

Abstract
Background
Gastric aspiration is a significant cause of acute lung injury and acute respiratory distress syndrome. Environmental risk factors, such as a diet high in pro-inflammatory advanced glycation end-products (AGEs), may render some patients more susceptible to lung injury after aspiration. We hypothesized that high dietary AGEs increase its pulmonary receptor, RAGE, producing an amplified pulmonary inflammatory response in the presence of high mobility group box 1 (HMGB1), a RAGE ligand and an endogenous signal of epithelial cell injury after aspiration.
Materials and Methods
CD-1 mice were fed either a low AGE (LAGE) or high AGE (HAGE) diet for 4 weeks. After aspiration injury with acidified small gastric particles, bronchial alveolar lavage (BAL) and whole-lung tissue samples were collected at 5 minutes, 1 hour, 5 hours, and 24 hours post-injury. RAGE, soluble RAGE (sRAGE), HMGB1, cytokine and chemokine concentrations, albumin levels, neutrophil influx, and lung myeloperoxidase (MPO) activity were measured.
Results
We observed that HAGE-fed mice exhibited greater pulmonary RAGE levels before aspiration and increased BAL sRAGE levels after aspiration compared to LAGE-fed mice. Lavage HMGB1 levels rose immediately after aspiration, peaking at 1 hour, and strongly correlated with sRAGE levels in both dietary groups. HAGE-fed mice demonstrated higher cytokine and chemokine levels with increased pulmonary MPO activity over 24 hours versus LAGE-fed mice.
Conclusions
This study indicates that high dietary AGEs can increase pulmonary RAGE, augmenting the inflammatory response to aspiration in the presence of endogenous damage signals such as HMGB1.
Keywords: Advanced glycation end-products, receptor for advanced glycation end-products, high mobility group box 1, gastric aspiration, acute lung injury
Introduction
Aspiration is an important cause of direct acute lung injury (ALI) and acute respiratory distress syndrome (ARDS), affecting thousands of patients each year [1–4]. In a recent multicenter study to accurately identify patients at high risk for ALI, aspiration was one of the strongest predictors of ALI development [5]. Gastric aspiration is of particular concern in the trauma and critical care setting. These patients frequently suffer from altered levels of consciousness and impaired airway protection reflexes, two major risk factors for aspiration. As current therapeutic strategies for ALI/ARDS remain limited to mostly supportive care, focus has turned towards preventative practices [6]. From recent work, we identified a potentially modifiable risk factor, dietary advanced glycation end-products (AGEs). We demonstrated that the pre-injury accumulation of high dietary AGEs led to a more pronounced inflammatory lung injury after gastric aspiration [7].
AGEs are proteins and lipids that are non-enzymatically glycated and oxidized in the presence of reducing sugars. These glycated structures accumulate in the body with age and are abundant in processed foods. AGEs can bind and cross-link extracellular structures, altering tissue function, and activating pro-inflammatory pathways through engagement of the AGE receptor, RAGE [8, 9]. RAGE is a multi-ligand, pattern recognition receptor (PRR) that is abundantly expressed in the lung on type I alveolar epithelial cells and is upregulated in the presence of its ligands [10].
Based on our previous study, we proposed that the AGE-RAGE axis is important to moderating the pulmonary inflammatory response and subsequent lung injury after aspiration [7]. Schmidt et al. hypothesized a “two-hit model” of tissue inflammation and injury mediated by RAGE and its ligands[10]. In our aspiration model, increased expression of RAGE in the lung, upregulated by high dietary AGEs, constitutes the ‘first hit’. The ‘second hit’ comes from the low-pH cellular injury of gastric aspiration, releasing a variety of endogenous danger signals known as damage associated molecular patterns (DAMPs) [11].
DAMPS are endogenous molecules that are released upon cell death, stimulating a number of PRRs to activate the innate immune system. We recently reported on the early release of mitochondrial DNA (a DAMP) in a murine model of acid aspiration [12]. High mobility group box 1 (HMGB1) is another DAMP released from its normally intranuclear location upon cell necrosis. HMGB1 is a prototypical RAGE ligand, acting as a pro-inflammatory cytokine through RAGE engagement. While HMGB1 can directly stimulate pulmonary inflammation, our understanding of its interaction with RAGE in ALI is limited and it has not been studied in aspiration-induced lung injury [11, 13].
The purpose of this study was to investigate the mechanism by which high dietary AGEs amplify the acute inflammatory response to gastric aspiration. We hypothesized that high dietary AGEs increase pulmonary RAGE, and in the presence of HMGB1 released from injured respiratory epithelial cells after aspiration, is associated with an enhanced pro-inflammatory state. We primarily focused on defining the kinetics of RAGE, its soluble receptor (sRAGE), and HMGB1 in the early phase of acute lung injury, for which a paucity of information is available. Using our unique in vivo murine model of acute aspiration pneumonitis, which combines small gastric particles and acid to best simulate clinical aspiration events, we hope these results provide new insight into the complex mechanics of this highly morbid lung injury.
Materials and Methods
Experimental animal model
Male CD-1 mice (4 weeks old) were purchased and housed according to protocols approved by the Institutional Animal Care and Use Committees of the University at Buffalo and the Veterans Administration Western New York Healthcare System. Mice were randomized to a group receiving either a low AGE (LAGE) or high AGE (HAGE) diet ad libitum for 4 weeks. The details of the diet preparation are described in our previous publication [7]. After 4 weeks, the animals in each dietary designation were randomly assigned to one of five groups (n=9–15/group): uninjured control animals or gastric aspiration injury with samples collected at 5 minutes, 1 hour, 5 hours, or 24 hours post-injury. Injury and sample collection were performed under general anesthesia using isoflurane in 100% oxygen, delivered via nose cone.
Gastric-aspiration induced lung injury
The aspirate injury solution was a combination of 20 mg/ml of small gastric food particles and hydrochloric acid, adjusted to a pH of 1.25. The small, non-acidified gastric particles were obtained from the stomachs of necropsied CD-1 mice and prepared according to previously published protocol [7]. After anesthesia induction, a midline tracheotomy was performed and a 22-gauge needle containing the aspirate solution (volume of 3.6 ml/kg of body weight + 0.2 ml air bolus) was inserted into the trachea. The chest wall was then compressed and rapidly released following aspirate instillation, maximizing pulmonary distribution. Once the incision was closed and anesthesia ceased, the mice received 100% oxygen and were monitored for recovery of regular, spontaneous breathing patterns. Mice were then returned to their respective cages, breathing room air only, until their pre-specified post-injury time point when they were sacrificed under anesthesia for bronchoalveolar lavage (BAL) sampling and lung tissue harvesting.
BAL sampling and processing procedures
After puncturing the diaphragm and performing a sternotomy, the pulmonary vasculature was flushed via the right ventricle with 5 ml of Hank’s balanced salt solution. BAL was performed by instilling five 1 ml aliquots of Hank’s balanced salt solution through a tracheal cannula, and recovering the pooled aliquots with a syringe. The BAL was kept on ice until processing, beginning with centrifugation at 1,500×g for 5 min at 4°C to pellet cells. The resulting supernatant was stored at −80°C for analysis of sRAGE, HMGB1, albumin, and cytokines/chemokines. The cell pellet was then suspended in 3 ml of phosphate-buffered normal saline plus 1 ml of 0.1% sodium azide to determine the BAL-recovered number of total leukocytes using a Multisizer 3 Coulter Counter. Slides were prepared from the cell suspension and a differential count was determined by light microscopy.
Whole-lung tissue homogenate processing and MPO activity measurement
Harvested lungs were homogenized on ice with a Polytron TP-2000 tissue homogenizer (Brinkman Instruments, Westbury, NY). The homogenate was centrifuged at 40,000xg for 10 min @ 4°C and the whole-lung supernatant was aliquoted for RAGE analysis. The remaining pellet was processed for MPO extraction. After resuspending in 2 ml of phosphate buffer (0.5% hexadecyltrimethylammonium bromide and 5 mM EDTA, pH 6.0), the pellet was sonicated on ice for 1 min using a Sonifer Cell Disruptor (Branson Ultrasonics, Danbury, CT), incubated at 55°C for 2 hr and centrifuged again at 40,000×g for 10 min @ 4°C. MPO activity was determined from the resultant supernatant. 10μl of extracted sample was combined with 300μl of assay buffer (50 mM KH2PO4, 176 mM H2O2, 52.5 mM o-dianisidine dihydrochloride, pH 6.0) in a 96-well microplate with absorbance measured at 460 nm for 90 seconds at 2 second intervals on a SPECTRAmax spectrophotometer (Molecular Devices, Sunnyvale, CA). MPO activity is expressed as absorbance change per min over the linear portion of the curve.
ELISA analysis of RAGE, sRAGE, HMGB1, and cytokines/chemokines
Concentrations of sRAGE, along with various cytokines (tumor necrosis factor-alpha (TNF-a), TNF soluble receptor I (TNFsRI), interleukin 1-beta (IL-1β), and IL-6) and chemokines (keratinocyte chemoattractant (KC), macrophage inflammatory protein 2 (MIP-2), and monocyte chemoattractant protein 1 (MCP-1)) were measured in cell-free BAL by sandwich ELISA methodology. RAGE concentrations were measured in the supernatant from the whole lung tissue homogenate by this same method. HMGB1 concentrations in cell-free BAL were determined by indirect ELISA protocol. Reagents were obtained from Bethyl Laboratories (Montgomery, TX). All values are reported in picograms per milliliter unless otherwise specified.
Albumin concentrations in cell-free BAL
As an index of pulmonary injury and alveolar-capillary barrier dysfunction, albumin concentrations were measured in cell-free BAL. Sandwich ELISA methodology was employed using reagents from BD Biosciences Pharminegen (San Diego, CA). Values are reported in micrograms per milliliter.
Statistical analysis
Data are reported as mean ± SEM unless otherwise specified. Data were analyzed by two-way ANOVA to assess the relative contribution of aspiration, diet, and their possible interaction on indices of inflammation and injury. Post-hoc multiple comparisons (Tukey) were used to analyze group differences between multiple time points. Pearson correlation analysis was applied to assess the association between sRAGE and HMGB1 at each time point. All statistical analysis was performed using GraphPad Prism 6 (GraphPad Software, La Jolla, CA). Significance was set at P < 0.05.
Results
Pulmonary RAGE after 4 weeks of HAGE diet
To assess the ability of dietary AGEs to increase pulmonary RAGE, we assayed RAGE concentrations in the whole-lung homogenate supernatant. We initially focused on two times post-aspiration: the peak of the early inflammatory response (5 hours) and the early phase of injury resolution (24 hours) [14, 15]. Uninjured mice fed a HAGE diet demonstrated increased pulmonary RAGE levels compared to mice fed a LAGE diet (555.1 ± 61.6 vs. 393.1 ± 62.6 μg/ml; P < 0.05) (Fig. 1). A similar relationship was present at 5 hours post-aspiration (553.4 ± 71.1 vs. 406.2 ± 37.6 μg/ml; P < 0.05), but not at 24 hours. Pulmonary RAGE levels did not significantly change over time.
Figure 1. Whole-lung homogenate RAGE concentrations in mice fed either a LAGE or HAGE diet for 4 weeks.

Mice fed a HAGE diet showed elevated pulmonary RAGE levels compared with mice fed a LAGE diet, both prior to injury and at 5 hr post-injury (*P < 0.05).
Release of HMGB1 over 24 hours after gastric aspiration
To identify HMGB1 and define its relationship to the inflammatory response after gastric aspiration, we assayed for cell-free BAL levels of HMGB1. Time points at 5 minutes and 1 hour after injury were added to identify the anticipated early, transient release of HMGB1 from injured respiratory cells. Low levels of HMGB1 were detectable in the airways of uninjured mice, but did not differ by dietary group. 5 minutes after injury, HMGB1 levels increased markedly in both dietary groups versus uninjured controls (P < 0.0001) (Fig. 2). No dietary effect was observed. At 1 hour, HMGB1 levels in the HAGE-fed mice were significantly lower than in the LAGE-fed mice (729.9 ± 85.9 vs. 1176 ± 181.1 pg/ml; P < 0.05). HMGB1 levels did not differ by diet at either 5 hours or 24 hours after injury. However, after returning to near basal levels at 5 hours, HMGB1 levels were elevated again at 24 hours versus uninjured controls (P < 0.05).
Figure 2. HMGB1 levels in cell-free BAL over 24 hours after gastric aspiration in mice fed either a LAGE or HAGE diet for 4 weeks.

Aspiration injury led to a rise in HMGB1 levels in both dietary groups at 5 min (P < 0.0001 vs. uninjured controls). HMGB1 levels were significantly higher in the LAGE-fed mice at 1 hr compared to HAGE-fed mice (*P < 0.05). After returning to basal levels at 5 hr, HMGB1 increased again at 24 hours in both diet groups (P < 0.05 vs. uninjured controls).
sRAGE levels in cell-free BAL
sSoluble RAGE (sRAGE) is a biomarker for acute lung injury and can act as a decoy receptor for RAGE ligands[16]. At 5 minutes after aspiration injury, BAL sRAGE levels increased in both diet groups versus uninjured controls (P < 0.0001), with a greater increase present in the HAGE group compared to the LAGE group (1536.0 ± 324.7 vs. 841.6 ± 114.0 ng/ml; P < 0.05) (Fig. 3). No diet effect was present at 1 hour. By 5 hours, sRAGE levels in both groups were much lower compared to the initial rise observed immediately upon injury, but greater still in the HAGE group (589.0 ± 130.4 vs. 238.4 ± 76.1 ng/ml; P < 0.05). 24 hours after aspiration, sRAGE levels in the HAGE group were trending higher compared to the LAGE group (P = 0.089).
Figure 3. Soluble RAGE (sRAGE) levels in cell-free BAL over 24 hours after gastric aspiration in mice fed either a LAGE or HAGE diet for 4 weeks.

sRAGE levels increased in both groups at 5 minutes post-injury (P < 0.0001 vs. uninjured controls), but to a greater degree in HAGE-fed mice vs. LAGE-fed mice (*P < 0.05). By 5 hr, sRAGE levels decreased in both groups, though still greater in the HAGE-fed mice (*P < 0.05).
We also assessed the correlation between sRAGE and HMGB1 at each time point by dietary group. A strong positive association was evident at 5 minutes (LAGE: r = 0.87; P < 0.01 and HAGE: r = 0.72; P < 0.05) and at 1 hour after aspiration (LAGE: r = 0.93; P < 0.01 and HAGE: r = 0.75; P < 0.05) (Fig. 4). At 5 hours, only the HAGE-fed mice showed a significant positive correlation between sRAGE and HMGB1 levels (r = 0.90; P < 0.001). No significant correlation was evident at 24 hours.
Figure 4. Correlation between sRAGE and HMGB1 levels in BAL at 1 hour after gastric aspiration.

sRAGE levels strongly correlated with HMGB1 levels in both dietary groups at 1 hr post-injury (LAGE: r = 0.93; P < 0.01 and HAGE: r = 0.75; P < 0.05).
Cytokine production over 24 hours after gastric aspiration, differentiated by diet
We observed no effect of dietary AGE content on pro-inflammatory cytokine levels of either IL-1β or IL-6 in our 24 hour timeline (Fig. 5). However, IL-1β levels were significantly elevated in injured mice at 1 hour post-injury compared to uninjured controls (P < 0.01) with no significant differences between injured groups observed at 5 hours and 24 hours. IL-6 levels were greater in injured mice at 5 hours compared to 1 hour post-injury (P < 0.0001) and uninjured controls (P < 0.0001). IL-6 levels peaked at 5 hours post-injury, significantly decreasing by 24 hours (P < 0.01).
Figure 5. BAL concentrations of IL-1β and IL-6 after gastric aspiration in mice fed a LAGE or HAGE diet.
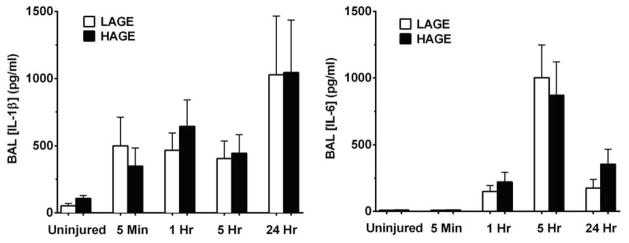
Dietary AGE content had no effect on IL-1β or IL-6 concentrations at any time post-aspiration. Pro-inflammatory cytokine levels were significantly greater in injured mice starting at 1 hr compared to uninjured controls (IL-1β: P < 0.01, IL-6: P < 0.0001)
TNF-alpha levels in the HAGE-fed mice were greater at 1 hour (266.1 ± 66.3 vs. 123.3 ± 18.1 pg/ml; P < 0.05) and 24 hours (138.3 ± 12.4 vs. 92.04 ± 8.12 pg/ml; P < 0.01) compared to LAGE-fed mice (Fig. 6). BAL levels of the soluble TNF receptor I (TNFsRI) were also greater after aspiration in the HAGE-fed mice at 5 hours (754.7 ± 93.3 vs. 445.6 ± 32.1 pg/ml; P < 0.01) and 24 hours (1171.0 ± 62.7 vs. 705.6 ± 126.7 pg/ml; P < 0.01) (Fig. 7).
Figure 6. TNF-α levels in cell-free BAL over 24 hours after gastric aspiration in mice fed either a LAGE or HAGE diet.

TNF-α levels significantly increased in mice fed a HAGE vs. LAGE diet at 1 hr after aspiration (*P < 0.05). Levels peaked at 5 hr, showing no statistically significant dietary effect. At 24 hr, though near basal levels, TNF-α was greater in the HAGE vs. LAGE diet mice (**P < 0.01).
Figure 7. TNFsRI levels in cell-free BAL over 24 hours after gastric aspiration in mice fed either a LAGE or HAGE diet.

TNFsRI levels were significantly greater in the HAGE vs. LAGE diet mice at both 5 hr and 24 hr after aspiration (**P < 0.01).
Chemokine production over 24 hours after gastric aspiration, differentiated by diet
Chemokine-mediated leukocyte recruitment is a critical event in the pathogenesis of ALI [17]. HAGE-fed mice exhibited increased BAL levels of the neutrophil CXC chemokine, MIP-2, at 1 hour (528.9 ± 159.0 vs. 179.8 ± 40.0 pg/ml; P < 0.05) and 5 hours post-injury (1386.0 ± 173.5 vs. 849.5 ± 127.3 pg/ml; P <0.05) compared to LAGE-fed mice (Fig. 8). Concentrations of the CXC PMN chemokine, KC, and of the monocyte chemoattractant, MCP-1, were elevated 1 hour after gastric aspiration compared to uninjured controls (P < 0.001), but no dietary effect was observed at any one time (Fig. 9). KC levels peaked at 5 hours, while MCP-1 levels were greatest at 24 hours.
Figure 8. MIP-2 levels in cell-free BAL over 24 hours after gastric aspiration in mice fed either a LAGE or HAGE diet.

HAGE-fed mice demonstrated greater MIP-2 levels than LAGE-fed mice at 1 hr and 5 hr post-injury (*P < 0.05). Levels returned to near baseline at 24 hr in both groups.
Figure 9. BAL concentrations of KC and MCP-1 after gastric aspiration in mice fed a LAGE or HAGE diet.
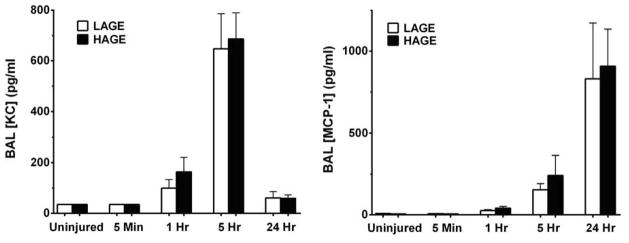
KC levels were not affected by dietary AGEs, significantly increasing by 1 hr (P < 0.001 vs. uninjured controls) and peaking at 5hr after injury in both groups. MCP-1 levels also showed no dietary effect, significantly increasing by 5 hr (P < 0.0001 vs. uninjured controls).
BAL albumin levels as a marker for injury to the alveolar-capillary barrier
Impairment of the alveolar-capillary barrier leading to pulmonary edema is a hallmark of acute lung injury, which we assessed by BAL albumin levels after gastric aspiration. Beginning at 5 minutes post-injury, we observed a greater increase in airway albumin levels of injured versus uninjured HAGE-fed mice (277.9 ± 77.6 vs. 116.5 ± 10.9 μg/ml; P < 0.05), a relationship absent in the LAGE diet group (Fig. 10). By 1 hour, albumin levels had increased in both dietary groups compared to uninjured controls (P < 0.0001). Thereafter, albumin levels were significantly greater at 5 hours and 24 hours compared to 1 hour post-injury. At 5 hours, BAL albumin levels were trending higher in the HAGE-fed mice (P = 0.087), with no dietary effect evident by 24 hours.
Figure 10. Albumin concentration in cell-free BAL over 24 hours after gastric aspiration in mice fed either a LAGE or HAGE diet.

As a marker of alveolar-capillary barrier dysfunction, BAL albumin concentrations significantly increased in both diet groups by 1 hr (P < 0.0001). At 5hr, albumin was trending toward greater concentrations in the HAGE-fed vs. LAGE-fed mice (#P = 0.0874). Albumin levels were also higher in both groups at 5 hr and 24 hr compared to 1 hr post-aspiration (LAGE: P <0.05 at 5 hr, P <0.001 at 24 hr; HAGE: P <0.0001 at 5 hr, P <0.001 at 24hr). No significant difference in albumin was evident between 5 hr and 24 hr.
Pulmonary neutrophil sequestration
Neutrophil accumulation and activation in the lung is a central process defining aspiration-induced lung injury. 1 hour after aspiration, BAL neutrophil counts increased in both dietary groups versus uninjured controls (LAGE: 18,986 ± 5,486 vs. 1,593 ± 1,174; HAGE: 17,067 ± 5193 vs. 60.91 ± 60.91 cell/ml; P < 0.01) (Fig. 11). Neutrophil counts in the HAGE-fed mice significantly increased from 1 hour to 5 hours (P < 0.01), but did not significantly differ from 5 hours to 24 hours after aspiration. In the LAGE-fed mice, neutrophil counts demonstrated a similar relationship, with a significant increase from 1 hr to 5 hr (P < 0.01) only.
Figure 11. Neutrophil counts in BAL over 24 hours after gastric aspiration in mice fed either a LAGE or HAGE diet.

PMN counts significantly increased in both diet groups by 1 hr (P < 0.01 vs. uninjured controls). Though further elevated by 5 hr post-injury (P < 0.01 vs. 1 hr), PMN counts were not affected by diet.
MPO activity in lung tissue over 24 hours
As a marker for neutrophil influx into the lung tissue after aspiration, MPO activity in the lung homogenate was assayed. No difference in MPO activity was observed between the uninjured dietary groups (Fig. 12). 1 hour after aspiration, MPO activity increased in both groups versus uninjured controls (P < 0.0001), but to a greater extent in the HAGE-fed mice compared to LAGE-fed mice (83.2 ± 10.8 vs. 57.12 ± 7.8 absorbance units/min per 10 μl; P < 0.05). We also observed this increased MPO activity in the HAGE-fed mice at 5 hours after aspiration (241.3 ± 28.3 vs. 180.6 ± 25.1 absorbance units/min per 10 μl; P < 0.05). No dietary effect on MPO activity was observed by 24 hours.
Figure 12. Myeloperoxidase activity in whole-lung homogenate over 24 hours after gastric aspiration in mice fed either a LAGE or HAGE diet.

Lung MPO activity significantly increased by 1 hr in both diet groups (P < 0.0001 vs. uninjured controls). HAGE-fed mice exhibited even greater MPO activity at 1 hr and 5 hr vs. LAGE-fed mice (*P < 0.05)
Discussion
Understanding the distinct mechanisms contributing to the inflammatory response following aspiration-induced lung injury is an important step in developing new preventative and therapeutic strategies for ALI/ARDS. Our findings indicate that high dietary AGE intake is associated with elevated pulmonary RAGE levels, establishing the ‘first hit’ of RAGE-ligand engagement. The ‘second-hit’ came after aspiration injury with a marked rise in HMGB1 levels, a signal of tissue injury and a RAGE ligand. What followed was a more pronounced inflammatory response in the HAGE-fed mice, evidenced by increased cytokine/chemokine levels and enhanced neutrophil activity. These findings support our hypothesis that pulmonary RAGE levels are increased in mice fed a high AGE diet prior to aspiration, rendering them susceptible to an exacerbated inflammatory response following low pH cellular injury and HMGB1 release into the airways.
The balance between RAGE, sRAGE, and RAGE ligands is complex, given the defining characteristic of RAGE as a pattern recognition receptor (PRR) with multiple roles in tissue homeostasis, inflammation, and repair [18]. Accumulations of its ligands can upregulate RAGE expression, increasing the potential for RAGE-mediated signaling through interaction with DAMPs after injury [10, 19–21]. Consistent with this observation, we found that pulmonary RAGE levels were increased in mice fed a high AGE diet for 4 weeks. We speculate that this priming event of AGE accumulation and RAGE upregulation is the ‘first-hit’ in the previously discussed ‘two-hit’ model of tissue inflammation and injury. The ‘second-hit’ is the low-pH injury of gastric aspiration, releasing DAMPs that can interact with RAGE and other PRRs [16, 22].
HMGB1 is a DAMP that is passively released from damaged and necrotic cell and is capable of initiating an innate immune response by binding and activating RAGE, as well as the Toll-like receptors 2, 4 and 9 [23–25]. HMGB1 is also actively secreted from immune cells, acting as a cytokine and late inflammatory mediator in models of endotoxin- and hemorrhagic shock-induced ALI [26]. Here, our study focused on the initial release of HMGB1 from injured cells within the first hour prior to the acute pro-inflammatory response to gastric aspiration, which peaks at 4 to 6 hours post-insult in our model [3]. Our findings demonstrate that HMGB1 levels markedly rise immediately after caustic cellular damage, reaching peak levels at 1 hour after aspiration, and then decreasing to near basal levels at 5 hours. The early increase in this DAMP indicates passive release of HMGB1 from respiratory epithelial cells following low-pH damage. HMGB1 is therefore available immediately following acid aspiration to ligate RAGE and other PRRs, activating pro-inflammatory signaling pathways [13, 27]. At 24 hours, we also identified an increase in HMGB1 levels in both dietary groups compared to uninjured controls. This increase in HMGB1 most likely represents its active secretion as a late mediator of inflammation [13].
Additionally, we observed a significant rise in HMGB1 in the LAGE-fed mice at 1 hour. This may be explained by two mechanisms. Firstly, HMGB1 ligation with membrane-bound RAGE, which was increased in HAGE-fed mice, would not be detected in the cell-free BAL of these animals. Secondly, increased HMGB1 oxidation and clearance may also occur in the lungs of HAGE-fed mice. Previous study has shown that the extracellular environment and ligand binding can alter the oxidation and clearance kinetics of HMGB1 [28]. The presence of greater amounts of sRAGE in the HAGE-fed mice may have facilitated enhanced HMGB1 clearance.
Soluble RAGE (sRAGE) is cleaved from the membrane-bound RAGE following ligation. This decoy receptor is a biomarker for both type I alveolar cell injury and the severity of ALI [29–31]. Levels of sRAGE also correlate well with AGE levels, reflecting the degree of RAGE expression in the respiratory epithelium [32]. Our results show that BAL levels of sRAGE rise at 5 minutes after aspiration, much like HMGB1, but to a greater concentration in the high-AGE diet mice. These findings suggest that increased pulmonary RAGE expression secondary to high dietary AGEs is responsible for the enhanced sRAGE generation. Increased sRAGE levels in the high-AGE mice were also observed at 5 hours following acid aspiration during the peak acute inflammatory response as assessed by increased cytokine/chemokine levels and lung tissue MPO activity. Moreover, the strong, positive correlation between HMGB1 and sRAGE in the early time period following acid aspiration suggests their possible interaction. HMGB1 released from injured cells is available to bind to both RAGE, activating inflammatory signaling cascades, as well as the ligation-generated sRAGE that then functions as a decoy receptor to limit HMGB1-RAGE signaling [16, 33]. However, the RAGE to sRAGE ratio clearly favored an enhanced pro-inflammatory state during the acute phase of aspiration injury. To our knowledge, this is the first study examining the axis of RAGE, sRAGE, and HMGB1 production following low pH aspiration-induced lung injury. A number of other studies have examined the role of these moieties in ALI/ARDS, but not following the direct low pH insult to the airways observed after acid aspiration. Additionally, the kinetics of RAGE, sRAGE, and HMGB1 during the acute phase of direct cell injury and innate immunity activation has not been previously described [34–39].
The inflammatory response to gastric aspiration has been studied extensively in our lab [15, 40–42]. In CD-1 mice, BAL levels of TNF-α and MIP-2 measured 5 hours after aspiration and TNF-α and MCP-1 measured 24 hours after aspiration are the inflammatory mediators that best correlate with the degree of pulmonary inflammation and injury [15]. From our results, TNF-α and its soluble type I surface receptor (TNFsRI) are elevated in the airways of HAGE-fed mice over the 24 hours following aspiration compared to LAGE-fed mice. Although, TNF-α levels alone did not demonstrate a dietary effect at 5 hours peak injury time, which is consistent with our previous findings [7]. Including the level of soluble receptors with the early TNF-α response provided a more dynamic assessment of this early pro-inflammatory cytokine in mediating aspiration-induced ALI. TNFsRI in particular has been shown to play a crucial role in alveolar epithelial dysfunction after acid aspiration, while also blocking and attenuating the biologic effects of excessive TNF-α [43, 44]. This may help explain our observation of TNF-α levels at 5 hours following low pH gastric aspiration, owing to a more pronounced cytoprotective response by TNFsRI in the HAGE-fed mice.
The neutrophil murine chemoattractants that are IL-8 homologues, KC and MIP-2, were elevated by 1 hour after aspiration in both diet groups. However, HAGE-fed mice demonstrated greater MIP-2 production during the acute phase of inflammation. The early appearance of these CXC chemokines appropriately preceded the large neutrophil influx observed at 5 hours. The peak levels of KC and MIP-2 observed at 5 hours is consistent with our prior work that the early acute inflammatory response occurs from 4 to 6 hours post-insult [41]. Additionally, these findings support early RAGE signaling in resident airway macrophages and respiratory epithelium by such inflammatory mediators as HMGB1 [11, 45]. However, compared to our previous study, we did not observe lower levels of the assayed chemokines in the HAGE mice compared to the LAGE animals following aspiration [7]. Different concentrations of gastric food particles in the aspirate though can produce varied levels of chemokines in mice [41].
Myeloperoxidase activity is both a marker of neutrophil presence and activation in the lung airways and parenchyma [46]. MPO activity in the lung tissue is increased beginning at 1 hour following low pH gastric aspiration, coincident with the arrival of increased neutrophil numbers into the airways. Furthermore, greater MPO activity is observed in the HAGE-fed mice at both 1 hour and 5 hours. These results are consistent with an earlier, augmented inflammatory response putatively caused by the increased release of HMGB1 and its subsequent ligation with increased pulmonary RAGE from the high AGE diet. Although we did not observe a corresponding increase in BAL neutrophil counts in our HAGE-fed mice, lung MPO activity does correlate well with pulmonary leukocyte infiltration observed in acute lung injury [47].
One limitation of this study was employment of a reduced aspiration insult specifically to improve survival at 24 hours following the low pH insult. This change in the amount of aspirated material resulted in a less pronounced injury with somewhat different variations in the chemokine response than was previously observed [7]. As a result, differences in the neutrophil counts are not as pronounced either in this model. However, MPO activity has proven to be a more sensitive marker for neutrophil presence in this decreased aspiration injury, especially in the first hour. Additionally, we did not evaluate gas exchange following the aspiration insult. This functional parameter, when combined with measures of pulmonary edema, neutrophil sequestration, hypoxemia and histopathology, provides a more complete picture of the pulmonary inflammatory injury [48]. Measuring impaired oxygenation may be particularly important in further assessing our model of gastric aspiration, which was affected by changes to gastric particle concentration. However, the primary objective of this study was to assess temporal interaction between dietary AGEs, RAGE, and HMGB1 as an initial step to understanding our previous observations. Finally, we did not definitively assess the causal role of pulmonary RAGE/sRAGE following low pH gastric aspiration. The use of RAGE-KO mice could be employed to establish this relationship, but again the primary focus of this study was to assess the temporal relationship between HMGB1, RAGE and sRAGE in the injured lung.
In summary, this study supports our hypothesis that high levels of dietary AGEs increase pulmonary RAGE, which when followed by HMGB1 release after gastric aspiration, is associated with an augmented acute pulmonary inflammatory response and injury. Decreasing RAGE expression through a reduction in dietary AGE consumption and accumulation may attenuate lung inflammation following low pH gastric aspiration. This is especially important in high-risk trauma and critical care patients where employing every indicated preventative strategy is crucial to their outcomes. These findings also suggest that pharmacotherapy employing sRAGE as a decoy receptor for ligands of RAGE, which can also activate other PRR pathways, may help decrease pro-inflammatory signaling and subsequent lung injury. Additional studies are warranted to further our understanding of the role RAGE and DAMP ligands play on pulmonary inflammation following direct, non-infectious lung injury as is observed with low pH gastric aspiration.
Footnotes
Authors’ contributions
PS – conception and design, data collection, analysis and interpretation, manuscript writing; WG - conception and design, data collection, analysis and interpretation, critical revision of manuscript, obtaining funding; BD – conception and design, analysis and interpretation, critical revision of manuscript, obtaining funding; BM – experimental design, data collection, analysis and interpretation, critical revision of manuscript; JH – data collection, analysis and interpretation, critical revision of manuscript; PK – experimental design, analysis and interpretation, critical revision of manuscript, obtaining funding. All authors critically read and approved the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- 1.Erickson SE, et al. Recent trends in acute lung injury mortality: 1996–2005. Crit Care Med. 2009;37(5):1574–9. doi: 10.1097/CCM.0b013e31819fefdf. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Marik PE. Pulmonary aspiration syndromes. Curr Opin Pulm Med. 2011;17(3):148–54. doi: 10.1097/MCP.0b013e32834397d6. [DOI] [PubMed] [Google Scholar]
- 3.Raghavendran K, et al. Aspiration-induced lung injury. Crit Care Med. 2011;39(4):818–26. doi: 10.1097/CCM.0b013e31820a856b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Walkey AJ, et al. Acute respiratory distress syndrome: epidemiology and management approaches. Clin Epidemiol. 2012;4:159–69. doi: 10.2147/CLEP.S28800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gajic O, et al. Early identification of patients at risk of acute lung injury: evaluation of lung injury prediction score in a multicenter cohort study. Am J Respir Crit Care Med. 2011;183(4):462–70. doi: 10.1164/rccm.201004-0549OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Litell JM, et al. Acute lung injury: prevention may be the best medicine. Respir Care. 2011;56(10):1546–54. doi: 10.4187/respcare.01361. [DOI] [PubMed] [Google Scholar]
- 7.Guo WA, et al. Effect of high advanced glycation end-product diet on pulmonary inflammatory response and pulmonary function following gastric aspiration. Shock. 2012;38(6):677–84. doi: 10.1097/SHK.0b013e318273982e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Thallas-Bonke V, et al. Targeting the AGE-RAGE axis improves renal function in the context of a healthy diet low in advanced glycation end-product content. Nephrology (Carlton) 2013;18(1):47–56. doi: 10.1111/j.1440-1797.2012.01665.x. [DOI] [PubMed] [Google Scholar]
- 9.Schmidt AM, et al. RAGE: a novel cellular receptor for advanced glycation end products. Diabetes. 1996;45(Suppl 3):S77–80. doi: 10.2337/diab.45.3.s77. [DOI] [PubMed] [Google Scholar]
- 10.Schmidt AM, et al. The multiligand receptor RAGE as a progression factor amplifying immune and inflammatory responses. J Clin Invest. 2001;108(7):949–55. doi: 10.1172/JCI14002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tolle LB, Standiford TJ. Danger-associated molecular patterns (DAMPs) in acute lung injury. J Pathol. 2013;229(2):145–56. doi: 10.1002/path.4124. [DOI] [PubMed] [Google Scholar]
- 12.Davidson BA, et al. NADPH oxidase and Nrf2 regulate gastric aspiration-induced inflammation and acute lung injury. J Immunol. 2013;190(4):1714–24. doi: 10.4049/jimmunol.1202410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Abraham E, et al. HMG-1 as a mediator of acute lung inflammation. J Immunol. 2000;165(6):2950–4. doi: 10.4049/jimmunol.165.6.2950. [DOI] [PubMed] [Google Scholar]
- 14.Kennedy TP, et al. Acute acid aspiration lung injury in the rat: biphasic pathogenesis. Anesth Analg. 1989;69(1):87–92. [PubMed] [Google Scholar]
- 15.Knight PR, et al. Progressive, severe lung injury secondary to the interaction of insults in gastric aspiration. Exp Lung Res. 2004;30(7):535–57. doi: 10.1080/01902140490489162. [DOI] [PubMed] [Google Scholar]
- 16.Guo WA, Knight PR, Raghavendran K. The receptor for advanced glycation end products and acute lung injury/acute respiratory distress syndrome. Intensive Care Med. 2012;38(10):1588–98. doi: 10.1007/s00134-012-2624-y. [DOI] [PubMed] [Google Scholar]
- 17.Abraham E. Neutrophils and acute lung injury. Crit Care Med. 2003;31(4 Suppl):S195–9. doi: 10.1097/01.CCM.0000057843.47705.E8. [DOI] [PubMed] [Google Scholar]
- 18.Maillard-Lefebvre H, et al. Soluble receptor for advanced glycation end products: a new biomarker in diagnosis and prognosis of chronic inflammatory diseases. Rheumatology (Oxford) 2009;48(10):1190–6. doi: 10.1093/rheumatology/kep199. [DOI] [PubMed] [Google Scholar]
- 19.Chen J, et al. Advanced glycation endproducts alter functions and promote apoptosis in endothelial progenitor cells through receptor for advanced glycation endproducts mediate overpression of cell oxidant stress. Mol Cell Biochem. 2010;335(1–2):137–46. doi: 10.1007/s11010-009-0250-y. [DOI] [PubMed] [Google Scholar]
- 20.Sorci G, et al. RAGE in tissue homeostasis, repair and regeneration. Biochim Biophys Acta. 2013;1833(1):101–9. doi: 10.1016/j.bbamcr.2012.10.021. [DOI] [PubMed] [Google Scholar]
- 21.Soman S, et al. A multicellular signal transduction network of AGE/RAGE signaling. J Cell Commun Signal. 2013;7(1):19–23. doi: 10.1007/s12079-012-0181-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Creagh-Brown BC, et al. The RAGE axis in systemic inflammation, acute lung injury and myocardial dysfunction: an important therapeutic target? Intensive Care Med. 2010;36(10):1644–56. doi: 10.1007/s00134-010-1952-z. [DOI] [PubMed] [Google Scholar]
- 23.Yamada S, Maruyama I. HMGB1, a novel inflammatory cytokine. Clin Chim Acta. 2007;375(1–2):36–42. doi: 10.1016/j.cca.2006.07.019. [DOI] [PubMed] [Google Scholar]
- 24.Dumitriu IE, et al. HMGB1: guiding immunity from within. Trends Immunol. 2005;26(7):381–7. doi: 10.1016/j.it.2005.04.009. [DOI] [PubMed] [Google Scholar]
- 25.Rovere-Querini P, et al. HMGB1 is an endogenous immune adjuvant released by necrotic cells. EMBO Rep. 2004;5(8):825–30. doi: 10.1038/sj.embor.7400205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fan J, et al. Hemorrhagic shock induces NAD(P)H oxidase activation in neutrophils: role of HMGB1-TLR4 signaling. J Immunol. 2007;178(10):6573–80. doi: 10.4049/jimmunol.178.10.6573. [DOI] [PubMed] [Google Scholar]
- 27.Wolfson RK, Chiang ET, Garcia JG. HMGB1 induces human lung endothelial cell cytoskeletal rearrangement and barrier disruption. Microvasc Res. 2011;81(2):189–97. doi: 10.1016/j.mvr.2010.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zandarashvili L, et al. Real-time Kinetics of High-mobility Group Box 1 (HMGB1) Oxidation in Extracellular Fluids Studied by in Situ Protein NMR Spectroscopy. J Biol Chem. 2013;288(17):11621–7. doi: 10.1074/jbc.M113.449942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Briot R, et al. Elevated levels of the receptor for advanced glycation end products, a marker of alveolar epithelial type I cell injury, predict impaired alveolar fluid clearance in isolated perfused human lungs. Chest. 2009;135(2):269–75. doi: 10.1378/chest.08-0919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Uchida T, et al. Receptor for advanced glycation end-products is a marker of type I cell injury in acute lung injury. Am J Respir Crit Care Med. 2006;173(9):1008–15. doi: 10.1164/rccm.200509-1477OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yamakawa N, et al. Proteolytic release of the receptor for advanced glycation end products from in vitro and in situ alveolar epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2011;300(4):L516–25. doi: 10.1152/ajplung.00118.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nakamura K, et al. Serum levels of soluble form of receptor for advanced glycation end products (sRAGE) are correlated with AGEs in both diabetic and non-diabetic subjects. Clin Exp Med. 2007;7(4):188–90. doi: 10.1007/s10238-007-0146-7. [DOI] [PubMed] [Google Scholar]
- 33.Bierhaus A, et al. Understanding RAGE, the receptor for advanced glycation end products. J Mol Med (Berl) 2005;83(11):876–86. doi: 10.1007/s00109-005-0688-7. [DOI] [PubMed] [Google Scholar]
- 34.Cohen MJ, et al. Early release of high mobility group box nuclear protein 1 after severe trauma in humans: role of injury severity and tissue hypoperfusion. Crit Care. 2009;13(6):R174. doi: 10.1186/cc8152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cohen MJ, et al. Early release of soluble receptor for advanced glycation endproducts after severe trauma in humans. J Trauma. 2010;68(6):1273–8. doi: 10.1097/TA.0b013e3181db323e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kim JY, et al. HMGB1 contributes to the development of acute lung injury after hemorrhage. Am J Physiol Lung Cell Mol Physiol. 2005;288(5):L958–65. doi: 10.1152/ajplung.00359.2004. [DOI] [PubMed] [Google Scholar]
- 37.Ogawa EN, et al. Contribution of high-mobility group box-1 to the development of ventilator-induced lung injury. Am J Respir Crit Care Med. 2006;174(4):400–7. doi: 10.1164/rccm.200605-699OC. [DOI] [PubMed] [Google Scholar]
- 38.Su X, et al. Receptor for advanced glycation end-products (RAGE) is an indicator of direct lung injury in models of experimental lung injury. Am J Physiol Lung Cell Mol Physiol. 2009;297(1):L1–5. doi: 10.1152/ajplung.90546.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhou Y, et al. HMGB1 and RAGE levels in induced sputum correlate with asthma severity and neutrophil percentage. Hum Immunol. 2012;73(11):1171–4. doi: 10.1016/j.humimm.2012.08.016. [DOI] [PubMed] [Google Scholar]
- 40.Davidson BA, et al. The role of tumor necrosis factor-alpha in the pathogenesis of aspiration pneumonitis in rats. Anesthesiology. 1999;91(2):486–99. doi: 10.1097/00000542-199908000-00024. [DOI] [PubMed] [Google Scholar]
- 41.Raghavendran K, et al. Acid and particulate-induced aspiration lung injury in mice: importance of MCP-1. Am J Physiol Lung Cell Mol Physiol. 2005;289(1):L134–43. doi: 10.1152/ajplung.00390.2004. [DOI] [PubMed] [Google Scholar]
- 42.Segal BH, et al. Acid aspiration-induced lung inflammation and injury are exacerbated in NADPH oxidase-deficient mice. Am J Physiol Lung Cell Mol Physiol. 2007;292(3):L760–8. doi: 10.1152/ajplung.00281.2006. [DOI] [PubMed] [Google Scholar]
- 43.Patel BV, et al. TNF-Induced Death Signaling Triggers Alveolar Epithelial Dysfunction in Acute Lung Injury. J Immunol. 2013;190(8):4274–82. doi: 10.4049/jimmunol.1202437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Van Zee KJ, et al. Tumor necrosis factor soluble receptors circulate during experimental and clinical inflammation and can protect against excessive tumor necrosis factor alpha in vitro and in vivo. Proc Natl Acad Sci U S A. 1992;89(11):4845–9. doi: 10.1073/pnas.89.11.4845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kokkola R, et al. RAGE is the major receptor for the proinflammatory activity of HMGB1 in rodent macrophages. Scand J Immunol. 2005;61(1):1–9. doi: 10.1111/j.0300-9475.2005.01534.x. [DOI] [PubMed] [Google Scholar]
- 46.Schmekel B, et al. Myeloperoxidase in human lung lavage. I. A marker of local neutrophil activity. Inflammation. 1990;14(4):447–54. doi: 10.1007/BF00914095. [DOI] [PubMed] [Google Scholar]
- 47.Goldblum SE, Wu KM, Jay M. Lung myeloperoxidase as a measure of pulmonary leukostasis in rabbits. J Appl Physiol. 1985;59(6):1978–85. doi: 10.1152/jappl.1985.59.6.1978. [DOI] [PubMed] [Google Scholar]
- 48.Reiss LK, Uhlig U, Uhlig S. Models and mechanisms of acute lung injury caused by direct insults. Eur J Cell Biol. 2012;91(6–7):590–601. doi: 10.1016/j.ejcb.2011.11.004. [DOI] [PubMed] [Google Scholar]