

Abstract
During neuropathic pain, glial cells (mainly astrocytes and microglia) become activated and initiate a series of signaling cascades that modulate pain processing at both spinal and supraspinal levels. It has been generally accepted that glial cell activation contributes to neuropathic pain because glia release proinflammatory cytokines, chemokines, and factors such as calcitonin gene-related peptide, substance P, and glutamate, which are known to facilitate pain signaling. However, recent research has shown that activation of glia also leads to some beneficial outcomes. Glia release anti-inflammatory factors that protect against neurotoxicity and restore normal pain. Accordingly, use of glial inhibitors might compromise the protective functions of glia in addition to suppressing their detrimental effects. With a better understanding of how different conditions affect glial cell activation, we may be able to promote the protective function of glia and pave the way for future development of novel, safe, and effective treatments of neuropathic pain.
Keywords: Pain, Glia, Astrocytes, Microglia
1. Introduction
Injury to the somatosensory nervous system can produce chronic neuropathic pain characterized by abnormal sensations such as allodynia (pain produced by normally non-painful stimuli) and hyperalgesia (excessive pain from noxious stimuli) (Sorge et al., 2012). Chronic pain can persist for several months and can be very difficult to manage or treat. In fact, only 40% to 60% of patients achieve even partial relief (Dworkin et al., 2007; Finnerup et al., 2010). Pain processing is a dynamic system that can be modulated at multiple levels of the nervous system (Milligan and Watkins, 2009). For many decades, most research on neuropathic pain has revolved around neuronal mechanisms. However, recent studies have demonstrated that neighboring astrocytes and microglia also act as powerful modulators of pain (Milligan and Watkins, 2009; Ji et al., 2013).
Glia are non-neuronal cells that maintain homeostasis, form myelin, and provide support and protection to neurons in both central and peripheral nervous systems (Jessen and Mirsky, 1980; Clarke et al., 2013). They also play an important role in the synthesis, release, and uptake of various neurotransmitters (Watkins et al., 2007). Glia account for 70% of central nervous system (CNS) cells (Nakajima and Kohsaka, 2001; Watkins et al., 2007) and consist mainly of two types, the microglia and the macroglia. Macroglia include astrocytes, oligodendrocytes, and radial cells.
For over a century, it was believed that the glial cells did not play a role in neurotransmission. However, it is now known that they indeed play important roles in physiological processes and in assisting neurons to form synaptic connections (Gourine et al., 2010). The involvement of glia in neuropathic pain was first suggested in the mid-1990s (Colburn et al., 1997; Colburn et al., 1999). It is now well established that nerve damage leads to activation of glial cells, particularly astrocytes and microglia, and that this activation can cause neuroanatomical and neurochemical transformations in the CNS that result in neuropathic pain (Colburn et al., 1999; Woolf and Mannion, 1999).
Both astrocytes and microglia play important roles in mediating neuropathic pain by releasing proinflammatory cytokines and chemokines and other factors known to facilitate pain signaling, such as calcitonin gene-related peptide (CGRP), substance P, and glutamate (Wieseler-Frank et al., 2005; Milligan and Watkins, 2009; Ji et al., 2013). In preclinical studies, mechanical allodynia and hyperalgesia have been associated with early increased levels of proinflammatory cytokines such as tumor necrosis factor (TNF)-α, interleukin (IL)-1β, and IL-6, and delayed expression of IL-10 (Chiang et al., 2007; Vallejo et al., 2010). Cytokines are very potent small proteins produced by immune (macrophages or helper T cells) and non-immune cells (endothelial cells or Schwann cells). They function as cellular communicators. Under normal conditions, the production of both pro- and anti-inflammatory cytokines aids the immune system in destroying pathogens and healing damaged tissue. However, release of proinflammatory cytokines for prolonged periods may lead to pathological conditions such as chronic pain (Kawasaki et al., 2008).
Recent studies suggest that activation of glia can also have beneficial effects, including release and maintenance of anti-inflammatory factors that protect against neurotoxicity and restore normal pain signaling (Milligan and Watkins, 2009). Despite the extensive research in this field, preclinical findings have not translated into improved therapeutic strategies for patients with chronic pain. Thus, an understanding of the mechanisms involved in both beneficial and pathological roles of activated glia is necessary for the development of novel, safe, and effective pain therapies.
2. Neuron-glia interactions in neuropathic pain
Neuron-glia interactions play critical roles in the development and maintenance of neuropathic pain (Scholz and Woolf, 2007; Watkins et al., 2007, Ji et al., 2013). Some fundamental questions about neuron-glia interactions in pain have been addressed, such as the signals that lead to glial activation after injury and how glial cells affect neuronal activity and promote hyperalgesia (Ren and Dubner, 2008). For example, Coull and colleagues (Coull et al., 2005) proposed that ATP-stimulated microglia send signals to pain-projection neurons in the dorsal horn of the spinal cord. Neuron-derived ATP then activates purinergic ionotropic receptors (P2X4) on microglia, causing further release of microglial ATP and brain-derived neurotrophic factor in spinal lamina neurons (Fig. 1). Another factor that contributes to neuropathic pain is the activation of microglial p38 mAPK (mitogen-activated protein kinase), which enhances Ca2+-sensitive intracellular signaling and thereby leads to production of proinflammatory cytokines. TNFα and matrix metalloproteinases (MMPs) also trigger p38 mAPK activation of microglia in the dorsal horn of spinal cord during neuropathic pain (Svensson et al., 2005). During the onset and early stages of neuropathic pain, MMP-9–induced pro-IL-1β cleavage leads to p38 mAPK activation in microglia, whereas during the ongoing and later stages of neuropathic pain, MMP-2 induces pro-IL-1β cleavage that leads to astrocyte activation (Kawasaki et al., 2008) (Fig. 2). These findings suggest that glial activation can be initiated by neurotransmitters and neuromodulators through neuron-to-glia signaling (Fig. 3).
Fig. 1.

Flow chart depicting how peripheral nerve injury leads to chronic pain. ATP: adenosine triphosphate; CGRP: calcitonin gene-related peptide; NO: nitric oxide; mAPK: mitogen-activated protein kinase.
Fig. 2.
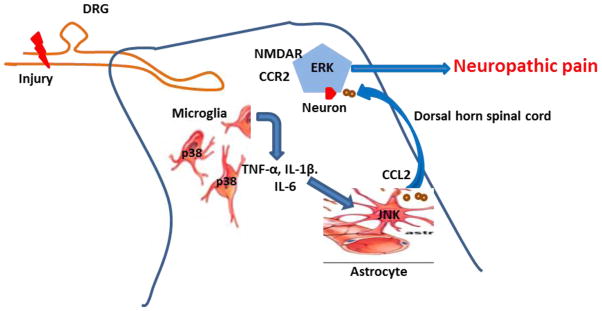
Astrocyte-neuron cross talk. After nerve injury, proinflammatory cytokines such as tumor necrosis factor-α (TNF-α), interleukin (IL)-1β, and IL-6 are released from microglia and activate JNK in astrocytes. JNK activation induces astrocytes to produce CCL2. Binding of CCL2 to its receptor, CCR2, in spinal cord neurons produces central sensitization via ERK-mediated activation of NMDA receptors, leading to an enhanced neuropathic pain state. CCL2: chemokine (C-C motif) ligand 2; DRG: dorsal root ganglion; ERK: extracellular signal-regulated kinases; JNK: c-Jun N-terminal kinases; NMDA: N-methyl-D-aspartate.
Fig. 3.
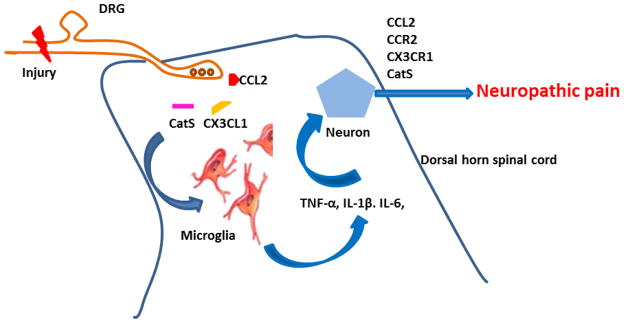
Microglia-neuron cross talk. Nerve injury leads to activation of chemokine receptors present on microglia, causing phosphorylation in microglia and leading to further production of proinflammatory cytokines and subsequent sensitization of dorsal horn neurons and neuropathic pain facilitation. Nerve injury also produces the protease cathepsin S (CatS) from microglia, which can cleave CX3CL1 from the cell surface of neurons (primary sensory and dorsal horn neurons) and astrocytes, leading to further activation of microglia. CX3CL1: chemokine (C-X3-C motif) ligand 1; IL-1β: interleukin-1β; IL-6: interleukin-6; TNF-α: tumor necrosis factor-α.
Chemokines play important roles in neuron-glia signaling during neuropathic pain. Expression of chemokines and their receptors is not limited to peripheral immune cells and glia; neurons can also express these factors (Tran and Miller, 2003). Studies have suggested that CCL2 (i.e., monocyte chemoattractant protein 1 [mCP1]) and its receptor, CCR2, are significantly upregulated in dorsal root ganglia and spinal cord after peripheral nerve injury (Zhang and De Koninck, 2006). CCL2 was reported to modulate thalamic nociceptive processing after spinal cord injury through remote activation of thalamic microglia (Zhao et al., 2007). Further, injection of CCL2 into the thalamus induced pain-related behavior in rats (Zhao et al., 2007). Spinal cord dorsal horn also expresses the lysosomal cysteine protease cathepsin S, an enzyme that may activate and induce fractalkine cleavage during neuropathic pain states (Clark et al., 2007) (Fig. 3). Thus, chemokines act as important neuron–glia signaling modulators during neuropathic pain.
Proinflammatory cytokines also have the ability to increase neural activity and mechanosensitivity of dorsal root ganglion neurons (Fig. 2). Cytokines can permeate the blood-brain barrier to reach the spinal cord, where they can activate microglia and astrocytes that facilitate pain transmission (Vallejo et al., 2010). Another mechanism by which cytokines contribute to chronic pain is by activating and increasing the number of AMPA and NMDA receptors on the surface of neurons (Stellwagen and Malenka, 2006). IL-1β induces the phosphorylation of a specific NMDA receptor subunit that allows for influx of Ca2+. In turn, the increased Ca2+ concentration leads to increased release of nitric oxide (NO) and prostaglandin E2, mediators involved in development and maintenance of neuropathic pain (Besson, 1999; Milligan and Watkins, 2009) (Fig. 1).
3. Another side of the coin: Protective role of glia
Historically, glial activation was thought to be undesirable, as it enhances the inflammatory response and leads to neurodegeneration. But recent evidence suggests that glial activation can also be beneficial, depending upon the type and stage of activation (Glezer et al., 2007; Milligan and Watkins, 2009; Kallendrusch et al., 2013). For example, immune responses in the CNS may benefit patients with Alzheimer’s disease (AD) by reducing the amount of amyloid deposition (Malm et al., 2005). The lipopolysaccharide (LPS) receptor CD14 seems to play an important role in the recognition of β-amyloid and the induction of phagocytosis by microglial cells (Liu et al., 2005). These data formed the basis for the first clinical trial to use active immunization in patients with mild to moderate AD. The treatment seemed quite effective in clearing the senile plaques, and postmortem analysis revealed strong microglial reactivity to β-amyloid and infiltration of T cells (Orgogozo et al., 2003). It is possible that vaccination caused a phenotype switch of brain microglia to dendritic-like (CD11c) cells. These findings are interesting as they suggest that activation of the systemic immune system can be a powerful approach to treat disease pathologies. Yet, nerve injury-induced signal that initiates glial activation is likely to differ from that induced during other disease pathologies such as AD, and the downstream signaling events may also be different.
4. Different microglial phenotypes and their roles
It turns out that microglia exist in three different phenotypic states. The classically activated state is responsible for the proinflammatory response and neurotoxic effects of microglia (El Khoury et al., 2007); alternatively activated microglia produce anti-inflammatory cytokines; and the acquired deactivation state is associated with the anti-inflammatory and functional repair (Gordon et al., 2003; Gordon and Taylor, 2005). These phenotypes can coexist in certain chronic inflammatory conditions and neurodegenerative disorders (Wynn et al., 2004). Thus, many questions arise. What dictates which microglia acquire a particular phenotype, and can these phenotypic states convert from one to another? If microglia can convert between states, what factors regulate their conversion from protective to destructive and back? Moreover, is it possible to regulate or at least stop the conversion to a destructive phenotype? If both pro- and anti-inflammatory cytokines exist at the same site simultaneously, the outcome will be determined by the balance among the various cytokines (Battista et al., 2006). It is also possible that the activated microglia are not pro- or anti-inflammatory but rather the balance of secretion between pro- and anti-inflammatory mediators influences the final outcome. However, it is difficult to predict under which conditions microglia will release pro- or anti-inflammatory cytokines, as the type of cytokine may be affected by multiple factors (e.g., type of stimulus/injury, stage of disease, and interaction with neighboring cells). Additional research is required to understand the functions of different activation states of microglia. However, it is clear that inhibiting all microglia without differentiating their state would not be an optimal strategy.
5. Factors that influence whether glial activation is neurotoxic or neuroprotective
Both in vitro and in vivo studies have shown that neuronal injury and degeneration are associated with glial activation. Microglia with an inflammatory phenotype release proinflammatory cytokines, neurotoxic factors, and reactive oxygen/nitrogen species that exacerbate neuronal injury (Watkins et al., 2007, Ji et al., 2013). Other studies have shown that microglia and astrocytes can mediate neuronal regeneration, repair, and neurogenesis through anti-inflammatory actions (Milligan and Watkins, 2009; Kallendrusch et al., 2013). However, these studies are difficult to compare directly, as they used different experimental setups that vary in terms of the stimulus used, timing of glial activation, and animal species and age (Luo and Chen, 2012). Thus, whether glial activation has positive or negative effects on neuronal function is controversial.
The nature of stimulation is an important factor that determines the pathological or protective role of glia. Microglia are very sensitive to even minor stimuli, and different stimuli may have different effects on their function; thus the result may be either benefit or harm to the neurons. In a neonatal mouse model in which striatal ethanol injection was used to induce brain injury, LPS-activated microglia were found to be neurotoxic. Systemic LPS administration in the ethanol-injury model also caused a marked increase in both the volume and number of lesions and degenerating neurons in the striatum (Sawada et al., 2010). In contrast, microglia activated by systemic administration of LPS were shown to be neuroprotective in an MPTP-induced brain injury model. Similarly, different types of pain may differentially activate microglia (Hald et al., 2009), and the load/intensity of stimuli may also determine whether microglia will release damaging or protective factors (Lai and Todd, 2008).
Another governing factor for determining glial function is the timing of glial activation. The communication between glia, neurons, and immune cells is very diversified and complex. Therefore, the timing of glial activation may lead to different outcomes related to the entire inflammatory episode. Inhibition of microglial activation during the induction of experimental allergic encephalomyelitis (EAE) markedly decreased EAE progression, whereas microglial activation before the onset of EAE promoted lower-level EAE and an earlier recovery from symptoms (Bhasin et al., 2007). Other evidence suggesting that the timing of glial activation is an influential factor comes from a multiple sclerosis model. Inhibition of microglial activation by knockout of tissue plasminogen activator led to delayed onset of the disease. However, microglial inhibition also increased the severity and delayed recovery from the neurological dysfunction, suggesting that microglial activation is harmful during the onset of the disease but beneficial in the recovery phase (Lu et al., 2002).
Although little is known about how astrocytes and microglia interact, some studies suggest that astrocytes play neuroprotective roles by modulating microglial activity and attenuating their cytotoxicity (von Bernhardi and Eugenin, 2004; Ramirez et al., 2005). Astrocytes also suppress expression of IL-12 and inducible NO synthase in activated microglia (Vincent et al., 1996). The communication between these two types of glial cells is bidirectional, as microglia both receive signals from and send signals to astrocytes. Proinflammatory cytokines released from microglia are known to inhibit gap junctions and downregulate connexin 43 expression in astrocytes (Meme et al., 2006). In many pathological conditions, including neuropathic pain, microglia are activated before astrocytes and then promote astrocytic activation through IL-1β. However, activated astrocytes not only facilitate activation of distant microglia via calcium signaling but also attenuate microglial activities.
Taken together, these findings suggest that whether glia are neuroprotective or neurotoxic depends upon several factors, including type and load of stimuli and timing of microglial activation. Additional studies are required to validate the effect of these and other potential factors on glia-related pain modulation.
6. Astrocytes as potential targets for pain therapy
Targeting specific types of glial activation to promote anti-inflammatory processes for therapeutic purposes is beginning to yield encouraging results. Anti-inflammatory factors released by astrocytes and microglia can increase the expression of self-associated proteins that facilitate the clearance of apoptotic cells and tissue debris and halt continued proinflammatory response. Type 1 and type 2 cannabinoid receptors (CBR1 and CBR2, respectively) are being explored as therapeutic targets for neuropathic pain. In particular, activation of CBR2 receptors, which are expressed on microglia and astrocytes, yields beneficial effects in animal models of neuropathic pain (Manzanares et al., 2006; Ashton and Glass, 2007). Activation of the cannabinoid system enhances anti-inflammatory processing by increasing expression of anti-inflammatory markers such as ED2 (Romero-Sandoval et al., 2008) (Fig. 4). Recently, Luongo et al. (2013) also suggested that palmitoylethanolamide (PEA), an endogenous cannabinoid-like compound, reduces formalin-induced pain behavior in mice by enhancing the anti-inflammatory (i.e., IL-10) potential of spinal glia cells. A randomized, double-blind, placebo-controlled clinical trial on nabilone, an oral cannabinoid, for the treatment of patients with diabetic peripheral neuropathic pain (DPN) suggested that the oral cannabinoid nabilone (1–4 mg/day) was effective in relieving DPN symptoms and improving disturbed sleep, quality of life, and overall patient status (Toth et al., 2012).
Fig. 4.
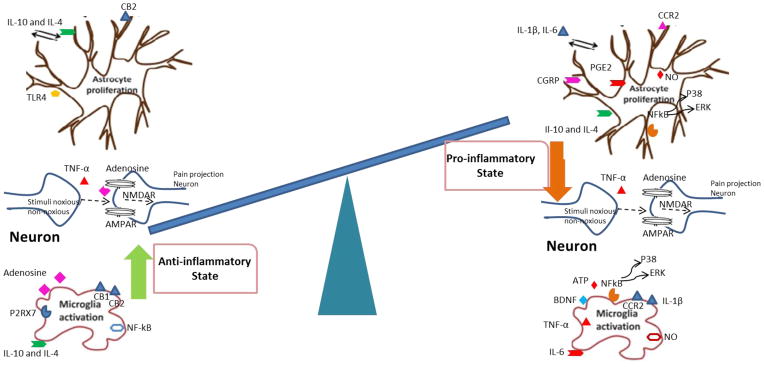
Pro- and anti-inflammatory states of glia. AMPAR: α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor; BDNF: brain-derived neurotropic factor; CB1: cannabinoid receptor-1; CB2: cannabinoid receptor-2; CGRP: calcitonin gene-related peptide; CCR2: chemokine receptor-2; ERK: extracellular signal-regulated kinases; IL-10: interleukin-10; IL-14: interleukin-14; NF-κB: nuclear factor kappa-light-chain-enhancer of activated B cells; NO: nitric oxide; PGE2: prostaglandin E2; P2RX7: P2X purinoceptor 7; NMDAR: N-methyl-D-aspartate receptor; TLR-4: toll-like receptor 4; TNF-α: tumor necrosis factor-alpha.
Activation of P2X4 receptor on microglia by ATP mediates neuropathic pain whereas activation of microglial P2RX7 may exert neuroprotective effects through the same signaling pathway (Inoue, 2006). Such findings suggest that the manners by which microglia are activated dictates the net outcome. IL-10 is a well-described anti-inflammatory cytokine that has been shown to prevent and reverse pathological pain, whereas IL-6 is a cytokine that has been shown to have both nociceptive and anti-nociceptive effects (Fig. 4). In rats with chronic constriction injury, one intrathecal injection of poly(lactic-co-glycolic) acid (PLGA) microparticles containing plasmid DNA encoding for IL-10 relieved neuropathic pain for more than 74 days (Soderquist et al., 2010). Recently, Lee et al. (2013) also reported the involvement of IL-10 signaling in the beneficial effect of gabapentin in attenuating neuropathic pain in rats. These results indicate that enhancing the anti-inflammatory actions of glial cells is a more powerful way to inhibit pain than is blocking the proinflammatory cytokines and preventing glial activation globally.
7. Novel therapies under development for the treatment of chronic pain
7.1. Cannabinoid receptor (CBR) type 2 agonist
Recently CBR2 has emerged as an attractive target for the treatment of chronic pain because it is associated with few side effects (Romero-Sandoval et al., 2008). CBR2 is expressed in immune cells (Klein et al. 2003) and keratinocytes (Ibrahim et al. 2005). It was originally believed to be present only in the periphery, but now it has been found in peripheral sensory neurons (Anand et al. 2008) and in the CNS (Van Sickle et al. 2005). CBR2 is expressed in microglia and perivascular cells in human and rat brain under normal conditions (Nunez et al. 2004) and in microglia and astrocytes during inflammation (Ramirez et al. 2005). Peripheral nerve injury induces CBR2 expression in peripheral nerve fibers and spinal cord (Beltramo et al. 2006; Anand et al. 2008). Activation of peripheral CBR2 induces anti-nociception by increasing keratinocytic release of beta-endorphin, which then acts at local neuronal mu-opioid receptors (Ibrahim et al. 2005). Prolonged administration of NESS400, a CBR2 agonist, was shown to prevent thermal hyperalgesia, alleviate mechanical allodynia, and facilitate the proliferation of anti-inflammatory microglial phenotype in the ipsilateral dorsal horn of the spinal cord in mice with spared nerve injury (Luongo et al., 2010). Microglial CBR2 activation is known to induce neuroprotection by modulating the microglial extracellular signal-regulated kinase (ERK) pathway and reducing NO production (Eljaschewitsch et al. 2006), halting TNF-α production (Ehrhart et al. 2005), decreasing microglial migration (Walter et al. 2003; Miller and Stella, 2008), and inducing production of anti-inflammatory factors from neurons and glia (Molina-Holgado et al. 2003). In vivo, spinal microglial CBR2 activation induces anti-nociception while reducing spinal Iba-1, CD11b (microglial marker), and glial fibrillary acidic protein (GFAP; astrocytic marker) expression in postoperative and neuropathic pain models (Romero-Sandoval et al. 2008). In addition, spinal microglial CBR2 activation increases anti-inflammatory factor ED2/CD163 in spinal perivascular microglia (Romero-Sandoval et al., 2008).
Racz et al. (2008a) found that when CBR2 knock-out mice and wild-type littermates underwent sciatic nerve injury, they developed similar hyperalgesia and allodynia in the ipsilateral hind paw. Surprisingly, however, CBR2 knock-out mice developed a contralateral mirror pain behavior that was associated with increases in microglial and astrocytic activity in the contralateral spinal cord. Sciatic nerve injury-induced hyperalgesia, allodynia, and microglial and astrocytic activity were attenuated in transgenic mice that overexpressed CBR2 receptors. These findings suggest that the CBR2 receptor has a crucial role in modulating glial activation in response to nerve injury (Racz et al., 2008a). In another study, Racz et al. (2008b) observed enhanced interferon-gamma (IFN-γ) response in the spinal cord of CBR2-deficient mice after nerve injury. However, nerve injury-induced neuropathic pain was completely abolished in mice that were deficient in both CBR2 and IFN-γ, indicating that CBR2 receptor-mediated control of neuropathic pain is IFN-γ dependent. Most interestingly, spinal CBR2 activation does not induce anti-nociceptive tolerance or cannabinoid-related neurologic side effects (Romero-Sandoval et al. 2008). Thus, microglial CBR2 activation is promising as a potential future treatment for chronic pain. However, currently available CBR2 agonists have some limitations that restrict their therapeutic potential, including short half-lives, poor selectivity over CBR1, and poor oral bioavailability. More selective CBR2 agonists with improved oral bioavailability and longer half-lives (Cheng et al. 2008; Ohta et al. 2008; Yao et al. 2009) are under development.
7.2. Phosphodiesterase (PDE) inhibitors
Ibudilast is a nonselective PDE inhibitor that shows potential for its anti-neuroinflammatory and neuroprotective effects (Rolan et al., 2009). Preclinical studies suggest that ibudilast can attenuate microglial activation and proinflammatory cytokines, inhibit toll-like receptor 4 (TLR4) signaling, and stimulate neurotrophic and anti-inflammatory factors (Suzumura et al., 1999). Very recently, Ellis et al. (2014) found that ibudilast, along with two other glial modulators [propentofylline and (+)-naltrexone], reversed below-level allodynia and spinal neuropathic avulsion pain in an animal model of central neuropathic pain. Ledeboer et al. (2006) found that in rats subjected to neuropathic pain from chronic constriction injury and spinal nerve ligation, ibudilast penetrates into the CNS, inhibits glial cell activation in brain and spinal cord, and attenuates pain hypersensitivity after intraperitoneal and oral administration (Ledeboer et al., 2006). Ibudilast was also found to act synergistically with morphine analgesia and to prevent the development of morphine tolerance and withdrawal symptoms (Ledeboer et al., 2006). In a clinical study of patients with DPN, patients who took ibudilast reported reduced pain and required less opioid than did those who took placebo (Rolan, 2007). In another clinical study, patients with multiple sclerosis who took ibudilast showed significant reduction in brain atrophy and reduction in persistent black holes seen on MRI (Barkhof et al., 2010; Fox, 2010). Overall, ibudilast seems to be a relatively long-acting, well-tolerated, and non-addictive drug, but additional research is warranted to elucidate its exact mechanism(s) of action. Based on encouraging preclinical data and its clinical safety, ibudilast has considerable potential for further clinical development as a therapy for neuropathic pain and opioid tolerance.
7.3. Anti-inflammatory cytokines
Subarachnoid (intrathecal) delivery of IL-10 leads to a robust and sustained reversal of neuropathic pain in rodent models (Ledeboer et al., 2007). Improving IL-10 gene delivery by using nonviral DNA encapsulated in the biologically safe copolymer PLGA leads to significantly improved therapeutic gene transfer in animal models and significantly extends pain relief (Milligan et al., 2012). Intrathecal administration of either IL-10 neutralizing antibody or the inflammatory HIV coat protein gp120 induces a rapid failure of the gene therapy (Sloane et al., 2008). These findings suggest that the IL-10 gene or other genes of regulatory cytokines that have the desired therapeutic efficacy may be harnessed to provide long-term pain relief.
7.4. Other strategies
Alternatively, glial proinflammatory responses can be regulated by targeting specific receptors that are already well defined, such as the adenosine receptor and TLR4. ATL313 is an adenosine 2A agonist that suppresses proinflammatory cytokines while enhancing the anti-inflammatory cytokine IL-10. One intrathecal injection has been shown to reverse neuropathic pain for 4 to 6 weeks in animal models of chronic constriction nerve injury, peripheral neuropathy, spinal cord injury, and central neuropathic pain (Loram et al., 2009, 2013). Orr et al. (2009) suggested that microglial motility can switch between chemoattraction and repulsion based on changes in cell surface receptor signaling. Very recently, Luongo et al. (2014) reported that selective stimulation of adenosine 1A receptor (A1AR) inhibits the morphological activation of microglia, possibly by suppressing the Ca2+ influx induced by ATP treatment. Pretreatment of cells with the A1AR agonist lowered their ability to facilitate the nociceptive neurons, as compared to cells treated with ATP alone (Luongo et al., 2014). TLR4 antagonists, such as naloxone, also offer a prominent target for the treatment of neuropathic pain and for increasing the efficacy of opioids (Hutchinson et al., 2008). Studies have shown that morphine and (+) naloxone administered together produce an increase in analgesia, and repeated dosing prevents the dependence and withdrawal effects that commonly occur with morphine. Studies have also shown that the TLR4 antagonist action of (+) naloxone also blocks the dose-limiting effects of morphine, such as constipation and respiratory depression (Sloan and Hamann 2006). In a case report, a patient with chronic refractory pain after laminectomy was treated with an intrathecal combination of morphine (2 mg) and ultralow-dose naloxone (20 ng). The patient reported dramatic pain relief within 20 minutes; the effect peaked at 1 hour and persisted with repeated infusions. Over a 3-year period of receiving this drug regimen daily, the patient reported 80% improvement in his pain (Hamann et al. 2008).
8. Potential caveats and challenges in translating glia-related pain research
Despite the many unanswered questions, glia-modulating agents have started moving into clinical trials for the treatment of neuropathic pain. The first such trial was conducted for propentofylline, a xanthine derivative that showed very good potential for the treatment of chronic pain in preclinical models. Propentofylline had previously progressed to a phase IIIb trial for AD, but was stopped in 2000 when patients on the drug failed to show improvement over placebo (Frampton et al., 2003). Although it is a PDE inhibitor, its precise mechanism of action is still unclear. Whether it acts on neurons, microglia, astrocytes, or another cell type is not known, but propentofylline is commonly accepted to attenuate glial activation (Sweitzer and De Leo, 2011). Based on the preclinical data for propentofylline, Solace Pharmaceuticals conducted a phase II clinical trial in 2009 to evaluate the efficacy of the drug in approximately 180 patients with postherpetic neuralgia. The drug failed to decrease self-reported pain in patients compared to placebo. Several reasons could account for the failure of the propentofylline trial, including drug dose, treatment duration, and even the disease pathology itself. Moreover, postherpetic neuralgia may not be a good candidate for treatment with this drug, as the role of glial responses in the pathophysiology of this disease is unknown. Consequently, the failure of propentofylline presents the scientific community with a major unanswered question: Is modulating glial activity a good strategy for pain therapy? According to DeLeo (Pain Research Forum, June 9, 2011), glia researchers need to think carefully about what they are doing rather than using propentofylline or any of the other “glial modulators” in an animal model, showing a decrease in expression of cell markers, and then concluding that obviously this drug is going to work in patients with neuropathic pain. There are too many disconnects between the laboratory and the patient in glial pain research.
Recent multicenter, randomized clinical trials on etanercept, a TNF-α inhibitor, for the treatment of pain in patients with subacute sciatica and postsurgical pain after inguinal hernia repair did not yield encouraging results (Cohen et al., 2012, 2013). Another recent double-blind, randomized, parallel-group, multicenter clinical trial with a CCR2 antagonist, AZD2423, failed to show significant effects in 133 patients with post-traumatic neuralgia compared to placebo (Kalliomäki et al., 2013). The failures may again result from multiple causes, including different pain conditions tested in rodents and humans (nerve injury-induced pain hypersensitivity vs. post-herpetic/traumatic neuralgia), different phase of disease (few weeks after nerve injury vs. many years of post-herpetic neuralgia), different methods of pain measurement in rodents and humans (evoked pain vs. spontaneous pain), and finally lack of translation from rodents to humans. Compared to the complete lack of effect of propentofylline, AZD2423 (150 mg) showed at least some trend toward reduction in paresthesia/dysesthesia and paroxysmal pain, indicating that the CCR2 antagonist may have some protective effects for some sensory components of pain (Kalliomäki et al., 2013). Notably, inhibition of glial responses in the CNS cannot be validated in this trial, because of the lack of effective imaging technique for detecting glial responses.
These recent clinical failures raise concerns about the current strategy of inhibiting all glial function without fully understanding its physiological or pathological roles. It is currently unknown how these glial mediators interact with glia-neuron machinery in vivo. Hence, more extensive research is needed in this area to uncover the beneficial effects of glia before additional clinical trials are conducted. With this approach, we might be able to improve clinical outcomes by promoting the protective function of glia rather than nonspecifically attenuating all glial activity.
9. Future scope of glia-related pain research
To date, modulation of glial activity has failed to prevent or treat neuropathic pain of patients in clinical trials. Because glial behavior is highly complex, it may be difficult to determine how to attenuate the proinflammatory activity of glia without altering their anti-inflammatory effects. Depending on the stimuli and associated factors, distinct patterns of microglial activation may occur. For example, glial activation by LPS leads to the release of proinflammatory cytokines that kill infected cells, whereas glia activated by growth factors or anti-inflammatory cytokines release protective factors that promote neuronal survival (Butovsky, 2001). Hence, suppressing glial functions nonspecifically could have adverse effects by silencing cells involved in pain resolution. Moreover, it is still unclear whether the effects of glial modulators are due to their actions on astrocytes, microglia, or neurons. Thus, specific inhibitors are needed that can selectively block some glial cells without affecting beneficial glia and neurons.
One challenge of translating promising preclinical findings to clinical trials is identifying a valid biological marker that can predict efficacy in human subjects. One possibility is the use of neuroimaging techniques, such as positron emission tomography (PET), to study pain processing and/or assess drug efficacy (Stephenson and Arneric, 2008). Numerous studies have suggested successful use of PET in both animal models and humans (Ji et al. 2008; Yasuno et al. 2008). It is difficult to predict the extent, time course, and presence/absence of spinal and/or supraspinal glial changes in patients with acute or chronic pain. By using neuroimaging and PET radiotracers, it may be possible to target drug therapy specifically to the patient and hence improve clinical efficacy (Imamoto et al., 2013). Thus, use of in vivo microglial imaging as a diagnostic tool to predict efficacy with novel glial modulating agents can open new avenues for pain research.
Although numerous studies have shown spinal and supraspinal glial changes in animal models of neuropathic pain, clinical evidence for such glial changes is almost nonexistent. In one published case study, levels of spinal GFAP- and CD68-positive microglia were elevated in autopsy tissue from a patient diagnosed with complex regional pain syndrome compared to those in healthy controls (Del Valle et al., 2009), suggesting that spinal glial reactivity observed in several animal models may also be present in patients with chronic pain syndrome. However, more needs to be known about human glia biology.
Furthermore, limited information is available on the role of human glia in pain control. Astrocytes from mice, monkeys, and humans are quite different in size, and the human brain contains subtypes of GFAP-positive astrocytes that are completely absent in rodents. Astrocytes in human cortex are more than twofold larger in diameter than those in rodents and extend 10-fold more GFAP-positive primary processes (Oberheim et al., 2009). Moreover, human glial progenitor cells implanted into neonatal immunodeficient mice propagate Ca2+ signals threefold faster than do their endogenous counterparts, and mice that receive these implants exhibit enhanced long-term potentiation and learning capability (Han et al., 2013). Hence, human astrocytes seem to be more sensitive and play a more sophisticated role than rodent astrocytes.
10. Conclusions
A major challenge of developing glia-dependent drug strategies for the treatment of neuropathic pain is blocking only glial cells that produce proinflammatory cytokines and chemokines while enhancing (or at least not inhibiting) their protective/anti-inflammatory potential. Understanding the molecular mechanisms that underlie both the positive and negative effects of glia on pain processing is very important for the development of novel, safe, and effective therapeutic modalities for the treatment of pain. Future research should also focus on imaging real-time glial activation in patients with acute and chronic pain, investigating the phenotypic changes of glia in postmortem tissues from patients with painful diseases, comparing the responses of human and rodent glia in cultures, and examining the effects of human glia transplantation in rodents. With a better understanding of the role of glia in pain, we can begin to develop novel therapeutic modalities that target the stage-dependent activation of glial cells during neuropathic pain.
Highlights.
During neuropathic pain glial cell activation modulate pain processing
Recent clinical failures raise concerns about inhibiting all glial function
Glia may lead to beneficial effect by releasing factors that restore normal pain
Enhancing protective potential of glia might be more beneficial approach
Acknowledgments
The authors thank Claire F. Levine, MS (scientific editor, Department of Anesthesiology/CCM, Johns Hopkins University), for editing the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Anand U, Otto WR, Sanchez-Herrera D, Facer P, Yiangou Y, Korchev Y, Birch R, Benham C, Bountra C, Chessell IP, Anand P. Cannabinoid receptor CB2 localisation and agonist-mediated inhibition of capsaicin responses in human sensory neurons. Pain. 2008;138(3):667–80. doi: 10.1016/j.pain.2008.06.007. [DOI] [PubMed] [Google Scholar]
- Ashton JC, Glass M. The cannabinoid CB2 receptor as a target for inflammation-dependent neurodegeneration. Curr Neuropharmacol. 2007;5(2):73–80. doi: 10.2174/157015907780866884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barkhof F, Hulst HE, Drulovic J, Uitdehaag BM, Matsuda K, Landin R MN166–001 Investigators. Ibudilast in relapsing-remitting multiple sclerosis: a neuroprotectant? Neurology. 2010 Mar 30;74(13):1033–40. doi: 10.1212/WNL.0b013e3181d7d651. [DOI] [PubMed] [Google Scholar]
- Battista D, Ferrari CC, Gage FH, Pitossi FJ. Neurogenic niche modulation by activated microglia: transforming growth factor beta increases neurogenesis in the adult dentate gyrus. Eur J Neurosci. 2006;23:83–93. doi: 10.1111/j.1460-9568.2005.04539.x. [DOI] [PubMed] [Google Scholar]
- Beltramo M, Bernardini N, Bertorelli R, Campanella M, Nicolussi E, Fredduzzi S, Reggiani A. CB2 receptor-mediated antihyperalgesia: possible direct involvement of neural mechanisms. Eur J Neurosci. 2006 Mar;23(6):1530–8. doi: 10.1111/j.1460-9568.2006.04684.x. [DOI] [PubMed] [Google Scholar]
- Besson JM. The neurobiology of pain. Lancet. 1999;353:1610–1615. doi: 10.1016/s0140-6736(99)01313-6. [DOI] [PubMed] [Google Scholar]
- Bhasin M, Wu M, Tsirka SE. Modulation of microglial/macrophage activation by macrophage inhibitory factor (TKP) or tuftsin (TKPR) attenuates the disease course of experimental autoimmune encephalomyelitis. BMC Immunol. 2007;8:10. doi: 10.1186/1471-2172-8-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckwalter MS, Yamane M, Coleman BS, Ormerod BK, Chin JT, Palmer T, Wyss-Coray T. Chronically increased transforming growth factor-beta1 strongly inhibits hippocampal neurogenesis in aged mice. Am J Pathol. 2006;169:154–164. doi: 10.2353/ajpath.2006.051272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butovsky O. Morphological aspects of spinal cord autoimmune neuroprotection: colocalization of T cells with B7–2 (CD86) and prevention of cyst formation. FASEB J. 2001;15:1065–1067. doi: 10.1096/fj.00-0550fje. [DOI] [PubMed] [Google Scholar]
- Cheng Y, Albrecht BK, Brown J, Buchanan JL, Buckner WH, DiMauro EF, Emkey R, et al. Discovery and optimization of a novel series of N-arylamide oxadiazoles as potent, highly selective and orally bioavailable cannabinoid receptor 2 (CB2) agonists. J Med Chem. 2008;51(16):5019–34. doi: 10.1021/jm800463f. [DOI] [PubMed] [Google Scholar]
- Chiang CY, Wang J, Xie YF, et al. Astroglial glutamate-glutamine shuttle is involved in central sensitization of nociceptive neurons in rat medullary dorsal horn. J Neurosci. 2007;27:9068–9076. doi: 10.1523/JNEUROSCI.2260-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark AK, Yip PK, Grist J, Gentry C, Staniland AA, Marchand F, Dehvari M, Wotherspoon G, Winter J, Ullah J, Bevan S, Malcangio M. Inhibition of spinal microglial cathepsin S for the reversal of neuropathic pain. Proc Natl Acad Sci U S A. 2007 Jun 19;104(25):10655–60. doi: 10.1073/pnas.0610811104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke CB, Suter MR, Gosselin RD. Glial cells and chronic pain: from the laboratory to clinical hope. Rev Med Suisse. 2013 Jun 26;9(392):1342–5. [PubMed] [Google Scholar]
- Cohen SP, Galvagno SM, Plunkett A, Harris D, Kurihara C, Turabi A, Rehrig S, Buckenmaier CC, 3rd, Chelly JE. A multicenter, randomized, controlled study evaluating preventive etanercept on postoperative pain after inguinal hernia repair. Anesth Analg. 2013 Feb;116(2):455–62. doi: 10.1213/ANE.0b013e318273f71c. [DOI] [PubMed] [Google Scholar]
- Cohen SP, White RL, Kurihara C, Larkin TM, Chang A, Griffith SR, Gilligan C, Larkin R, Morlando B, Pasquina PF, Yaksh TL, Nguyen C. Epidural steroids, etanercept, or saline in subacute sciatica: a multicenter, randomized trial. Ann Intern Med. 2012 Apr 17;156(8):551–9. doi: 10.7326/0003-4819-156-8-201204170-00397. [DOI] [PubMed] [Google Scholar]
- Colburn RW, DeLeo JA, Rickman AJ, Yeager MP, Kwon P, Hickey WF. Dissociation of microglial activation and neuropathic pain behaviors following peripheral nerve injury in the rat. J Neuroimmunol. 1997 Nov;79(2):163–75. doi: 10.1016/s0165-5728(97)00119-7. [DOI] [PubMed] [Google Scholar]
- Colburn RW, Rickman AJ, DeLeo JA. The effect of site and type of nerve injury on spinal glial activation and neuropathic pain behavior. Exp Neurol. 1999 Jun;157(2):289–304. doi: 10.1006/exnr.1999.7065. [DOI] [PubMed] [Google Scholar]
- Coull JA, Beggs S, Boudreau D, Boivin D, Tsuda M, Inoue K, Gravel C, Salter MW, De Koninck Y. BDNF from microglia causes the shift in neuronal anion gradient underlying neuropathic pain. Nature. 2005 Dec 15;438(7070):1017–21. doi: 10.1038/nature04223. [DOI] [PubMed] [Google Scholar]
- Del Valle L, Schwartzman RJ, Alexander G. Spinal cord histopathological alterations in a patient with longstanding complex regional pain syndrome. Brain Behav Immun. 2009 Jan;23(1):85–91. doi: 10.1016/j.bbi.2008.08.004. [DOI] [PubMed] [Google Scholar]
- Dworkin RH, O’Connor AB, Backonja M, et al. Pharmacologic management of neuropathic pain: evidence-based recommendations. Pain. 2007;132(3):237–51. doi: 10.1016/j.pain.2007.08.033. [DOI] [PubMed] [Google Scholar]
- Ehrhart J, Obregon D, Mori T, Hou H, Sun N, Bai Y, Klein T, Fernandez F, Tan J, Shytle RD. Stimulation of cannabinoid receptor 2 (CB2) suppresses microglial activation. J Neuroinflammation. 2005 Dec 12;2:29. doi: 10.1186/1742-2094-2-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Khoury J, Toft M, Hickman SE, Means TK, Terada K, Geula C, Luster AD. Ccr2 deficiency impairs microglial accumulation and accelerates progression of Alzheimer-like disease. Nat Med. 2007;13:432–438. doi: 10.1038/nm1555. [DOI] [PubMed] [Google Scholar]
- Ellis A1, Wieseler J2, Favret J2, Johnson KW3, Rice KC4, Maier SF2, Falci S5, Watkins LR2. Systemic administration of propentofylline, ibudilast, and (+)-naltrexone each reverses mechanical allodynia in a novel rat model of central neuropathic pain. J Pain. 2014;15(4):407–21. doi: 10.1016/j.jpain.2013.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eljaschewitsch E, Witting A, Mawrin C, Lee T, Schmidt PM, Wolf S, Hoertnagl H, Raine CS, Schneider-Stock R, Nitsch R, Ullrich O. The endocannabinoid anandamide protects neurons during CNS inflammation by induction of MKP-1 in microglial cells. Neuron. 2006 Jan 5;49(1):67–79. doi: 10.1016/j.neuron.2005.11.027. [DOI] [PubMed] [Google Scholar]
- Finnerup NB, Sindrup SH, Jensen TS. The evidence for pharmacological treatment of neuropathic pain. Pain. 2010 Sep;150(3):573–81. doi: 10.1016/j.pain.2010.06.019. [DOI] [PubMed] [Google Scholar]
- Fox RJ. Primary neuroprotection: the Holy Grail of multiple sclerosis therapy. Neurology. 2010 Mar 30;74(13):1018–9. doi: 10.1212/WNL.0b013e3181d6b165. [DOI] [PubMed] [Google Scholar]
- Frampton M, Harvey RJ, Kirchner V. Propentofylline for dementia. Cochrane Database Syst Rev. 2003;(2):CD002853. doi: 10.1002/14651858.CD002853. [DOI] [PubMed] [Google Scholar]
- Glezer I, Simard AR, Rivest S. Neuroprotective role of the innate immune system by microglia. Neuroscience. 2007 Jul 29;147(4):867–83. doi: 10.1016/j.neuroscience.2007.02.055. [DOI] [PubMed] [Google Scholar]
- Gordon S, Taylor PR. Monocyte and macrophage heterogeneity. Nat Rev Immunol. 2005;5:953–964. doi: 10.1038/nri1733. [DOI] [PubMed] [Google Scholar]
- Gordon S. Alternative activation of macrophages. Nat Rev Immunol. 2003;3:23–35. doi: 10.1038/nri978. [DOI] [PubMed] [Google Scholar]
- Gourine AV, Kasymov V, Marina N, Tang F, Figueiredo MF, Lane S, Teschemacher AG, Spyer KM, Deisseroth K, Kasparov S. Astrocytes control breathing through pH-dependent release of ATP. Science. 2010 Jul 30;329(5991):571–5. doi: 10.1126/science.1190721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hald A, Nedergaard S, Hansen RR, Ding M, Heegaard AM. Differential activation of spinal cord glial cells in murine models of neuropathic and cancer pain. Eur J Pain. 2009;13:138–145. doi: 10.1016/j.ejpain.2008.03.014. [DOI] [PubMed] [Google Scholar]
- Hamann S, Sloan PA, Witt W. Low-dose intrathecal naloxone to enhance intrathecal morphine analgesia: a case report. J Opioid Manag. 2008;4(4):251–254. doi: 10.5055/jom.2008.0028. [DOI] [PubMed] [Google Scholar]
- Han X, Chen M, Wang F, Windrem M, Wang S, Shanz S, Xu Q, Oberheim NA, Bekar L, Betstadt S, Silva AJ, Takano T, Goldman SA, Nedergaard M. Forebrain engraftment by human glial progenitor cells enhances synaptic plasticity and learning in adult mice. Cell Stem Cell. 2013 Mar 7;12(3):342–53. doi: 10.1016/j.stem.2012.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchinson MR, Zhang Y, Brown K, Coats BD, Shridhar M, Sholar PW, Patel SJ, Crysdale NY, Harrison JA, Maier SF, Rice KC, Watkins LR. Non-stereoselective reversal of neuropathic pain by naloxone and naltrexone: involvement of toll-like receptor 4 (TLR4) Eur J Neurosci. 2008 Jul;28(1):20–9. doi: 10.1111/j.1460-9568.2008.06321.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibrahim MM, Porreca F, Lai J, Albrecht PJ, Rice FL, Khodorova A, Davar G, et al. CB2 cannabinoid receptor activation produces antinociception by stimulating peripheral release of endogenous opioids. Proc Natl Acad Sci U S A. 2005;102(8):3093–8. doi: 10.1073/pnas.0409888102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imamoto N, Momosaki S, Fujita M, Omachi S, Yamato H, Kimura M, Kanegawa N, Shinohara S, Abe K. [11C]PK11195 PET imaging of spinal glial activation after nerve injury in rats. Neuroimage. 2013 Oct 1;79:121–8. doi: 10.1016/j.neuroimage.2013.04.039. [DOI] [PubMed] [Google Scholar]
- Inoue K. The function of microglia through purinergic receptors: neuropathic pain and cytokine release. Pharmacol Therapeut. 2006;109:210–226. doi: 10.1016/j.pharmthera.2005.07.001. [DOI] [PubMed] [Google Scholar]
- Jessen KR, Mirsky R. Glial cells in the enteric nervous system contain glial fibrillary acidic protein. Nature. 1980 Aug 14;286(5774):736–7. doi: 10.1038/286736a0. [DOI] [PubMed] [Google Scholar]
- Ji B, Maeda J, Sawada M, Ono M, Okauchi T, Inaji M, Zhang MR, et al. Imaging of peripheral benzodiazepine receptor expression as biomarkers of detrimental versus beneficial glial responses in mouse models of Alzheimer’s and other CNS pathologies. J Neurosci. 2008;28(47):12255–67. doi: 10.1523/JNEUROSCI.2312-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji RR, Berta T, Nedergaard M. Glia and pain: Is chronic pain a gliopathy? Pain. 2013 Jun 20; doi: 10.1016/j.pain.2013.06.022. pii: S0304–3959(13)00330–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kallendrusch S, Kremzow S, Nowicki M, Grabiec U, Winkelmann R, Benz A, Kraft R, Bechmann I, Dehghani F, Koch M. The G Protein-Coupled Receptor 55 Ligand l-α-Lysophosphatidylinositol Exerts Microglia-Dependent Neuroprotection After Excitotoxic Lesion. Glia. 2013 Sep 3; doi: 10.1002/glia.22560. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- Kalliomäki J, Attal N, Jonzon B, Bach FW, Huizar K, Ratcliffe S, Eriksson B, Janecki M, Danilov A, Bouhassira D AZD2423 PTN Study Group. A randomized, double-blind, placebo-controlled trial of a chemokine receptor 2 (CCR2) antagonist in posttraumatic neuralgia. Pain. 2013 May;154(5):761–7. doi: 10.1016/j.pain.2013.02.003. [DOI] [PubMed] [Google Scholar]
- Kawasaki Y, Xu ZZ, Wang X, Park JY, Zhuang ZY, Tan PH, Gao YJ, Roy K, Corfas G, Lo EH, Ji RR. Distinct roles of matrix metalloproteases in the early- and late-phase development of neuropathic pain. Nat Med. 2008 Mar;14(3):331–6. doi: 10.1038/nm1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein TW, Newton C, Larsen K, Lu L, Perkins I, Nong L, Friedman H. The cannabinoid system and immune modulation. J Leukoc Biol. 2003;74(4):486–96. doi: 10.1189/jlb.0303101. [DOI] [PubMed] [Google Scholar]
- Lai AY, Todd KG. Differential regulation of trophic and proinflammatory microglial effectors is dependent on severity of neuronal injury. Glia. 2008;56:259–270. doi: 10.1002/glia.20610. [DOI] [PubMed] [Google Scholar]
- Ledeboer A, Hutchinson MR, Watkins LR, Johnson KW. Ibudilast (AV-411). A new class therapeutic candidate for neuropathic pain and opioid withdrawal syndromes. Expert Opin Investig Drugs. 2007 Jul;16(7):935–50. doi: 10.1517/13543784.16.7.935. [DOI] [PubMed] [Google Scholar]
- Ledeboer A, Liu T, Shumilla JA, Mahoney JH, Vijay S, Gross MI, Vargas JA, Sultzbaugh L, Claypool MD, Sanftner LM, Watkins LR, Johnson KW. The glial modulatory drug AV411 attenuates mechanical allodynia in rat models of neuropathic pain. Neuron Glia Biol. 2006 Nov;2(4):279–91. doi: 10.1017/S1740925X0700035X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee BS, Jun IG, Kim SH, Park JY. Intrathecal gabapentin increases interleukin-10 expression and inhibits pro-inflammatory cytokine in a rat model of neuropathic pain. J Korean Med Sci. 2013 Feb;28(2):308–14. doi: 10.3346/jkms.2013.28.2.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Walter S, Stagi M, Cherny D, Letiembre M, Schulz-Schaeffer W, Heine H, Penke B, Neumann H, Fassbender K. LPS receptor (CD14): A receptor for phagocytosis of Alzheimer’s amyloid peptide. Brain. 2005;128:1778–1789. doi: 10.1093/brain/awh531. [DOI] [PubMed] [Google Scholar]
- Loram LC, Harrison JA, Sloane EM, Hutchinson MR, Sholar P, Taylor FR, Berkelhammer D, Coats BD, Poole S, Milligan ED, et al. Enduring reversal of neuropathic pain by a single intrathecal injection of adenosine 2A receptor agonists: a novel therapy for neuropathic pain. J Neurosci. 2009 Nov 4;29(44):14015–25. doi: 10.1523/JNEUROSCI.3447-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loram LC, Taylor FR, Strand KA, Harrison JA, Rzasalynn R, Sholar P, Rieger J, Maier SF, Watkins LR. Intrathecal injection of adenosine 2A receptor agonists reversed neuropathic allodynia through protein kinase (PK)A/PKC signaling. Brain Behav Immun. 2013 Jun 28; doi: 10.1016/j.bbi.2013.06.004. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu W, Bhasin M, Tsirka SE. Involvement of tissue plasminogen activator in onset and effector phases of experimental allergic encephalomyelitis. J Neurosci. 2002;22:10781–10789. doi: 10.1523/JNEUROSCI.22-24-10781.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo XG, Chen SD. The changing phenotype of microglia from homeostasis to disease. Transl Neurodegener. 2012;1:9. doi: 10.1186/2047-9158-1-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luongo L, Guida F, Boccella S, Bellini G, Gatta L, Rossi F, de Novellis V, Maione S. Palmitoylethanolamide reduces formalin-induced neuropathic-like behaviour through spinal glial/microglial phenotypical changes in mice. CNS Neurol Disord Drug Targets. 2013 Feb 1;12(1):45–54. doi: 10.2174/1871527311312010009. [DOI] [PubMed] [Google Scholar]
- Luongo L1, Guida F, Imperatore R, Napolitano F, Gatta L, Cristino L, Giordano C, Siniscalco D, Di Marzo V, Bellini G, Petrelli R, Cappellacci L, Usiello A, de Novellis V, Rossi F, Maione S. The A1 adenosine receptor as a new player in microglia physiology. Glia. 2014;62:122–32. doi: 10.1002/glia.22592. [DOI] [PubMed] [Google Scholar]
- Luongo L, Palazzo E, Tambaro S, Giordano C, Gatta L, Scafuro MA, Rossi FS, Lazzari P, Pani L, de Novellis V, Malcangio M, Maione S. 1-(2′,4′-dichlorophenyl)-6-methyl-N-cyclohexylamine-1,4-dihydroindeno[1,2-c]pyrazole-3-carboxamide, a novel CB2 agonist, alleviates neuropathic pain through functional microglial changes in mice. Neurobiol Dis. 2010;37:177–85. doi: 10.1016/j.nbd.2009.09.021. [DOI] [PubMed] [Google Scholar]
- Malm TM, Koistinaho M, Pärepalo M, Vatanen T, Ooka A, Karlsson S, Koistinaho J. Bone-marrow-derived cells contribute to the recruitment of microglial cells in response to beta-amyloid deposition in APP/PS1 double transgenic Alzheimer mice. Neurobiol Dis. 2005 Feb;18(1):134–42. doi: 10.1016/j.nbd.2004.09.009. [DOI] [PubMed] [Google Scholar]
- Manzanares J, Julian M, Carrascosa A. Role of the cannabinoid system in pain control and therapeutic implications for the management of acute and chronic pain episodes. Curr Neuropharmacol. 2006 Jul;4(3):239–57. doi: 10.2174/157015906778019527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meme W, Calvo CF, Froger N, Ezan P, Amigou E, Koulakoff A, Giaume C. Proinflammatory cytokines released from microglia inhibit gap junctions in astrocytes: potentiation by beta-amyloid. Faseb J. 2006;20:494–496. doi: 10.1096/fj.05-4297fje. [DOI] [PubMed] [Google Scholar]
- Miller AM, Stella N. CB2 receptor-mediated migration of immune cells: it can go either way. Br J Pharmacol. 2008 Jan;153(2):299–308. doi: 10.1038/sj.bjp.0707523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milligan ED, Penzkover KR, Soderquist RG, Mahoney MJ. Spinal interleukin-10 therapy to treat peripheral neuropathic pain. Neuromodulation. 2012 Nov-Dec;15(6):520–6. doi: 10.1111/j.1525-1403.2012.00462.x. discussion 526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milligan ED, Watkins LR. Pathological and protective roles of glia in chronic pain. Nat Rev Neurosci. 2009;10:23–36. doi: 10.1038/nrn2533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molina-Holgado F, Pinteaux E, Moore JD, Molina-Holgado E, Guaza C, Gibson RM, Rothwell NJ. Endogenous interleukin-1 receptor antagonist mediates anti-inflammatory and neuroprotective actions of cannabinoids in neurons and glia. J Neurosci. 2003 Jul 23;23(16):6470–4. doi: 10.1523/JNEUROSCI.23-16-06470.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajima K, Kohsaka S. Microglia: activation and their significance in the central nervous system. J Biochem. 2001 Aug;130(2):169–75. doi: 10.1093/oxfordjournals.jbchem.a002969. [DOI] [PubMed] [Google Scholar]
- Núñez E, Benito C, Pazos MR, Barbachano A, Fajardo O, González S, Tolón RM, Romero J. Cannabinoid CB2 receptors are expressed by perivascular microglial cells in the human brain: an immunohistochemical study. Synapse. 2004 Sep 15;53(4):208–13. doi: 10.1002/syn.20050. [DOI] [PubMed] [Google Scholar]
- Oberheim NA, Takano T, Han X, He W, Lin JH, Wang F, Xu Q, Wyatt JD, Pilcher W, Ojemann JG, Ransom BR, Goldman SA, Nedergaard M. Uniquely hominid features of adult human astrocytes. J Neurosci. 2009 Mar 11;29(10):3276–87. doi: 10.1523/JNEUROSCI.4707-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohta H, Ishizaka T, Tatsuzuki M, Yoshinaga M, Iida I, Yamaguchi T, Tomishima Y, Futaki N, Toda Y, Saito S. Imine derivatives as new potent and selective CB2 cannabinoid receptor agonists with an analgesic action. Bioorg Med Chem. 2008;16(3):1111–24. doi: 10.1016/j.bmc.2007.10.087. [DOI] [PubMed] [Google Scholar]
- Orgogozo JM, Gilman S, Dartigues JF, Laurent B, Puel M, Kirby LC, et al. Subacute meningoencephalitis in a subset of patients with AD after Abeta42 immunization. Neurology. 2003;61:46–54. doi: 10.1212/01.wnl.0000073623.84147.a8. [DOI] [PubMed] [Google Scholar]
- Orr AG, Orr AL, Li XJ, Gross RE, Traynelis SF. Adenosine A(2A) receptor mediates microglial process retraction. Nat Neurosci. 2009;12:872–8. doi: 10.1038/nn.2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Racz I, Nadal X, Alferink J, Baños JE, Rehnelt J, Martín M, Pintado B, Gutierrez-Adan A, Sanguino E, Manzanares J, Zimmer A, Maldonado R. Crucial role of CB(2) cannabinoid receptor in the regulation of central immune responses during neuropathic pain. J Neurosci. 2008a;28:12125–35. doi: 10.1523/JNEUROSCI.3400-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Racz I, Nadal X, Alferink J, Banos JE, Rehnelt J, Martin M, Pintado B, Gutierrez-Adan A, Sanguino E, Bellora N, Manzanares J, Zimmer A, Maldonado R. Interferon-gamma is a critical modulator of CB(2) cannabinoid receptor signaling during neuropathic pain. J Neurosci. 2008b;28:12136–45. doi: 10.1523/JNEUROSCI.3402-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez G, Toro R, Dobeli H, von Bernhardi R. Protection of rat primary hippocampal cultures from A beta cytotoxicity by pro-inflammatory molecules is mediated by astrocytes. Neurobiol Dis. 2005;19:243–254. doi: 10.1016/j.nbd.2005.01.007. [DOI] [PubMed] [Google Scholar]
- Ren K, Dubner R. Neuron-glia crosstalk gets serious: role in pain hypersensitivity. Curr Opin Anaesthesiol. 2008 Oct;21(5):570–9. doi: 10.1097/ACO.0b013e32830edbdf. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rolan P, Hutchinson M, Johnson K. Ibudilast: a review of its pharmacology, efficacy and safety in respiratory and neurological disease. Expert Opin Pharmacother. 2009 Dec;10(17):2897–904. doi: 10.1517/14656560903426189. [DOI] [PubMed] [Google Scholar]
- Rolan P. Methods for targeting glial activation in the treatment of patients with chronic pain. Mechanisms and Treatment of Neuropathic Pain, annual meeting; Snowbird, Utah. Nov 2007. [Google Scholar]
- Romero-Sandoval A, Nutile-McMenemy N, DeLeo JA. Spinal microglial and perivascular cell cannabinoid receptor type 2 activation reduces behavioral hypersensitivity without tolerance after peripheral nerve injury. Anesthesiology. 2008 Apr;108(4):722–34. doi: 10.1097/ALN.0b013e318167af74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawada H, Suzuki H, Nagatsu T, Sawada M. Neuroprotective and neurotoxic phenotypes of activated microglia in neonatal mice with respective MPTP and ethanol-induced brain injury. Neurodegener Dis. 2010;7:64–67. doi: 10.1159/000285508. [DOI] [PubMed] [Google Scholar]
- Scholz J, Woolf CJ. The neuropathic pain triad: neurons, immune cells and glia. Nat Neurosci. 2007;10:1361–1368. doi: 10.1038/nn1992. [DOI] [PubMed] [Google Scholar]
- Sloan P, Hamann S. Ultra-low-dose opioid antagonists to enhance opioid analgesia. J Opioid Manag. 2006 Sep-Oct;2(5):295–304. doi: 10.5055/jom.2006.0044. [DOI] [PubMed] [Google Scholar]
- Sloane E, Langer S, Jekich B, Mahoney J, Hughes T, Frank M, Seibert W, Huberty G, Coats B, Harrison J, Klinman D, Poole S, Maier S, Johnson K, Chavez R, Watkins LR, Leinwand L, Milligan E. Immunological priming potentiates non-viral anti-inflammatory gene therapy treatment of neuropathic pain. Gene Ther. 2009 Oct;16(10):1210–22. doi: 10.1038/gt.2009.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soderquist RG, Sloane EM, Loram LC, Harrison JA, Dengler EC, Johnson SM, Amer LD, Young CS, Lewis MT, Poole S, Frank MG, Watkins LR, Milligan ED, Mahoney MJ. Release of plasmid DNA-encoding IL-10 from PLGA microparticles facilitates long-term reversal of neuropathic pain following a single intrathecal administration. Pharm Res. 2010 May;27(5):841–54. doi: 10.1007/s11095-010-0077-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorge RE, Trang T, Dorfman R, Smith SB, Beggs S, Ritchie J, Austin JS, Zaykin DV, Vander Meulen H, Costigan M, Herbert TA, Yarkoni-Abitbul M, Tichauer D, Livneh J, Gershon E, Zheng M, Tan K, John SL, Slade GD, Jordan J, Woolf CJ, Peltz G, Maixner W, Diatchenko L, Seltzer Z, Salter MW, Mogil JS. Genetically determined P2X7 receptor pore formation regulates variability in chronic pain sensitivity. Nat Med. 2012 Mar 25;18(4):595–9. doi: 10.1038/nm.2710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stellwagen D, Malenka RC. Synaptic scaling mediated by glial TNF-alpha. Nature. 2006 Apr 20;440(7087):1054–9. doi: 10.1038/nature04671. [DOI] [PubMed] [Google Scholar]
- Stephenson DT, Arneric SP. Neuroimaging of pain: advances and future prospects. J Pain. 2008 Jul;9(7):567–79. doi: 10.1016/j.jpain.2008.02.008. [DOI] [PubMed] [Google Scholar]
- Suzumura A, Ito A, Yoshikawa M, Sawada M. Ibudilast suppresses TNFalpha production by glial cells functioning mainly as type III phosphodiesterase inhibitor in the CNS. Brain Res. 1999 Aug 7;837(1–2):203–12. doi: 10.1016/s0006-8993(99)01666-2. [DOI] [PubMed] [Google Scholar]
- Svensson CI, et al. Spinal p38b isoform mediates tissue injury-induced hyperalgesia and spinal sensitization. J Neurochem. 2005;92:1508–1520. doi: 10.1111/j.1471-4159.2004.02996.x. [DOI] [PubMed] [Google Scholar]
- Sweitzer S, De Leo J. Propentofylline: glial modulation, neuroprotection, and alleviation of chronic pain. Handb Exp Pharmacol. 2011;(200):235–50. doi: 10.1007/978-3-642-13443-2_8. [DOI] [PubMed] [Google Scholar]
- Toth C, Mawani S, Brady S, Chan C, Liu C, Mehina E, Garven A, Bestard J, Korngut L. An enriched-enrolment, randomized withdrawal, flexible-dose, double-blind, placebo-controlled, parallel assignment efficacy study of nabilone as adjuvant in the treatment of diabetic peripheral neuropathic pain. Pain. 2012 Oct;153(10):2073–82. doi: 10.1016/j.pain.2012.06.024. [DOI] [PubMed] [Google Scholar]
- Tran PB, Miller RJ. Chemokine receptors: signposts to brain development and disease. Nat Rev Neurosci. 2003 Jun;4(6):444–55. doi: 10.1038/nrn1116. [DOI] [PubMed] [Google Scholar]
- Vallejo R, Tilley DM, Vogel L, Benyamin R. The role of glia and the immune system in the development and maintenance of neuropathic pain. Pain Pract. 2010 May Jun;10(3):167–84. doi: 10.1111/j.1533-2500.2010.00367.x. [DOI] [PubMed] [Google Scholar]
- Van Sickle MD, Duncan M, Kingsley PJ, Mouihate A, Urbani P, Mackie K, Stella N, et al. Identification and functional characterization of brainstem cannabinoid CB2 receptors. Science. 2005;310(5746):329–32. doi: 10.1126/science.1115740. [DOI] [PubMed] [Google Scholar]
- Vincent VA, Van Dam AM, Persoons JH, Schotanus K, Steinbusch HW, Schoffelmeer AN, Berkenbosch F. Gradual inhibition of inducible nitric oxide synthase but not of interleukin-1 beta production in rat microglial cells of endotoxin-treated mixed glial cell cultures. Glia. 1996;17:94–102. doi: 10.1002/(SICI)1098-1136(199606)17:2<94::AID-GLIA2>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- von Bernhardi R, Eugenin J. Microglial reactivity to beta-amyloid is modulated by astrocytes and proinflammatory factors. Brain Res. 2004;1025:186–193. doi: 10.1016/j.brainres.2004.07.084. [DOI] [PubMed] [Google Scholar]
- Watkins LR, Hutchinson MR, Milligan ED, Maier SF. “Listening” and “talking” to neurons: implications of immune activation for pain control and increasing the efficacy of opioids. Brain Res Rev. 2007;56:148–169. doi: 10.1016/j.brainresrev.2007.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wieseler-Frank J, Maier SF, Watkins LR. Central proinflammatory cytokines and pain enhancement. Neurosignals. 2005;14:166–74. doi: 10.1159/000087655. [DOI] [PubMed] [Google Scholar]
- Woolf CJ, Mannion RJ. Neuropathic pain: aetiology, symptoms, mechanisms, and management. Lancet. 1999 Jun 5;353(9168):1959–64. doi: 10.1016/S0140-6736(99)01307-0. [DOI] [PubMed] [Google Scholar]
- Wynn TA, Thompson RW, Cheever AW, Mentink-Kane MM. Immunopathogenesis of schistosomiasis. Immunol Rev. 2004;201:156–167. doi: 10.1111/j.0105-2896.2004.00176.x. [DOI] [PubMed] [Google Scholar]
- Yao BB, Hsieh G, Daza AV, Fan Y, Grayson GK, Garrison TR, El Kouhen O, et al. Characterization of a cannabinoid CB2 receptor-selective agonist, A-836339 [2,2,3,3-tetramethyl-cyclopropanecarboxylic acid [3-(2-methoxy-ethyl)-4,5-dimethyl-3H-thiazol-(2Z)-ylidene]-amide], using in vitro pharmacological assays, in vivo pain models, and pharmacological magnetic resonance imaging. J Pharmacol Exp Ther. 2009;328(1):141–51. doi: 10.1124/jpet.108.145011. [DOI] [PubMed] [Google Scholar]
- Yasuno F, Ota M, Kosaka J, Ito H, Higuchi M, Doronbekov TK, Nozaki S, et al. Increased binding of peripheral benzodiazepine receptor in Alzheimer’s disease measured by positron emission tomography with [11C]DAA 1106. Biol Psychiatry. 2008;64(10):835–41. doi: 10.1016/j.biopsych.2008.04.021. [DOI] [PubMed] [Google Scholar]
- Zhang J, De Koninck Y. Spatial and temporal relationship between monocyte chemoattractant protein-1 expression and spinal glial activation following peripheral nerve injury. J Neurochem. 2006 May;97(3):772–83. doi: 10.1111/j.1471-4159.2006.03746.x. [DOI] [PubMed] [Google Scholar]
- Zhao P, Waxman SG, Hains BC. Modulation of thalamic nociceptive processing after spinal cord injury through remote activation of thalamic microglia by cysteine cysteine chemokine ligand 21. J Neurosci. 2007 Aug 15;27(33):8893–902. doi: 10.1523/JNEUROSCI.2209-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]