with inflammatory bowel disease. Ned Tijdschr Geneeskd. 2007;151:2787–91. [PubMed] [Google Scholar]
- 45.Svrcek M, Cosnes J, Beaugerie L, et al. Colorectal neoplasia in Crohn’s colitis: a retrospective comparative study with ulcerative colitis. Histopathology. 2007;50:574–83. doi: 10.1111/j.1365-2559.2007.02663.x. [DOI] [PubMed] [Google Scholar]
- 46.Rutter M, Saunders B, Wilkinson K, et al. Severity of inflammation is a risk factor for colorectal neoplasia in ulcerative colitis. Gastroenterology. 2004;126:451–9. doi: 10.1053/j.gastro.2003.11.010. [DOI] [PubMed] [Google Scholar]
- 47.Hardy RG, Meltzer SJ, Jankowski JA. ABC of colorectal cancer. Molecular basis for risk factors. BMJ. 2000;321:886–9. doi: 10.1136/bmj.321.7265.886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Soderlund S, Brandt L, Lapidus A, et al. Decreasing time-trends of colorectal cancer in a large cohort of patients with inflammatory bowel disease. Gastroenterology. 2009;136:1561–7. doi: 10.1053/j.gastro.2009.01.064. [DOI] [PubMed] [Google Scholar]
- 49.Winther KV, Jess T, Langholz E, Munkholm P, Binder V. Long-term risk of cancer in ulcerative colitis: a population-based cohort study from Copenhagen County. Clin Gastroenterol Hepatol. 2004;2:1088–95. doi: 10.1016/s1542-3565(04)00543-9. [DOI] [PubMed] [Google Scholar]
- 50.Surova O, Zhivotovsky B. Various modes of cell death induced by DNA damage. Oncogene. 2013;32:3789–97. doi: 10.1038/onc.2012.556. [DOI] [PubMed] [Google Scholar]
- 51.Asaithamby A, Chen DJ. Mechanism of cluster DNA damage repair in response to high-atomic number and energy particles radiation. Mutat Res. 2011;711:87–99. doi: 10.1016/j.mrfmmm.2010.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sanford KK, Price FM, Brodeur C, et al. Deficient DNA repair in chronic ulcerative colitis. Cancer Detect Prev. 1997;21:540–5. [PubMed] [Google Scholar]
- 53.Meyer JN, Bess AS. Involvement of autophagy and mitochondrial dynamics in determining the fate and effects of irreparable mitochondrial DNA damage. Autophagy. 2012:8. doi: 10.4161/auto.21741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shrotriya S, Deep G, Gu M, et al. Generation of reactive oxygen species by grape seed extract causes irreparable DNA damage leading to G2/M arrest and apoptosis selectively in head and neck squamous cell carcinoma cells. Carcinogenesis. 2012;33:848–58. doi: 10.1093/carcin/bgs019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Weinberger M, Feng L, Paul A, et al. DNA replication stress is a determinant of chronological lifespan in budding yeast. PLoS One. 2007;2:e748. doi: 10.1371/journal.pone.0000748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dykens JA, Baginski TJ. Urinary 8-hydroxydeoxyguanosine excretion as a non-invasive marker of neutrophil activation in animal models of inflammatory bowel disease. Scand J Gastroenterol. 1998;33:628–36. doi: 10.1080/00365529850171918. [DOI] [PubMed] [Google Scholar]
- 57.Kelly GL, Strasser A. The essential role of evasion from cell death in cancer. Adv Cancer Res. 2011;111:39–96. doi: 10.1016/B978-0-12-385524-4.00002-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tagawa Y, Sanders JD, Uchida I, et al. Genetic and functional analysis of Haemophilus somnus high molecular weight-immunoglobulin binding proteins. Microb Pathog. 2005;39:159–70. doi: 10.1016/j.micpath.2005.08.002. [DOI] [PubMed] [Google Scholar]
- 59.Cory JG. Ribonucleotide reductase as a chemotherapeutic target. Adv Enzyme Regul. 1988;27:437–55. doi: 10.1016/0065-2571(88)90030-1. [DOI] [PubMed] [Google Scholar]
- 60.Adams CW, Fornwald JA, Schmidt FJ, et al. Gene organization and structure of the Streptomyces lividans gal operon. J Bacteriol. 1988;170:203–12. doi: 10.1128/jb.170.1.203-212.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Croce CM, Tsujimoto Y, Erikson J, Nowell P. Chromosome translocations and B cell neoplasia. Lab Invest. 1984;51:258–67. [PubMed] [Google Scholar]
- 62.Vaux DL, Cory S, Adams JM. Bcl-2 gene promotes haemopoietic cell survival and cooperates with c-myc to immortalize pre-B cells. Nature. 1988;335:440–2. doi: 10.1038/335440a0. [DOI] [PubMed] [Google Scholar]
- 63.Fiers W, Beyaert R, Declercq W, Vandenabeele P. More than one way to die: apoptosis, necrosis and reactive oxygen damage. Oncogene. 1999;18:7719–30. doi: 10.1038/sj.onc.1203249. [DOI] [PubMed] [Google Scholar]
- 64.Shen H, Liu J, Wang Y, et al. Aflatoxin G-induced Oxidative stress causes DNA damage and triggers apoptosis through MAPK signaling pathway in A549 cells. Food Chem Toxicol. 2013;62:661–9. doi: 10.1016/j.fct.2013.09.030. [DOI] [PubMed] [Google Scholar]
- 65.Choi PM, Zelig MP. Similarity of colorectal cancer in Crohn’s disease and ulcerative colitis: implications for carcinogenesis and prevention. Gut. 1994;35:950–4. doi: 10.1136/gut.35.7.950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Terzic J, Grivennikov S, Karin E, Karin M. Inflammation and colon cancer. Gastroenterology. 2010;138:2101–14. e5. doi: 10.1053/j.gastro.2010.01.058. [DOI] [PubMed] [Google Scholar]
- 67.Eaden JA, Abrams KR, Mayberry JF. The risk of colorectal cancer in ulcerative colitis: a meta-analysis. Gut. 2001;48:526–35. doi: 10.1136/gut.48.4.526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gilson TP, Sollenberger LL. Adenocarcinoma of an ileostomy in a patient with familial adenomatous polyposis. Report of a case. Dis Colon Rectum. 1992;35:261–5. doi: 10.1007/BF02051020. [DOI] [PubMed] [Google Scholar]
- 69.Gillen CD, Walmsley RS, Prior P, et al. Ulcerative colitis and Crohn’s disease: a comparison of the colorectal cancer risk in extensive colitis. Gut. 1994;35:1590–2. doi: 10.1136/gut.35.11.1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Rezaie A, Parker RD, Abdollahi M. Oxidative stress and pathogenesis of inflammatory bowel disease: an epiphenomenon or the cause? Dig Dis Sci. 2007;52:2015–21. doi: 10.1007/s10620-006-9622-2. [DOI] [PubMed] [Google Scholar]
- 71.Tsunada S, Iwakiri R, Ootani H, et al. Redox imbalance in the colonic mucosa of ulcerative colitis. Scand J Gastroenterol. 2003;38:1002–3. doi: 10.1080/00365520310005055. [DOI] [PubMed] [Google Scholar]
- 72.Kruidenier L, Kuiper I, Van Duijn W, et al. Imbalanced secondary mucosal antioxidant response in inflammatory bowel disease. J Pathol. 2003;201:17–27. doi: 10.1002/path.1408. [DOI] [PubMed] [Google Scholar]
- 73.Koch TR, Yuan LX, Stryker SJ, et al. Total antioxidant capacity of colon in patients with chronic ulcerative colitis. Dig Dis Sci. 2000;45:1814–9. doi: 10.1023/a:1005517824877. [DOI] [PubMed] [Google Scholar]
- 74.Mueller S. Sensitive and nonenzymatic measurement of hydrogen peroxide in biological systems. Free Radic Biol Med. 2000;29:410–5. doi: 10.1016/s0891-5849(00)00261-6. [DOI] [PubMed] [Google Scholar]
- 75.Togashi H, Shinzawa H, Matsuo T, et al. Analysis of hepatic oxidative stress status by electron spin resonance spectroscopy and imaging. Free Radic Biol Med. 2000;28:846–53. doi: 10.1016/s0891-5849(99)00280-4. [DOI] [PubMed] [Google Scholar]
- 76.Faulkner K, Fridovich I. Luminol and lucigenin as detectors for O2. Free Radic Biol Med. 1993;15:447–51. doi: 10.1016/0891-5849(93)90044-u. [DOI] [PubMed] [Google Scholar]
- 77.Kruidenier L, Kuiper I, Lamers CB, Verspaget HW. Intestinal oxidative damage in inflammatory bowel disease: semi-quantification, localization, and association with mucosal antioxidants. J Pathol. 2003;201:28–36. doi: 10.1002/path.1409. [DOI] [PubMed] [Google Scholar]
- 78.Cao W, Vrees MD, Kirber MT, et al. Hydrogen peroxide contributes to motor dysfunction in ulcerative colitis. Am J Physiol Gastrointest Liver Physiol. 2004;286:G833–43. doi: 10.1152/ajpgi.00414.2003. [DOI] [PubMed] [Google Scholar]
- 79.Chiarpotto E, Scavazza A, Leonarduzzi G, et al. Oxidative damage and transforming growth factor beta 1 expression in pretumoral and tumoral lesions of human intestine. Free Radic Biol Med. 1997;22:889–94. doi: 10.1016/s0891-5849(96)00481-9. [DOI] [PubMed] [Google Scholar]
- 80.Holmes EW, Yong SL, Eiznhamer D, Keshavarzian A. Glutathione content of colonic mucosa: evidence for oxidative damage in active ulcerative colitis. Dig Dis Sci. 1998;43:1088–95. doi: 10.1023/a:1018899222258. [DOI] [PubMed] [Google Scholar]
- 81.Szanto I, Rubbia-Brandt L, Kiss P, et al. Expression of NOX1, a superoxide-generating NADPH oxidase, in colon cancer and inflammatory bowel disease. J pathol. 2005;207:164–76. doi: 10.1002/path.1824. [DOI] [PubMed] [Google Scholar]
- 82.Buffinton GD, Doe WF. Depleted mucosal antioxidant defences in inflammatory bowel disease. Free Radic Biol Med. 1995;19:911–8. doi: 10.1016/0891-5849(95)94362-h. [DOI] [PubMed] [Google Scholar]
- 83.Spitz DR, Azzam EI, Li JJ, Gius D. Metabolic oxidation/reduction reactions and cellular responses to ionizing radiation: a unifying concept in stress response biology. Cancer Metastasis Rev. 2004;23:311–22. doi: 10.1023/B:CANC.0000031769.14728.bc. [DOI] [PubMed] [Google Scholar]
- 84.Hendrickson BA, Gokhale R, Cho JH. Clinical aspects and pathophysiology of inflammatory bowel disease. Clin Microbiol Rev. 2002;15:79–94. doi: 10.1128/CMR.15.1.79-94.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Nguyen T, Sherratt PJ, Pickett CB. Regulatory mechanisms controlling gene expression mediated by the antioxidant response element. Anl Revi of Pharmacol Toxicol. 2003;43:233–60. doi: 10.1146/annurev.pharmtox.43.100901.140229. [DOI] [PubMed] [Google Scholar]
- 86.Motohashi H, Yamamoto M. Nrf2-Keap1 defines a physiologically important stress response mechanism. Trends Mol Med. 2004;10:549–57. doi: 10.1016/j.molmed.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 87.Wakabayashi N, Dinkova-Kostova AT, Holtzclaw WD, et al. Protection against electrophile and oxidant stress by induction of the phase 2 response: fate of cysteines of the Keap1 sensor modified by inducers. Proc Natl Acad Sci U S A 2004. 2004;101:2040–5. doi: 10.1073/pnas.0307301101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.McMahon M, Thomas N, Itoh K, et al. Redox-regulated turnover of Nrf2 is determined by at least two separate protein domains, the redox-sensitive Neh2 degron and the redox-insensitive Neh6 degron. J Biol Chem. 2004;279:31556–67. doi: 10.1074/jbc.M403061200. [DOI] [PubMed] [Google Scholar]
- 89.Nguyen T, Nioi P, Pickett CB. The Nrf2-antioxidant response element signaling pathway and its activation by oxidative stress. J Biol Chem. 2009;284:13291–5. doi: 10.1074/jbc.R900010200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Itoh K, Wakabayashi N, Katoh Y, et al. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999;13:76–86. doi: 10.1101/gad.13.1.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kalthoff S, Ehmer U, Freiberg N, et al. Interaction between oxidative stress sensor Nrf2 and xenobiotic-activated aryl hydrocarbon receptor in the regulation of the human phase II detoxifying UDP-glucuronosyltransferase 1A10. J Biol Chem. 2010;285:5993–6002. doi: 10.1074/jbc.M109.075770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Annese V, Bassotti G, Napolitano G, et al. Gastrointestinal motility disorders in patients with inactive Crohn’s disease. Scand J Gastroenterol. 1997;32:1107–17. doi: 10.3109/00365529709002989. [DOI] [PubMed] [Google Scholar]
- 93.Winston JH, Li Q, Sarna SK. Paradoxical regulation of ChAT and nNOS expression in animal models of Crohn’s colitis and ulcerative colitis. Am J Physiol Gastrointest Liver Physiol. 2013;305:G295–302. doi: 10.1152/ajpgi.00052.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Snape WJ, Jr, Williams R, Hyman PE. Defect in colonic smooth muscle contraction in patients with ulcerative colitis. Am J Physiol. 1991;261:G987–91. doi: 10.1152/ajpgi.1991.261.6.G987. [DOI] [PubMed] [Google Scholar]
- 95.Vermillion DL, Huizinga JD, Riddell RH, Collins SM. Altered small intestinal smooth muscle function in Crohn’s disease. Gastroenterology. 1993;104:1692–9. doi: 10.1016/0016-5085(93)90647-u. [DOI] [PubMed] [Google Scholar]
- 96.Vrees MD, Pricolo VE, Potenti FM, Cao W. Abnormal motility in patients with ulcerative colitis: the role of inflammatory cytokines. Arch Surg. 2002;137:439–45. doi: 10.1001/archsurg.137.4.439. [DOI] [PubMed] [Google Scholar]
- 97.Mukhopadhyay S, Sekhar KR, Hale AB, et al. Loss of NRF2 impairs gastric nitrergic stimulation and function. Free Radic Biol Med. 2011;51:619–25. doi: 10.1016/j.freeradbiomed.2011.04.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ramakrishna BS, Varghese R, Jayakumar S, Mathan M, Balasubramanian KA. Circulating antioxidants in ulcerative colitis and their relationship to disease severity and activity. J Gastroenterol Hepatol. 1997;12:490–4. doi: 10.1111/j.1440-1746.1997.tb00471.x. [DOI] [PubMed] [Google Scholar]
- 99.Fang J, Seki T, Maeda H. Therapeutic strategies by modulating oxygen stress in cancer and inflammation. Adv Drug Deliv Rev. 2009;61:290–302. doi: 10.1016/j.addr.2009.02.005. [DOI] [PubMed] [Google Scholar]
- 100.Virgili F, Marino M. Regulation of cellular signals from nutritional molecules: a specific role for phytochemicals, beyond antioxidant activity. Free Radic Biol Med. 2008;45:1205–16. doi: 10.1016/j.freeradbiomed.2008.08.001. [DOI] [PubMed] [Google Scholar]
- 101.Surh YJ, Kundu JK, Na HK, Lee JS. Redox-sensitive transcription factors as prime targets for chemoprevention with anti-inflammatory and antioxidative phytochemicals. J Nutr. 2005;135:2993S–3001S. doi: 10.1093/jn/135.12.2993S. [DOI] [PubMed] [Google Scholar]
- 102.Imlay JA. Pathways of oxidative damage. Annu Rev Microbiol. 2003;57:395–418. doi: 10.1146/annurev.micro.57.030502.090938. [DOI] [PubMed] [Google Scholar]
- 103.Vainshtein BK, Melik-Adamyan WR, Barynin VV, et al. Three-dimensional structure of the enzyme catalase. Nature. 1981;293:411–2. doi: 10.1038/293411a0. [DOI] [PubMed] [Google Scholar]
- 104.Chelikani P, Fita I, Loewen PC. Diversity of structures and properties among catalases. Cellular and molecular life sciences : CMLS. 2004;61:192–208. doi: 10.1007/s00018-003-3206-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Yasmineh WG, Theologides A. Catalase as a roving scavenger of hydrogen peroxide: a hypothesis. J Lab Clin Med. 1993;122:110–4. [PubMed] [Google Scholar]
- 106.Giuffrida MA. Type II error and statistical power in reports of small animal clinical trials. J Am Vet Med Assoc. 2014;244:1075–80. doi: 10.2460/javma.244.9.1075. [DOI] [PubMed] [Google Scholar]
- 107.Armstrong RA. When to use the Bonferroni correction. Ophthalmic Physiol Opt. 2014 doi: 10.1111/opo.12131. [DOI] [PubMed] [Google Scholar]
- 108.Chu FF, Esworthy RS, Chu PG, et al. Bacteria-induced intestinal cancer in mice with disrupted Gpx1 and Gpx2 genes. Cancer Res. 2004;64:962–8. doi: 10.1158/0008-5472.can-03-2272. [DOI] [PubMed] [Google Scholar]
- 109.Loguercio C, D’Argenio G, Delle Cave M, et al. Glutathione supplementation improves oxidative damage in experimental colitis. Dig Liver Dis. 2003;35:635–41. doi: 10.1016/s1590-8658(03)00379-7. [DOI] [PubMed] [Google Scholar]
- 110.M’Koma AE. Serum biochemical evaluation of patients with functional pouches ten to 20 years after restorative proctocolectomy. Int J Colorectal Dis. 2006;21:711–20. doi: 10.1007/s00384-005-0076-3. [DOI] [PubMed] [Google Scholar]
- 111.M’Koma AE, Lindquist K, Liljeqvist L. Biochemical laboratory data in patients before and after restorative proctocolectomy. A study on 83 patients with a follow-up of 36 months. Ann Chir. 1994;48:525–34. [PubMed] [Google Scholar]
- 112.M’Koma AE. Follow-up results of hematology data before and after restorative proctocolectomy. Clinical outcome. Dis Colon Rectum. 1994;37:932–7. doi: 10.1007/BF02052601. [DOI] [PubMed] [Google Scholar]
- 113.Nicholls RJ, Pezim ME. Restorative proctocolectomy with ileal reservoir for ulcerative colitis and familial adenomatous polyposis: a comparison of three reservoir designs. Br J Surgery. 1985;72:470–4. doi: 10.1002/bjs.1800720622. [DOI] [PubMed] [Google Scholar]
- 114.Christie PM, Hill GL. Return to normal body composition after ileoanal J-pouch anastomosis for ulcerative colitis. Dis Colon Rectum. 1990;33:584–6. doi: 10.1007/BF02052211. [DOI] [PubMed] [Google Scholar]
- 115.Geerling BJ, Badart-Smook A, Stockbrugger RW, Brummer RJ. Comprehensive nutritional status in recently diagnosed patients with inflammatory bowel disease compared with population controls. Eur J Clin Nutr. 2000;54:514–21. doi: 10.1038/sj.ejcn.1601049. [DOI] [PubMed] [Google Scholar]
- 116.Esworthy RS, Aranda R, Martin MG, et al. Mice with combined disruption of Gpx1 and Gpx2 genes have colitis. Am J Physiol Gastrointest Liver Physiol. 2001;281:G848–55. doi: 10.1152/ajpgi.2001.281.3.G848. [DOI] [PubMed] [Google Scholar]
- 117.Forman HJ, Fridovich I. Superoxide dismutase: a comparison of rate constants. Arch Biochem Biophys. 1973;158:396–400. doi: 10.1016/0003-9861(73)90636-x. [DOI] [PubMed] [Google Scholar]
- 118.Dincer Y, Erzin Y, Himmetoglu S, et al. Oxidative DNA damage and antioxidant activity in patients with inflammatory bowel disease. Dig Dis Sci. 2007;52:1636–41. doi: 10.1007/s10620-006-9386-8. [DOI] [PubMed] [Google Scholar]
- 119.Abraham C, Cho JH. Inflammatory bowel disease. N Engl J Med. 2009;361:2066–78. doi: 10.1056/NEJMra0804647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Pezim ME, Pemberton JH, Beart RW, Jr, et al. Outcome of “indeterminant” colitis following ileal pouch-anal anastomosis. Dis Colon Rectum. 1989;32:653–8. doi: 10.1007/BF02555768. [DOI] [PubMed] [Google Scholar]
- 121.Bousvaros A, Antonioli DA, Colletti RB, et al. Differentiating ulcerative colitis from Crohn disease in children and young adults. J Pediatr Gastroenterol Nutr. 2007;44:653–74. doi: 10.1097/MPG.0b013e31805563f3. [DOI] [PubMed] [Google Scholar]
- 122.Knights D, Lassen KG, Xavier RJ. Advances in inflammatory bowel disease pathogenesis: linking host genetics and the microbiome. Gut. 2013;62:1505–10. doi: 10.1136/gutjnl-2012-303954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Jones DT, Osterman MT, Bewtra M, Lewis JD. Passive smoking and inflammatory bowel disease: a meta-analysis. Am J Gastroenterol. 2008;103:2382–93. doi: 10.1111/j.1572-0241.2008.01999.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Mikhailov TA, Furner SE. Breastfeeding and genetic factors in the etiology of inflammatory bowel disease in children. World J Gastroenterol. 2009;15:270–9. doi: 10.3748/wjg.15.270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Brandtzaeg P, Haraldsen G, Rugtveit J. Immunopathology of human inflammatory bowel disease. Springer Semin Immunopathol. 1997;18:555–89. doi: 10.1007/BF00824058. [DOI] [PubMed] [Google Scholar]
- 126.Loftus EV, Jr, Silverstein MD, Sandborn WJ, et al. Ulcerative colitis in Olmsted County, Minnesota, 1940–1993: incidence, prevalence, and survival. Gut. 2000;46:336–43. doi: 10.1136/gut.46.3.336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Munkholm P, Langholz E, Nielsen OH, et al. Incidence and prevalence of Crohn’s disease in the county of Copenhagen, 1962–87: a sixfold increase in incidence. Scand J Gastroenterol. 1992;27:609–14. doi: 10.3109/00365529209000127. [DOI] [PubMed] [Google Scholar]
- 128.Gasparetto M, Guariso G. Highlights in IBD Epidemiology and Its Natural History in the Paediatric Age. Gastroenterol Res Pract. 2013;2013:829040. doi: 10.1155/2013/829040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Shikhare G, Kugathasan S. Inflammatory bowel disease in children: current trends. J Gastroenterol. 2010;45:673–82. doi: 10.1007/s00535-010-0241-5. [DOI] [PubMed] [Google Scholar]
- 130.Adamiak T, Walkiewicz-Jedrzejczak D, Fish D, et al. Incidence, clinical characteristics, and natural history of pediatric IBD in Wisconsin: a population-based epidemiological study. Inflamm Bowel Dis. 2013;19:1218–23. doi: 10.1097/MIB.0b013e318280b13e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Chouraki V, Savoye G, Dauchet L, et al. The changing pattern of Crohn’s disease incidence in northern France: a continuing increase in the 10- to 19-year-old age bracket (1988–2007) Aliment Pharmacol Ther. 2011;33:1133–42. doi: 10.1111/j.1365-2036.2011.04628.x. [DOI] [PubMed] [Google Scholar]
- 132.Nosti PA, Stahl TJ, Sokol AI. Surgical repair of rectovaginal fistulas in patients with Crohn’s disease. Eur J Obstet Gynecol Reprod Biol. 2013;171:166–70. doi: 10.1016/j.ejogrb.2013.08.011. [DOI] [PubMed] [Google Scholar]
- 133.Yheulon CG, Derosa DC, Gagliano RA. Retained pill camera at an entero-uracho-vesical fistula site in a patient with Crohn’s disease. Hawaii J Med Public Health. 2013;72:186–9. [PMC free article] [PubMed] [Google Scholar]
- 134.Nielsen OH, Rogler G, Hahnloser D, Thomsen OO. Diagnosis and management of fistulizing Crohn’s disease. Nat Clin Pract Gastroenterol Hepatol. 2009;6:92–106. doi: 10.1038/ncpgasthep1340. [DOI] [PubMed] [Google Scholar]
- 135.Klineberg EO, James SP, Dunkin BJ. Crohn’s disease complicated by a urachoenteric fistula. Dig Dis Sci. 2002;47:1728–31. doi: 10.1023/a:1016428108363. [DOI] [PubMed] [Google Scholar]
- 136.Bergman R, Sloots CE. Diagnostic image (246). A man with faecal production of the umbilicus. Ned Tijdschr Geneeskd. 2005;149:1940. [PubMed] [Google Scholar]
- 137.Kavanagh DO, Carter MC, Keegan D, et al. Management of colorectal cancer in patients with inflammatory bowel disease. Tech Coloproctol. 2014;18:23–8. doi: 10.1007/s10151-013-0981-3. [DOI] [PubMed] [Google Scholar]
- 138.Devroede GJ, Taylor WF, Sauer WG, et al. Cancer risk and life expectancy of children with ulcerative colitis. N Engl J Med. 1971;285:17–21. doi: 10.1056/NEJM197107012850103. [DOI] [PubMed] [Google Scholar]