

Introduction
Ischemic heart disease (IHD) is one of the major cardiovascular disorders and reported to be the single largest cause of death in the developed countries and a main contributor to the disease burden in developing countries.[1] A majority of drugs used against myocardial ischemia such as nitrates, β blockers, calcium channel blockers and angiotensin converting enzyme inhibitors reduce imbalance between oxygen consumption and oxygen supply. Other drugs like anti-atherosclerotic and thrombolytic drugs reduces the chances of reoccurrence of the events and improve hemodynamic parameters, however, none prevent direct cell death that occurs during the ischemic injury. Reperfusion of viable myocardium would be beneficial for survival of the heart cells; paradoxically, however, the reperfusion of ischemic myocardium may lead to arrhythmias, myocardial stunning (i.e., reversible contractile dysfunction), or lethal reperfusion injury. Reperfusion injury is mediated in part by oxygen free radicals, neutrophil–endothelium interactions, apoptosis, and intracellular calcium overload.[2-4] The current drugs used in the management of IHD, however, have no ability to prevent cell death that occurs during the ischemic reperfusion injury. The better management of IHD, therefore, warrants the development of novel agents.
Epoxyeicosatrienoic acids (EETs) are endogenous oxylipids reported to play an important role in cardioprotection. The cardiovascular effects of EETs range from vasodilation, anti-hypertension, pro-angiogenesis, anti-atherosclerosis, and anti-inflammatory to cytoprotection. [5] The ability of EETs to prevent cell death during reperfusion injury is attributed to their anti-apoptotic, pro-mitogenic, and anti-migration effects at the cellular levels.[5, 6] EETs are reported to reduce calcium overload during cardiac ischemia by activating ATP-sensitive K+ channels (KATP).[7-9] Opening of the KATP ATP channels allows a K+ efflux, resulting in an increased rate of repolarization and a decreased period of Ca2+ influx during the plateau phase of the action potential.[10, 11] In addition EETs are reported to be pro-mitogenic and attenuate reactive oxygen species level, mitochondrial dysfunction, caspase activation, cell migration and apoptosis. All the above effects of EETs are reported to play an important role in their cardioprotection against reperfusion injury.[12-15]
The cytochrome P450 system catalyzes the formation of EETs from arachidonic acid. The EETs are later converted to biologically inactive or less active diols by soluble epoxide hydrolase (sEH) enzyme.[16] One of the novel strategies, therefore, is to inhibit the enzyme sEH and thereby promote the cardioprotective benefits of EETs. A diverse class of agents such as amides, ureas, thioamides, thioureas, carbamates, acyl hydrazones, chalcone oxdies, etc., have been reported to possess potential as both in vitro and in vivo sEH inhibitors.[17] Among these, compounds containing either urea or amide moiety as central pharmacophore are reported to be the most potent inhibitors.[18] Hammock et al., have synthesized and reported the structural activity relationships of various N, N’-disubstituted ureas.[19, 20] They report the poor solubility and rapid in vivo metabolism of the early sEH inhibitors resulted in reduced in vivo activity.[21] They have also carried out various modifications in the structure to improve their pharmacokinetics. Positioning of polar residues such as esters, sulfones, amides and carbamates roughly 5-7 Å from the central pharmacophore has been reported to improve binding without reducing the potency, however, some of these molecules were found to be metabolically unstable.[20] To overcome these problems, conformationally restricted N, N’-disubstituted ureas having polar substitutions have been synthesized and evaluated for their in vitro sEH inhibition activity and also for their bioavailability. These derivatives have reported to show improved bioavailability and in vitro activity.[22, 23] In the present study, one such molecule, trans-4-{4-[3-(4-trifluoromethoxyphenyl)ureido]cyclohexyloxy}benzoic acid (t-TUCB), synthesized by this group has been chosen for the evaluation of its in vivo cardioprotective activity against isoproterenol (ISO) induced myocardial infarction in rats. This molecule has a central N, N’-disubstituted urea pharmacophore in which a cyclohexane ring serves as a linker (between a urea and a polar group) and also as a template to restrict flexibility of the structure (Figure 1).
Figure 1.
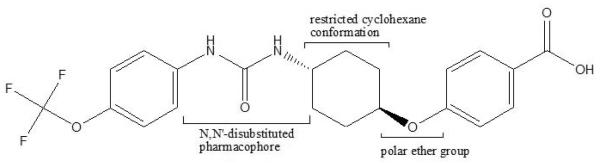
trans-4-{4-[3-(4-Trifluoromethoxyphenyl)ureido]cyclohexyloxy}benzoic acid (t-TUCB)
Materials and Methods
Animals
Male Wistar albino rats and Swiss albino mice were procured from in-house animal facility of J.S.S College of Pharmacy, Udhagamandalam. The animals were housed under laboratory conditions of temperature (18-24 °C) and relative humidity (55-75%) with a 12:12 h light-dark cycle. The animals were fed with standard pellet diet and water ad libitum. All the experiments were performed with a prior approval from the Institutional Animal Ethics Committee of J.S.S College of Pharmacy, Udhagamandalam (Approval no. JSSCP/IAEC/M.PHARM/PH.COLOGY/01/2012-13).
Chemicals and reagents
The soluble epoxide hydrolase inhibitor, t-TUCB, was synthesized as previously described.[19] Isoproterenol (ISO) was from Sigma Aldrich Ltd., India. The creatine kinase (CK-MB), lactate dehydrogenase (LDH) and calcium kits were from Labkit Ltd., India. All other chemicals and reagents used were of analytical grade.
Preparation of test compound
t-TUCB stock solutions (1/10 of the dose) were prepared in 20% PEG-400 in saline and administered at a dose volume of 10 ml/kg, body weight.
Acute oral toxicity study
Acute oral toxicity of t- TUCB was evaluated in Swiss albino mice as per OECD 423 guidelines. A limit test at a dose of 2000 mg/kg, body weight was carried out using 6 mice (3 per step). Following overnight fasting the first set of 3 animals were dosed with the test compound and observed for clinical signs and mortality at 30, 60, 120 and 240 min on day 0, and thereafter daily once for a period of 14 days. Based on 24h observation results of the first set, the second set of 3 animals were dosed with the same dose and observed. Body weights were recorded prior to administration and again on days 7 and 14. On day 15, all the animals were culled by deep ether anesthesia and subjected to gross necropsy analysis.
In vivo cardioprotective activity
Forty male Wistar rats were divided into 5 groups of 8 each. The group 1 and 2 animals received vehicle (10 mL/kg, p.o.) and served as normal and control, respectively. The group 3-5 animals received t-TUCB at 3, 10 and 30 mg/kg, p.o., respectively. On day 13 and 14 all the groups except the group 1 received subcutaneous ISO injection at a dose of 150 mg/kg, body weight. 24h after the last ISO injections EKG parameters were monitored from all the groups using PowerLab (AD Instruments, Australia). Later, blood was collected from retro-orbital sinus under light ether anesthesia and used for the estimation of the biochemical parameters (CKMB and LDH). Thereafter, rats were sacrificed by cervical dislocation and hearts were excised, weighed and immediately frozen in liquid nitrogen for the analysis of infarct size, myocardial calcium and antioxidant enzymes.
In vivo electrocardiographic (EKG) recordings
All electrocardiographic recordings were done with Animal Bio Amp (FE136, AD instruments, Australia). For in vivo recordings of EKG, rats were anaesthetized with ketamine (100 mg/kg, i.p.) and xylazine (15 mg/kg, i.m.). The positive and negative lead I electrodes were inserted into left and right forearms, respectively. The reference electrode was grounded to right hind limb. All the parameters of EKG were recorded using LabChart 7 Pro software (AD instruments, Australia).
Estimation of serum CK-MB and LDH
Blood was allowed to clot and serum was separated by centrifuging at 3000 rpm for 10 min in a cold centrifuge. The serum CK-MB and LDH levels were estimated in Merck-200 semi auto analyzer (Merck Ltd, Darmstadt, Germany) using Labkit diagnostic kits.
Infarct size analysis
Briefly, hearts were cut into thin cross sectional slices (1-2 mm) and kept in 1% solution of 2,3,5-triphenyltetrazolium chloride (TTC) dissolved in phosphate buffer (0.1 M, pH 7.4) at 37 °C for 20 min. The individual slices were digitally photographed and the area of infarction was quantified by measuring stained (red, live tissue) and unstained (white, necrotic) regions using Image J, version 1.47 (NIH, USA) and the percentage infarct size was calculated.
Estimation of cardiac calcium and antioxidant parameters
One part of heart tissue was homogenized with 5 parts of phosphate buffer (0.1 M, pH 7.4) in Elvenjan homogenizer fitted with a Teflon plunger at 600 rpm for 20 minutes. The homogenate was then centrifuged at 10,000 rpm for 20 min at 4°C. The supernatant was used to estimate Ca2+ levels and antioxidant parameters.
Calcium was estimated in tissue homogenate using calcium kit as per the manufacturer’s protocol. Calcium with arsenazo III (1, 8-Dihydroxy-3, 6-disulpho-2, 7-naphthalene-bis (azo)-dibenzenearsonic acid), at neutral pH, yields a blue colored complex. The intensity of the color formed is proportional to the calcium concentration in the sample. All the glass ware was pretreated with concentrated nitric acid overnight followed by extensive rinsing before the estimation. Lipid peroxide level in tissue homogenate was estimated by the method of Okhawa.[24] Catalase was estimated according to the method of Beers and Sizer [25] and SOD in tissue homogenate was estimated as previously described.[26]
Statistical analysis
Values are expressed as the mean ± SEM. Statistical significance was determined by one way ANOVA followed by Dunnett’s multiple comparison test to assess differences between the groups. Values were considered significant, if p<0.05.
Results
Acute oral toxicity study
All the six mice treated with t-TUCB at 2000 mg/kg, p.o., showed no abnormal clinical signs during the study period and there were no mortalities. All the animals gained 1-2 g of body weight during the study period (Table 1). The gross necropsy analysis carried out at the end of the study showed no abnormal findings. Based on the above results the acute oral LD50 of t-TUCB was, therefore, considered greater than 2000mg/kg body weight.
Table 1.
Acute oral toxicity study results of t-TUCB in female Swiss mice
| Animal no. |
Body weight (g) |
Gross necropsy findings |
||
|---|---|---|---|---|
| Initial | Day7 | Day14 | ||
| 1. | 28 | 28 | 29 | NAD |
| 2. | 26 | 27 | 27 | NAD |
| 3. | 26 | 28 | 28 | NAD |
| 4. | 25 | 25 | 26 | NAD |
| 5. | 27 | 27 | 28 | NAD |
| 6. | 24 | 26 | 26 | NAD |
NAD: No Abnormalities Detected
In vivo cardioprotective activity
Effect of t-TUCB on EKG
The results are given in Table 2 and Figure 2. Pretreatment with t-TUCB at 3, 10 and 30 mg/kg, p.o., significantly prevented ISO induced EKG abnormalities such as prolongation of QTc interval, ST height depression, pathological Q wave formation and T-wave inversion (p<0.05).
Table 2.
Effect of t-TUCB treatment on ISO induced EKG changes in rats
| Groups | QTc interval (s) | ST height (mV) | Q wave (mV) | T wave (mV) |
|---|---|---|---|---|
| Normal | 0.147±0.015 | 0.036±0.032 | 0.023±0.010 | 0.206±0.040 |
| Control | 0.206±0.008* | −0.157±0.032* | −0.160±0.109* | −0.010±0.07* |
| t-TUCB (3 mg/kg) | 0.191±0.009 | 0.015±0.002† | 0.059±0.007† | 0.221±0.04† |
| t-TUCB (10 mg/kg) | 0.164±0.006† | 0.050±0.021† | 0.034±0.008† | 0.223±0.05† |
| t-TUCB (30 mg/kg) | 0.163±0.010† | −0.013±0.021† | −0.100±0.121† | 0.161±0.043† |
Values represent mean ± SEM; n=6 per group;
p<0.05 vs. Normal;
p<0.05 vs. Control rats. One way ANOVA followed by Dunnett’s multiple comparison test.
Figure 2. Effect of sEH inhibitor, t-TUCB, in preventing EKG abnormalities in rats.

Representative images of EKG recordings in a) normal, b) control, c) t-TUCB (3 mg/kg), d) t-TUCB (10 mg/kg) and e) t-TUCB (30 mg/kg) groups using LabChart pro 7.0.
Effect of t-TUCB on relative heart weight and serum CK-MB and LDH
The results are given in Table 3. Pretreatment with t-TUCB at 10 and 30 mg/kg, p.o., significantly prevented ISO induced increase in heart weight and elevation of CK-MB and LDH levels (p<0.05), indicating its cardioprotective effect against ISO induced cardiac injury. At 3 mg/kg, p.o. t-TUCB only showed significant protection against heart weight changes.
Table 3.
Effect of t-TUCB treatment on ISO induced changes in relative heart weight and CK-MB and LDH levels.
| Groups | Heart to body weight (mg/g) |
CK-MB (IU/L) | LDH (U/L) |
|---|---|---|---|
| Normal | 4.165±0.487 | 76.14±13.38 | 699.2±55.8 |
| Control | 5.461±0.716* | 163.90±15.50* | 1612.1±215.7* |
| t-TUCB (3 mg/kg) | 4.586±0.410† | 116.70±17.68 | 1318.2±179.6 |
| t-TUCB (10 mg/kg) | 4.553±0.345† | 96.29±12.11† | 874.4±97.8† |
| t-TUCB (30 mg/kg) | 4.606±0.538† | 98.71±14.07† | 968.6±144.3† |
Values represent mean ± SEM; n=6 per group;
p<0.05 vs. Normal;
p<0.05 vs. Control rats. One way ANOVA followed by Dunnett’s multiple comparison test.
Effect of t-TUCB on infarct size
The results are given in Table 4 and Figure 3. Pretreatment with t-TUCB at 3, 10 and 30 mg/kg, p.o., significantly reduced the ISO induced infarct size (p<0.05) when compared to control. The calculated percentage reductions for these doses were found to by 15.90, 46.60 and 40.44%, respectively.
Table 4.
Effect of t-TUCB treatment on ISO induced infarct size
| Groups | % infarct size | % protection fold in reducing infarct |
|---|---|---|
| Normal | --- | --- |
| Control | 44.5±4.7 | 0.0 |
| t-TUCB (3 mg/kg) | 37.4±4.7* | 15.9 |
| t-TUCB (10 mg/kg) | 23.7±3.0*** | 46.6 |
| t-TUCB (30 mg/kg) | 26.5±5.2*** | 40.4 |
Values represent mean ± SEM; n=3 per group;
p<0.05;
p<0.001 vs. Control rats. One way ANOVA followed by Dunnett’s multiple comparison test.
Figure 3. Effect of sEH inhibitor, t-TUCB, in ameliorating infarct size.

a) Quantification of infarct size and b) Representative images of TTC staining in t-TUCB treated and control hearts. Surviving tissue stained red with TTC and infarcted white tissue. Bars represent mean ± SEM; n=3 per group; *p<0.05; ***p<0.001 vs. Control rats. One way ANOVA followed by Dunnett’s multiple comparison test.
Effect of t-TUCB on myocardial calcium and antioxidant parameters
The results are given in Table 5. ISO treatment significantly increased the intracellular calcium and lipid peroxide levels, and decreased the SOD and catalase levels (p<0.05). Pretreatment with t-TUCB at 3, 10 and 30 mg/kg, p.o., significantly prevented the above changes induced by ISO (p<0.05), except, low dose showing a non-significant protection against lipid peroxide, catalase and SOD levels.
Table 5.
Effect of t-TUCB treatment on ISO induced changes in intracellular Ca2+ ions and antioxidants levels.
| Groups | Intracellular Ca2+ concentration (mg/g tissue) |
Lipid peroxidation (nM MDA/mg tissue) |
Catalase (U/min/mg tissue) |
SOD (U/min/mg tissue) |
|---|---|---|---|---|
| Normal | 0.374±0.003 | 0.109±0.010 | 1.266±0.086 | 2.327±0.285 |
| Control | 0.454±0.012* | 0.521±0.016* | 0.499±0.089* | 1.126±0.156* |
| t-TUCB (3 mg/kg) | 0.419±0.011† | 0.381±0.067 | 0.668±0.102 | 1.366±0.172 |
| t-TUCB (10 mg/kg) | 0.419±0.005† | 0.332±0.067† | 1.015±0.159† | 1.938±0.169† |
| t-TUCB (30 mg/kg) | 0.418±0.006† | 0.325±0.041† | 1.006±0.128† | 1.920±0.142† |
Values represent mean ± SEM; n=3 per group;
p<0.05 vs. Normal;
p<0.05 vs. Control rats. One way ANOVA followed by Dunnett’s multiple comparison test.
Discussion
t-TUCB, is a potent in vitro sEH inhibitor (IC50=0.9 nM), and is reported to show improved bioavailability and metabolic stability than previous urea derivatives.[22, 27] It is in formal clinical trials in cats, dogs and horses, and a preliminary case study for a successful treatment of Equine laminitis has been published.[28] In the current study we, therefore, selected this molecule for the investigation of its cardioprotective effects against ISO induced myocardial infarction in rats. ISO-induced MI injury model is a well-developed model for ischemia related ventricular remodeling and cardiac hypertrophy. Also, high dose treatment with ISO had been seen to produce myocardial infarction in multiple animal models.[29-31] High dose ISO treatment triggers various cellular pathways and leads to activation of several apoptotic events. Most of its effects are attributed due to its ability to cause functional hypoxia and ischemia, coronary insufficiency, alterations in metabolism, decreased level of high-energy phosphate stores, intracellular Ca2+ overloading, changes in electrolyte contents and oxidative stress.
In this study, the typical changes induced with ISO treatment were EKG related abnormalities (QTc interval prolongation, ST height depression, appearance of pathological Q waves and T wave inversion), increased cardiac mass, infarct size, CK-MB and LDH levels in serum, myocardial Ca2+ and free radical generation. Our results demonstrated that treatment with t-TUCB significantly abolished development of EKG abnormalities, reduced the cardiac hypertrophy, infarct size, intracellular Ca2+ overloading, myocardial damage and inflammation and free radicals generation. Moreover, the use of sEHIs resulted in the prevention of electrical remodeling post MI and in so doing, prevents the propensity for the development of life threatening cardiac arrhythmias as assessed by electrophysiological studies in vivo.
In vivo EKG recordings revealed a dose dependent prevention of QTc interval prolongation and ST height depression, appearance of pathological Q waves and T wave inversion with 10 and 30 mg/kg doses being statistically significant for all the measured parameters while a non-significant protection was seen at 3 mg/kg dose. Ischemia presents a condition where there is abnormality in distribution of ions and ion channels. Prolonged ischemia leads to inactivation of delayed rectifier K+ channels. These channels are responsible for repolarization current after initiation of an action potential in ventricular cardiomyocytes.[32] QT prolongation reflects a decreased repolarization following depolarization of the cardiac ventricles, whereas ST height abnormality provides a measure of ischemic injury. In acute myocardial infarction, ST-segment depression is often seen in leads opposite to the affected area. The ST segment may be depressed in subendocardial infarction (an infarction that does not involve the full thickness of the left ventricle) as in case of ISO induced MI, or during an exercise stress test in a patient suspected of having coronary ischemia. .
Cardiac biomarkers such as CK-MB and LDH have long been used as markers of cardiac injury and inflammation. We correlated our drug efficacy in terms of levels of these marker enzymes in serum. Animals treated with ISO showed marked increase in serum levels of these enzymes. When pre-treated with t-TUCB, for a period of 2 weeks, serum levels of these enzymes were not significantly elevated as that seen with ISO alone. Previous studies have suggested that the progressive deterioration in cardiac function post MI is associated with cellular apoptosis. In our study, t-TUCB has shown beneficial effects in preventing cellular apoptosis by decreasing Ca2+ overloading in cells and preventing the formation of oxygen free radicals. Our results show a dose dependent decrease in formation of lipid peroxide and increase in catalase and SOD levels. Also, protection against injury was seen by reduced infarct size in t-TUCB treated hearts at all doses.
Conclusions
Electrocardiographic, biochemical and histological changes associated with ISO treatment is well co-related in this study and treatment with our inhibitor, t-TUCB was able to protect the ischemic heart against all of these changes. Hence, the use of sEHIs appears to be an attractive opportunity to prevent post-ischemic injury.. In conclusion, our findings provide a strong evidence for in vivo efficacy of newer soluble epoxide hydrolase inhibitor, t-TUCB, in ameliorating myocardial ischemic injury.
Acknowledgments
Funding This work was partially supported by National Institute of Environmental Health Sciences (NIEHS) grant. [Grant number: ES002710]. Bruce D Hammock is a George and Judy Marcus senior fellow of the American Asthma Foundation.
Footnotes
Conflict of Interest: Sung Hee Hwang and Bruce D Hammock are authors on University of California patents on soluble epoxide hydrolase inhibitors.
References
- 1.Gaziano T, et al. Cardiovascular Disease. In: Jamison DT, et al., editors. Disease Control Priorities in Developing Countries. Washington (DC): 2006. [Google Scholar]
- 2.Toldo S, et al. Inhibition of apoptosis signal-regulating kinase 1 reduces myocardial ischemia-reperfusion injury in the mouse. J Am Heart Assoc. 2012;5:e002360. doi: 10.1161/JAHA.112.002360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Penna C, et al. Cardioprotection: a radical view Free radicals in pre and postconditioning. Biochim Biophys Acta. 2009;7:781–793. doi: 10.1016/j.bbabio.2009.02.008. [DOI] [PubMed] [Google Scholar]
- 4.Hoffman JW, Jr., et al. Myocardial reperfusion injury: etiology, mechanisms, and therapies. J Extra Corpor Technol. 2004;4:391–411. [PubMed] [Google Scholar]
- 5.Xu X, et al. The roles of CYP450 epoxygenases and metabolites, epoxyeicosatrienoic acids, in cardiovascular and malignant diseases. Advanced drug delivery reviews. 2011;8:597–609. doi: 10.1016/j.addr.2011.03.006. [DOI] [PubMed] [Google Scholar]
- 6.Nithipatikom K, Gross GJ. Review article: epoxyeicosatrienoic acids: novel mediators of cardioprotection. J Cardiovasc Pharmacol Ther. 2010;2:112–119. doi: 10.1177/1074248409358408. [DOI] [PubMed] [Google Scholar]
- 7.Gross GJ, et al. Mechanisms by which epoxyeicosatrienoic acids (EETs) elicit cardioprotection in rat hearts. J Mol Cell Cardiol. 2007;3:687–691. doi: 10.1016/j.yjmcc.2006.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lu T, et al. Stereospecific activation of cardiac ATP-sensitive K(+) channels by epoxyeicosatrienoic acids: a structural determinant study. Mol Pharmacol. 2002;5:1076–1083. doi: 10.1124/mol.62.5.1076. [DOI] [PubMed] [Google Scholar]
- 9.Lu T, et al. Activation of ATP-sensitive K(+) channels by epoxyeicosatrienoic acids in rat cardiac ventricular myocytes. J Physiol. 2001;(Pt 3):811–827. doi: 10.1111/j.1469-7793.2001.00811.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Theroux P. Protection of the myocardial cell during ischemia. Am J Cardiol. 1999;10A:3G–9G. doi: 10.1016/s0002-9149(99)00246-5. [DOI] [PubMed] [Google Scholar]
- 11.Gross GJ, Auchampach JA. Role of ATP dependent potassium channels in myocardial ischaemia. Cardiovasc Res. 1992;11:1011–1016. doi: 10.1093/cvr/26.11.1011. [DOI] [PubMed] [Google Scholar]
- 12.Liu L, et al. Epoxyeicosatrienoic acids attenuate reactive oxygen species level, mitochondrial dysfunction, caspase activation, and apoptosis in carcinoma cells treated with arsenic trioxide. J Pharmacol Exp Ther. 2011;2:451–463. doi: 10.1124/jpet.111.180505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhao G, et al. Epoxyeicosatrienoic acids protect rat hearts against tumor necrosis factor-alpha-induced injury. J Lipid Res. 2012;3:456–466. doi: 10.1194/jlr.M017319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yan G, et al. Activation of sphingosine kinase-1 mediates induction of endothelial cell proliferation and angiogenesis by epoxyeicosatrienoic acids. Cardiovasc Res. 2008;2:308–314. doi: 10.1093/cvr/cvn006. [DOI] [PubMed] [Google Scholar]
- 15.Fleming I. Epoxyeicosatrienoic acids, cell signaling and angiogenesis. Prostaglandins Other Lipid Mediat. 2007;1-4:60–67. doi: 10.1016/j.prostaglandins.2006.05.003. [DOI] [PubMed] [Google Scholar]
- 16.Spector AA, et al. Epoxyeicosatrienoic acids (EETs): metabolism and biochemical function. Prog Lipid Res. 2004;1:55–90. doi: 10.1016/s0163-7827(03)00049-3. [DOI] [PubMed] [Google Scholar]
- 17.Shen HC. Soluble epoxide hydrolase inhibitors: a patent review. Expert Opin Ther Pat. 2010;7:941–956. doi: 10.1517/13543776.2010.484804. [DOI] [PubMed] [Google Scholar]
- 18.Anandan SK, et al. Non-urea functionality as the primary pharmacophore in soluble epoxide hydrolase inhibitors. Bioorg Med Chem Lett. 2009;4:1066–1070. doi: 10.1016/j.bmcl.2009.01.013. [DOI] [PubMed] [Google Scholar]
- 19.Hwang SH, et al. Orally bioavailable potent soluble epoxide hydrolase inhibitors. J Med Chem. 2007;16:3825–3840. doi: 10.1021/jm070270t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim IH, et al. Design, synthesis, and biological activity of 1,3-disubstituted ureas as potent inhibitors of the soluble epoxide hydrolase of increased water solubility. J Med Chem. 2004;8:2110–2122. doi: 10.1021/jm030514j. [DOI] [PubMed] [Google Scholar]
- 21.Watanabe T, et al. In vitro metabolism of the mammalian soluble epoxide hydrolase inhibitor, 1-cyclohexyl-3-dodecyl-urea. Drug Metab Dispos. 2003;7:846–853. doi: 10.1124/dmd.31.7.846. [DOI] [PubMed] [Google Scholar]
- 22.Wagner K, et al. Comparative efficacy of 3 soluble epoxide hydrolase inhibitors in rat neuropathic and inflammatory pain models. Eur J Pharmacol. 2013;1-3:93–101. doi: 10.1016/j.ejphar.2012.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu JY, et al. Pharmacokinetic optimization of four soluble epoxide hydrolase inhibitors for use in a murine model of inflammation. Br J Pharmacol. 2009;2:284–296. doi: 10.1111/j.1476-5381.2008.00009.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ohkawa H, et al. Assay for lipid peroxides in animal tissues by thiobarbituric acid reaction. Anal Biochem. 1979;2:351–358. doi: 10.1016/0003-2697(79)90738-3. [DOI] [PubMed] [Google Scholar]
- 25.Beers RF, Jr., Sizer IW. A spectrophotometric method for measuring the breakdown of hydrogen peroxide by catalase. J Biol Chem. 1952;1:133–140. [PubMed] [Google Scholar]
- 26.Saggu H, et al. A selective increase in particulate superoxide dismutase activity in parkinsonian substantia nigra. J Neurochem. 1989;3:692–697. doi: 10.1111/j.1471-4159.1989.tb11759.x. [DOI] [PubMed] [Google Scholar]
- 27.Liu JY, et al. Substituted phenyl groups improve the pharmacokinetic profile and anti-inflammatory effect of urea-based soluble epoxide hydrolase inhibitors in murine models. Eur J Pharm Sci. 2013;4-5:619–627. doi: 10.1016/j.ejps.2012.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Guedes AG, et al. Use of a soluble epoxide hydrolase inhibitor as an adjunctive analgesic in a horse with laminitis. Vet Anaesth Analg. 2013;4:440–448. doi: 10.1111/vaa.12030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yogeeta SK, et al. Synergistic interactions of ferulic acid with ascorbic acid: its cardioprotective role during isoproterenol induced myocardial infarction in rats. Mol Cell Biochem. 2006;1-2:139–146. doi: 10.1007/s11010-006-2494-0. [DOI] [PubMed] [Google Scholar]
- 30.Khatua TN, et al. Garlic provides protection to mice heart against isoproterenol-induced oxidative damage: role of nitric oxide. Nitric Oxide. 2012;1:9–17. doi: 10.1016/j.niox.2012.03.004. [DOI] [PubMed] [Google Scholar]
- 31.Gupta P, et al. Cardioprotective effect of ritonavir, an antiviral drug, in isoproterenol induced myocardial necrosis: a new therapeutic implication. J Transl Med. 2013:80. doi: 10.1186/1479-5876-11-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Batchu SN, et al. Epoxyeicosatrienoic acid prevents postischemic electrocardiogram abnormalities in an isolated heart model. J Mol Cell Cardiol. 2009;1:67–74. doi: 10.1016/j.yjmcc.2008.09.711. [DOI] [PubMed] [Google Scholar]