

Abstract
tert-Butoxyacetylene is shown to undergo Sonogashira coupling with aryl iodides to yield aryl-substituted tert-butyl ynol ethers. These intermediates participate in a [1,5]-hydride shift, which results in the extrusion of isobutylene and the generation of aryl ketenes. The ketenes are trapped in situ with multiple nucleophiles or undergoelectrocyclic ring closure to yield hydroxynaphthalenes and quinolines.
Keywords: Ketene, Sonogashira coupling, naphthol, ynol
Benzyl ketones and aryl acetic acid derivatives are found in many biologically active natural products anddrug candidates (Scheme 1). For example, benzyl ketones or their derivatives appear within actinoplanone A, the epilepsy drug oxcarbazepine, and the platelet inhibitor prasugrel. Likewise, aryl acetic acid derivatives appear in many natural products and pharmaceutical agents, such as (−)-curvularin and penicillin G. For these reasons, we were motivated to develop a mild and generalcatalytic method to access these substructures. Herein we report a novel cross coupling between aryl iodides and ynol ethers that yields aryl ketenes (1) and therefore provides a general catalytic synthesis of benzyl carbonyl compounds.
Scheme 1.

Coupling of ynol ethers with aryl halides to yield aryl ketenes.
Previous catalytic approaches to aryl acetic acid derivatives have relied on arylation of enolates. Ni and Pd catalyze couplings of esters and amides with aryl halides. These protocols have proven relatively general, and, in some cases, enantioselective.1 Nonetheless, enolate couplings require strongly basic conditions, elevated temperatures and/or independent preparation of the zinc enolates. They frequently involve specialized ligands, and achieving selective mono-arylation can be challenging. Alternative catalytic approaches to the α-arylation of carbonyl compounds include α -arylation with iodonium salts2 or activated sulfoxides,3 addition of aldehydes to in situ-generated quinone derivatives,4 and a recent oxidative coupling of ketones with nitroarenes.5
We hypothesized that aryl ketenes (1) could give rise to esters, amides, carboxylic acids, ketones and thioesters from the same intermediate. In contrast, the alternative approaches outlined above require separate reaction conditions for each of these products, if they are accessible at all. To date, no direct methods have been reported to couple metalated ketene (2) with an aromatic ring, and such a reaction appeared implausible. An alternative presented itself, however, in the [1,5]-hydride shift of ynol ethers. This process is accompanied by the sigmatropic extrusion of an olefin, and occurs under thermal conditions.6 Finally, the requisite aryl-substituted ynol ethers (3) could arise from the currently unknown coupling of an alkoxyacetylene with aryl halides. In this way, an alkoxyacetylene could serve as a ketene surrogate, so we refer to the process as the ketene surrogate coupling.7,8
Thermal generation of ketenes from ynol ethers has been exploited previously in the synthesis of complex molecules, which indicates that this transformation is reliable and occurs under mild conditions.9 In contrast, the cross coupling of alkoxyacetylenes remains poorly developed. In this context, Stille coupling of alkoxyethynyltin reagents has been described,10 but Sonogashira coupling could directly connect the aryl iodides and ynol ethers without necessitating pre-functionalization of the acetylene motif. While the Sonogashira coupling between menthol-derived ynol ethers and terminal vinyl iodides has been investigated,11 the only Sonogoshara coupling between an ynol ether and an aryl halide of which we are aware proceeded in only 11% yield.12,13
To develop the ketene surrogate coupling, we first investigated the Pd-catalyzed coupling of alkyl ynol ethers with aryl iodides. 4-Cyano-iodobenzene 4a was combined with ethoxy acetylene, Pd(PPh3)4 and CuI in triethyl amine (entry 1). No desired product was isolated, and the alkyne appeared to have polymerized under the reaction conditions. Reasoning that a more sterically hindered alkyne might be less prone to polymerization, we repeated the experiment using tert-butoxy acetylene.14 In addition to being more stable, tert-butoxyacetylenes rearrange to ketenes at around 80 °C compared to the 120 °C that is required for conversion of ethoxyacetylenes to ketenes.15 We were encouraged when alkyne 5a was formed in 54% yield when tert-butoxyacetylene was used (entry 2). The reaction also generated products arising from hydration and hydrolysis of 5a (6a and 7a, respectively) and enyne 8a, a 2:1 adduct of tert-butoxy acetylene and the aryl iodide.
To minimize the formation of ester and carboxylic acid side products, we included molecular sieves in the reaction. Additionally, the use of a bulkier base, iPr2NEt, suppressed the generation of enynol ether 8a (entry 3). Under these conditions, the aryl-substituted ynol ether 5a was formed in an improved 85% yield. Enynol 8a could arise from either dimerization of tert-butoxyacetylene followed by coupling with the aryl iodide or carbometalation of the Sonogashira product 5a. We favor the former hypothesis because tert-buytoxyacetylene did not add to isolated ynol ether 5a under the reaction conditions We speculate that a bulky amine prevents the (R3N)nCu(acetylide) complex from reacting with a second equivalent of tert-butoxyacetylene. Ultimately, the reproducibility of the reaction could be improved by forming Pd(PPh3)n in situ from Pd2(dba)3 and PPh3 (entry 4).16
Unfortunately, when these conditions were applied to an electron neutral substrate (4b), incomplete conversion was observed (entry 5). Changing to a secondary amine (iPr2NH) accelerated the coupling (entry 5) without introducing impurities. Under these conditions, aryl iodide 4b was completely consumed, and the aryl-substituted ynol ether 5b was formed in good yield (entry 6). Reactions with iPr2NH are generally faster than those containing iPr2NEt, but electron-poor arenes form small quantities of the corresponding tertiary amide (e.g. 9a) during the reaction. Accordingly, we generally recommend iPr2NEt for electron poor substrates and iPr2NH for electron rich and neutral substrates. Finally, sterically hindered substrates such as 4c benefited from an even more active catalyst. In particular, tri(2-furyl)phosphine (TFP) formed a competent catalyst in conjunction with Pd2(dba)3, promoting the coupling of a hindered aryl iodide in good yield (entry 8). To summarize, three closely related reaction conditions accommodate a wide variety of substrate classes: the couplings are usually more efficient with iPr2NH than with iPr2NEt, although with electron deficient substrates, we observed minor amounts of the amide 9 when iPr2NH was used. While these substrates perform admirably with inexpensive PPh3, challenging aryl iodides often necessitate the electron deficient phosphine TFP.
The coupling tolerates a wide range of electronic properties and functional groups (Table 2). In these experiments, Cu, Pd, and amines were removed by rapid chromatography over neutral Al2O3. In general, electron neutral or electron rich Sonogashira products could be isolated in high purity whereas electron poor congeners were prone to hydrolysis. For example, the ynol derived from 4-methyoxy-iodobenzene was stored for >1 year at 4 °C with no signs of decomposition. In contrast, the p-CN-substituted ynol 5a underwent hydrolysis to the extent of 5–10% upon attempted purification. Therefore, following filtration over Al2O3, the crude aryl-substituted ynol ethers 5 were heated in the presence of morpholine to generate, consecutively, the aryl ketene (1) and then the morpholine amides (10, Table 2).17 These amides were targeted because of their utility in the synthesis of ketones (see below). They behave similarly to Weinreb amides, but are more stable and less expensive.18 We found that ketones, esters, and nitro, cyano and trifluoromethyl groups were all compatible with the reaction conditions. Likewise, substrates featuring electron donating groups including 2-, 3- and 4-methyl (10q, 10r, 10h) and 2-, 3- and 4-methoxy (10s, 10t, 10k) provided the corresponding morpholine amides in good yield. Both 1- and 2-iodonaphthalene were excellent substrates (10v, 10m). Moreover, several heterocycles participated in the reaction yielding 3- and 4-substituted pyridines (10n, 10o) and 5- and 7-substitued indoles (10p, 10w). In the latter cases, the indole NH did not require protection. The thiazole 10x was formed in good yield, extending the chemistry to 5-membered heterocycles. Aryl bromides do not couple efficiently, but this characteristic allows for the selective coupling of 4-bromo-iodobenzene to form the 4-bromophenyl acetamide 10j in 74% yield. Additionally 4-fluoro-iodobenzene provided 10c in poor yield due to volatility of the ynol ether and instability of the amide.
Table 2.
Ketene Surrogate Coupling Scope
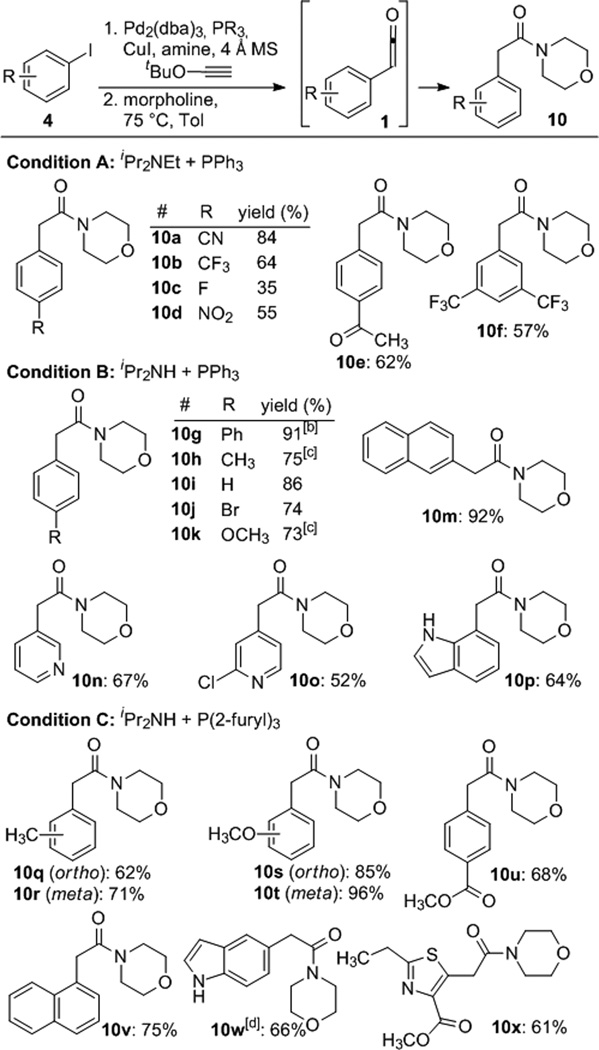 |
Isolated yields over 2 steps. Reactions were conducted on a 0.3 mmol scale unless otherwise noted, 0.25 M in 1:1 (v:v) amine:ynol ether with 5 mol% Pd2(dba)3, 20 mol% phosphine, and 13 mol% of CuI for 12–24 h at rt.
3 mmol scale.
1 mmol scale.
48 h reaction time.
The examples in Table 2 involve trapping the arylketene with morpholine, but we wanted to explore the reactivity of this intermediate more broadly. To this end, ynol ether 5g was synthesized on a 2.9 g scale and isolated in 88% yield (Scheme 2). Ketene 1g was then generated at 75 °C in the presence of a variety of trapping reagents. Oxygen-based nucleophiles reacted cleanly and efficiently. Specifically, water, methanol, and primary, secondary and tertiary alcohols yielded the carboxylic acid or esters 11a–11e in high yield. Likewise, pentafluorophenol and phenol formed the phenolic esters 11f and 11g. The former could serve as a useful acylating agent while the latter can participate in a Fries rearrangement (see below). In the special case of allyl alcohol, the intermediate allyl ester was exposed to soft enolization conditions to promote a Claisen rearrangement in a two-step, one pot procedure to give C-allyl ester 12.19 In related transformations, amines other than morpholine react with equal facility. Thus, Weinreb amides (13) emerge from trapping with (MeO)MeNH, most conveniently free-based in situ from the hydrochloride salt, and aniline reacted to form anilide 14 in nearly quantitative yield. Of note, HI and isobutylene are the only chemical waste products generated in these acylations.
Scheme 2.

Aryl acetic acid derivatives and benzyl ketones via ketene trapping
We next explored strategies to form ketones through C-C bond formation. To this end, ynol 5g was heated in the presence of the ylide derived from ethyl 2-bromopropionate (15), and the trisubstituted allene 16 was isolated in good yield.20 Similarly, exposure to alkyl-vinyl ethers initiated a [2+2]-cycloaddition to provide the cyclobutanone products 17 as single trans diastereomers.21 These last examples provide compelling evidence for the intermediacy of ketene 1g. While, in principle, most of the other products in Scheme 2 could have arisen from Nu-H addition across the ynol ether triple bond followed by hydrolysis of the tert-butyl enol ether, the formation of allenes and [2+2] adducts is more consistent with a ketene intermediate under these reaction conditions.
The facile synthesis of a variety of aryl acetic acid derivatives provided an entry into several other types of ketones. For example, the phenol ester 11g underwent Fries rearrangement to yield the aryl, benzyl ketone 18 as a single regioisomer.22 The thiol ester 19 was formed in good yield, and then converted into the ketone 20 under reaction conditions introduced by Liebeskind and coworkers.23 Finally, we exploited the ability of morpholine amides to react with hard nucleophiles to provide ketones.17 In the event, Grignard reagents proved too basic for these transformations, and enolate formation dominated the reaction. However, we found that LaCl3·2LiCl promoted these additions effectively. This additive, introduced by Knochel and colleagues,24 assists nucleophilic addition to acidic aldehydes,25 but this is the first report of its use to facilitate addition to morpholine amides.26 Thus, a primary and a secondary Grignard reagent performed well, as did a vinyl and phenyl reagent. No tertiary alcohols were observed from double addition. Taken together, these examples demonstrate that the ketene surrogate coupling provides efficient access to aryl, vinyl and alkyl ketones.
Finally, taking inspiration from the Danheiser benzannulation,27 we developed a new benzannulation protocol as outlined in Scheme 3.28 2-Iodostyrenes (22, X = CH) were found to couple with tert-butoxy electrocyclic ringacetylene, rearrange to the aryl ketene, and undergo 6 electrocyclic ring closure to provide the naphthols 23a–23c in good yield. The substituted styrenes leading to 23b and 22c were used as mixture of E and Z isomers, and both isomers appear to participate in the sigmatropic rearrangement. The annulation was also successful with 2-vinyl-3-iodopyridine to generate quinoline 23d, and even showed modest success with the imine derived from 2-iodoaniline (23e).
Scheme 3.

Benzannulation with tert-butoxyacetylene.
In summary, we have found that tert-butoxy acetylene serves the role of metalated ketene in cross-coupling reactions. It can undergo Sonogashira coupling with aryl iodides, and then transform into a ketene under mild thermal conditions. This ketene surrogate coupling leads to aryl acetic acid derivatives, ketones, allenes and cyclobutanone products in good yield. An advantageous characteristic of the ketone surrogate coupling is the ability to access a wide range of carbonyl compounds from a single intermediate. Moreover, an efficient benzannulation process has been developed to provide hydroxy naphthylenes and hydroxy quinolines.
Experimental Section
Representative procedure
Aryl iodide (0.3 mmol), Pd2(dba)3 (15.6 mg, 0.015 mmol), PPh3 (15.9 mg, 0.06 mmol), CuI (7.5 mg, 0.039 mmol), and 150 mg 4 Å molecular sieves were combined in a vial and purged with argon. Diisopropylethyl amine (0.6 mL) and tert-butoxyacetylene (0.6 mL) were added at room temperature. The reaction was stirred at room temperature until it reached completion (12–24 h), and was then directly loaded onto an Al2O3 plug and eluted with ethyl acetate and hexane (1:10). The crude ynol either was dissolved in toluene (2.0 mL), and morpholine (0.2 mL) was added. The reaction mixture was heated to 75 00B0;C for 3 h, cooled, and concentrated under reduced pressure. Pure morpholine amides were isolated following flash chromatography on silica gel.
Supplementary Material
Table 1.
Optimization of Coupling Conditions.a
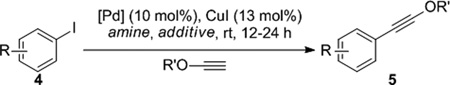 | ||||||
|---|---|---|---|---|---|---|
| entry | R | R' | catalyst | amine | additive | yield (%)[b] |
| 1 | 4-CN (4a) | Et | Pd(PPh3)4 | Et3N | - | <5 |
| 2 | 4-CN | tBu | Pd(PPh3)4 | Et3N | - | 54 |
| 3 | 4-CN | tBu | Pd(PPh3)4 | iPr2NEt | 4Å MS | 85 |
| 4 | 4-CN | tBu | Pd2(dba)3/PPh3[c] | iPr2NEt | 4Å MS | 95 |
| 5 | 4-Me (4b) | tBu | Pd2(dba)3/PPh3[c] | iPr2NEt | 4Å MS | 66 |
| 6 | 4-Me | tBu | Pd2(dba)3/PPh3[c] | iPr2NH | 4Å MS | 89 |
| 7 | 2-Me (4c) | tBu | Pd2(dba)3/PPh3[c] | iPr2NH | 4Å MS | 55 |
| 8 | 2-Me | tBu | Pd2(dba)3/TFP[c],[d] | iPr2NH | 4Å MS | 79 |
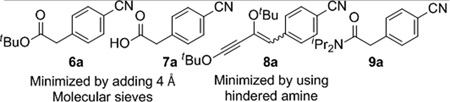 | ||||||
| Minimized by adding 4 Å Molecular sieves | Minimized by using hindered amine | |||||
Reactions were conducted on a 0.1 mmol scale, 0.25 M in 1:1 (v:v) amine:ynol ether.
NMR yields based on an internal standard.
20 mol% phosphine was used.
TFP = tri(2-furyl)phosphine.
Footnotes
Funding provided by the NIH (R01GM102403) and the Welch Foundation (I-1612). We thank Karen S. MacMillan (UT Southwestern) for preliminary experiments related to the synthesis of anilides and Jibao Xia (UT Southwestern) for independently repeating the synthesis of 10g, and Gary Sulikowski for suggesting the Fries rearrangement to synthesize ketone 18.
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/anie.201xxxxxx.
References
- 1.a) Palucki M, Buchwald SL. J. Am. Chem. Soc. 1997;119:11108–11109. [Google Scholar]; b) Culkin DA, Hartwig JF. Acc. Chem. Res. 2003;36:234–245. doi: 10.1021/ar0201106. [DOI] [PubMed] [Google Scholar]; c) Hama T, Culkin DA, Hartwig JF. J. Am. Chem. Soc. 2006;128:4976–4985. doi: 10.1021/ja056076i. [DOI] [PubMed] [Google Scholar]
- 2.a) Allen AE, MacMillan DWC. J. Am. Chem. Soc. 2011;133:4260–4263. doi: 10.1021/ja2008906. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Bigot Al, Williamson AE, Gaunt MJ. J. Am. Chem. Soc. 2011;133:13778–13781. doi: 10.1021/ja206047h. [DOI] [PubMed] [Google Scholar]; c) Harvey JS, Simonovich SP, Jamison CR, MacMillan DWC. J. Am. Chem. Soc. 2011;133:13782–13785. doi: 10.1021/ja206050b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.a) Huang X, Patil M, Farès C, Thiel W, Maulide N. J. Am. Chem. Soc. 2013;135:7312–7323. doi: 10.1021/ja4017683. [DOI] [PubMed] [Google Scholar]; b) Peng B, Geerdink D, Farès C, Maulide N. Angew. Chem. Int. Ed. 2014;53:5462–5466. doi: 10.1002/anie.201402229. [DOI] [PubMed] [Google Scholar]; Peng B, Geerdink D, Farès C, Maulide N. Angew. Chem. 2014;126:5566–5570. doi: 10.1002/anie.201402229. [DOI] [PubMed] [Google Scholar]
- 4.Jensen KL, Franke PT, Nielsen LT, Daasbjerg K, Jørgensen KA. Angew. Chem. Int. Ed. 2010;49:129–133. doi: 10.1002/anie.200904754. [DOI] [PubMed] [Google Scholar]; Jensen KL, Franke PT, Nielsen LT, Daasbjerg K, Jørgensen KA. Angew. Chem. 2010;122:133–137. doi: 10.1002/anie.200904754. [DOI] [PubMed] [Google Scholar]
- 5.Xu Q-L, Gao H, Yousufuddin M, Ess DH, Kurti L. J. Am. Chem. Soc. 2013;135:14048–14051. doi: 10.1021/ja4074563. [DOI] [PubMed] [Google Scholar]
- 6.Brandsma L, Bos HJT, Arena JF. In: Chemistry of Acetylenes. Viebe HG, editor. New York: Marcel Dekker; 1969. pp. 808–810. [Google Scholar]
- 7.α-Aryl acetamides from ynamides: Bhunia S, Chang C-J, Liu R-S. Org. Lett. 2012;14:5522–5525. doi: 10.1021/ol302621z.
- 8.Oxidation of aryl acetylenes to aryl acetic acid derivatives: Kim I, Lee C. Angew. Chem. Int. Ed. 2013;52:10023–10026. doi: 10.1002/anie.201303669. Kim I, Lee C. Angew. Chem. 2013;125:10207–10210.
- 9.a) Magriotis PA, Vourloumis D, Scott ME, Tarli A. Tetrahedron Lett. 1993;34:2071–2074. [Google Scholar]; b) Moslin RM, Jamison TF. J. Am. Chem. Soc. 2006;128:15106–15107. doi: 10.1021/ja0670660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.a) Sakamoto T, Yasuhara A, Kondo Y, Yamanaka H. Chem. Pharm. Bull. 1994;42:2032–2035. [Google Scholar]; b) Nelson HM, Gordon JR, Virgil SC, Stoltz BM. Angew. Chem. Int. Ed. 2013;52:6699–6703. doi: 10.1002/anie.201301212. [DOI] [PMC free article] [PubMed] [Google Scholar]; Nelson HM, Gordon JR, Virgil SC, Stoltz BM. Angew. Chem. 2013;125:6831–6835. doi: 10.1002/anie.201301212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dussault PH, Sloss DG, Symonsbergen DJ. Synlett. 1998:1387–1389. [Google Scholar]
- 12.a) Davies PW, Cremonesi A, Dumitrescu L. Angew. Chem. Int. Ed. 2011;50:8931–8935. doi: 10.1002/anie.201103563. [DOI] [PubMed] [Google Scholar]; Davies PW, Cremonesi A, Dumitrescu L. Angew. Chem. 2011;123:9093–9097. doi: 10.1002/anie.201103563. [DOI] [PubMed] [Google Scholar]; b) Tatlock JH. J. Org. Chem. 1995;60:6221–6223. [Google Scholar]
- 13.An efficient Sonogashira coupling with ynamides has been reported: Tracey MR, Zhang Y, Frederick MO, Mulder JA, Hsung RP. Org. Lett. 2004;6:2209–2212. doi: 10.1021/ol0493251.
- 14.van Daalen JJ, Kraak A, Arens JF. Recl. Trav. Chim. Pays-Bas. 1961;80:810–818. [Google Scholar]
- 15.a) Valenti E, Pericas MA, Serratosa F. J. Org. Chem. 1990;55:395–397. [Google Scholar]; b) Moyano AA, Pericas MA, Serratosa F, Valenti E. J. Org. Chem. 1987;52:5532–5538. [Google Scholar]
- 16.We found wide variations in the yields when different batches of Pd(PPh3)4 from SigmaAldrich were used, while those from Strem were more consistent. High reactivity could be rescued by increasing the copper loading to 20 mol%. It is possible that higher levels of PPh3 may poison the copper co-catalyst, which can be overcome with additional copper. Reactions with Pd2(dba)3 + PPh3 were uniformly reproducible.
- 17.a) MaGee DI, Ramaseshan M, Leach JD. Can. J. Chem. 1995;73:2111–2118. [Google Scholar]; b) Mak XY, Ciccolini RP, Robinson JM, Tester JW, Danheiser RL. J. Org. Chem. 2009;74:9381–9387. doi: 10.1021/jo9021875. [DOI] [PubMed] [Google Scholar]
- 18.Martin R, Romea P, Tey C, Urpi F, Vilarrasa J. Synlett. 1997:1414–1416. [Google Scholar]
- 19.Kobayashi M, Masumoto K, Nakai E-i, Nakai T. Tetrahedron Lett. 1996;37:3005–3008. [Google Scholar]
- 20.Lang RW, Hansen HJ. Helv. Chim. Acta. 1980;63:438–455. [Google Scholar]
- 21.Hyatt JA, Raynolds PW. Org. React. 1994;45:159–646. [Google Scholar]
- 22.Jeon I, Mangion IK. Synlett. 2012;23:1927–1930. [Google Scholar]
- 23.Liebeskind LS, Srogl J. J. Am. Chem. Soc. 2000;122:11260–11261. [Google Scholar]
- 24.Krasovskiy A, Kopp F, Knochel P. Angew. Chem. Int. Ed. 2006;45:497–500. doi: 10.1002/anie.200502485. [DOI] [PubMed] [Google Scholar]; Krasovskiy A, Kopp F, Knochel P. Angew. Chem. 2006;118:511–515. doi: 10.1002/anie.200502485. [DOI] [PubMed] [Google Scholar]
- 25.Wang Y, Wang C, Butler JR, Ready JM. Angew. Chem. Int. Ed. 2013;52:10796–10799. doi: 10.1002/anie.201304812. [DOI] [PMC free article] [PubMed] [Google Scholar]; Wang Y, Wang C, Butler JR, Ready JM. Angew. Chem. 2013;125:10996–10999. [Google Scholar]
- 26.Badioli M, Ballini R, Bartolacci M, Bosica G, Torregiani E, Marcantoni E. J. Org. Chem. 2002;67:8938–8942. doi: 10.1021/jo0263061. [DOI] [PubMed] [Google Scholar]
- 27.a) Danheiser RL, Gee SK. J. Org. Chem. 1984;49:1672–1674. [Google Scholar]; b) Turnbull P, Moore HW. J. Org. Chem. 1995;60:644–649. [Google Scholar]
- 28.a) Myers AG, Horiguchi Y. Tetrahedron Lett. 1997;38:4363–4366. [Google Scholar]; b) Araki K, Katagiri T, Inoue M. J. Fluorine Chem. 2014;157:41–47. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.