

Abstract
LAGLIDADG homing endonucleases (LHEs) are valuable tools for genome engineering, and our ability to alter LHE target site specificity is rapidly evolving. However, widespread use of these enzymes is limited due to the small number of available engineering scaffolds, each requiring extensive redesign to target widely varying DNA sequences. Here, we describe a technique for the chimerization of homologous I-OnuI family LHEs. Chimerization greatly expands the pool of unique starting scaffolds, thereby enabling more effective and efficient LHE redesign. I-OnuI family enzymes are divided into N- and C-terminal halves based on sequence alignments, and then combinatorially rejoined with a hybrid linker. The resulting chimeric enzymes are expressed on the surface of yeast where stability, DNA binding affinity, and cleavage activity can be assayed by flow cytometry.
Keywords: Homing endonuclease, Meganuclease, Chimera, Chimerization, Protein engineering, Yeast surface display, Flow cytometry, LAGLIDADG, Assembly PCR
1 Introduction
Significant advances have been made in engineering LAGLIDADG homing endonucleases (LHEs) to cleave novel DNA target sequences, but large-scale redesign remains a challenging task. Fortunately, the need for extensive engineering can be avoided when an existing native enzyme closely matches the final DNA target sequence desired. Expansion of the pool of available LHE design scaffolds may be the fastest and simplest means to overcome difficult engineering tasks. The recent identification of new enzymes from the monomeric I-OnuI family [1] provides a number of unique engineering scaffolds, but the list of target sites recognized by native enzymes is still dwarfed by the total number of target sites possible.
The dimeric/pseudodimeric architecture of LHEs provides an opportunity for expansion of our existing scaffold collection through the creation of chimeric enzymes (increasing the number of available scaffolds from N to N2). The feasibility of mixing the N- and C-terminal domains of various native LHEs was initially verified through the successful generation of an active chimera from the monomeric LHE I-DmoI and the homodimeric I-CreI [2, 3]. The structural dissimilarity of these two parent enzymes made necessary both computational modeling and in vitro selection to repack the chimeric interface. Based on this work, it was hypothesized and confirmed that the generation of chimeras from within a family of highly homologous monomeric LHEs could produce a large number of active enzymes with minimal engineering [4].
Here we describe an efficient and structure-independent method for the generation and analysis of chimeric LHEs from I-OnuI family homologues (Fig. 1). Break points defining the N- and C-terminal halves of each parent enzyme are determined through sequence alignment to the well-characterized I-OnuI endonuclease [1]. These N- and C-terminal halves are then recombined and joined with an artificial linker. In our experience, the choice of protein linker can have a significant effect on the stability and activity of the resulting chimera, so for the sake of simplicity and overall compatibility, we suggest using a “half-and-half” hybrid linker [4]. This strategy incorporates residues from both native parents to maintain important interactions with each respective domain, and introduces a flexible and minimally invasive tri-residue bridge at the point of fusion. The resulting chimeras are expressed on the surface of yeast under a galactose-inducible promoter, and flow cytometry is used to assess a variety of enzyme properties [5, 6]. Antibody staining of a C-terminal Myc epitope tag reports the presence of stable, full-length enzyme on the surface of yeast (Fig. 2), and introduction of fluorescently labeled DNA substrates allows for the rapid evaluation of both DNA binding and cleavage activities (Fig. 3). The activity observed by flow cytometry is validated by a complementary non-tethered in vitro cleavage assay (Fig. 4). Lastly, we provide a list of guidelines for further optimization of partially active chimeras.
Fig. 1.

Schematic outline of chimeric endonuclease generation. Sequence alignments are used to divide parent endonucleases into N- and C-terminal domains, and an -SGT-tripeptide linker is substituted at the center of the construct (Subheading 3.1 ). The resulting sequences are ordered as synthesized genes or generated by assembly PCR (Subheading 3.2). The pETCON yeast surface expression vector is digested to prepare open vector for cloning (Subheading 3.3), and the parent endonuclease sequences are digested into N- and C-terminal halves with the help of a restriction site in the SGT linker (Subheading 3.4). The endonucle-ase domains are then ligated into the open pETCON vector in the desired combinations (Subheading 3.5), and transformed into yeast for expression on the yeast surface (Subheadings 3.8 and 3.9). Finally, the stability and DNA binding/cleavage activities of the surface-expressed chimeras are assessed using flow cytometry (Subheadings 3.10 and 3.11)
Fig. 2.

Protein expression on the surface of yeast. (a) The pETCON vector expresses a cloned sequence on the surface of yeast with an N-terminal hemagglutinin (HA) tag and a C-terminal Myc tag. (b ) Fluorescent antibody staining of the N- and C-terminal epitope tags allows for detection of stable, full-length protein. The N-terminal HA tag is stained with streptavidin–phycoerythrin (SAV-PE) via a biotin–streptavidin bridge, and the C-terminal Myc tag is stained with fluorescein isothiocyanate (FITC) conjugated anti-Myc antibody. (c ) Sample FlowJo plot of surface expression staining. The y-axis measures signal from N-terminal biotin–streptavidin-linked PE fluo-rophore, while the x-axis measures signal from the C-terminal FITC fluorophore. Dual-stained cells in the upper right-hand quadrant represent stable surface expression of full-length protein
Fig. 3.

Schematic representation of the flow-cytometric DNA cleavage assay. (a ) An N-terminal hemagglutinin (HA) tag enables biotin–streptavidin tethering of a fluorescently labeled, double-stranded DNA substrate to the enzyme. (b ) In the presence of calcium, the enzyme can bind the DNA substrate but not cleave it; with the addition of magnesium, the enzyme is able to bind and cleave the target substrate. When cleavage occurs, the N-terminal tethered PE fluorescence will be maintained while the A647 signal on the opposite end of the DNA target is lost. (c) The tethered, intact target substrate produces a characteristic co-linear PE versus A647 fluo-rescence profile on the flow cytometer (left). Cleavage of the tethered substrate leads to a loss of fluorescence in the A647 channel. Superposition of PE versus A647 plots from the Ca2+ and Mg2+ samples can be used to roughly quantify cleavage activity (right)
Fig. 4.
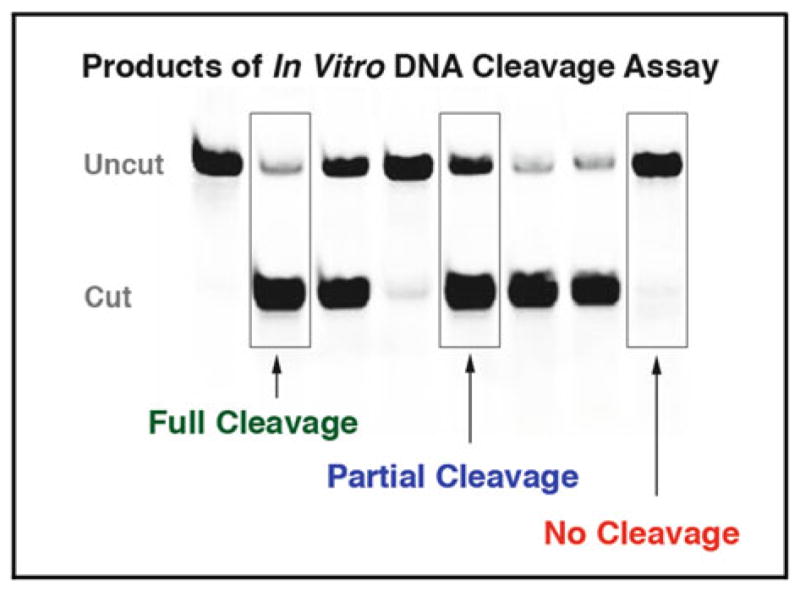
Sample gel of in vitro cleavage assay products. Double-stranded DNA target substrates with an incorporated A647 fluorophore are incubated at 37 °C in the presence of free enzyme released from the yeast surface by DTT. The resulting cleavage products are run on an acrylamide gel and visualized by a fluorescence imager. The sample lanes shown here represent fully cleaved, partially cleaved, and non-cleaved target substrate. The non-tethered nature of this in vitro assay provides an important validation of cleavage activity observed in the flow-cytometric cleavage assay. In this assay, the enzyme must have suffi-cient binding affinity for its DNA substrate to produce observable cleavage products
2 Materials
Prepare all solutions using ultrapure RNAse- and DNAse-free water (0.22 μm filtered, deionized water) and analytical grade reagents. Cultures and reagents may be prepared at the bench, but care should be taken to use sterile components and aseptic technique.
2.1 Assembly PCR
Set of assembly oligonucleotide primers (see explanation of design in Subheading 3.2).
Forward and reverse primers for the full-length desired product (see Subheading 3.2).
Ultrapure water.
1.5 mL microcentrifuge tubes.
Phusion® High-Fidelity DNA Polymerase with supplied 5× buffer (New England Biolabs).
dNTPs (10 mM).
Thermal cycler.
Agarose, molecular grade (Bioline).
Ethidium bromide solution (10 mg/mL) (Sigma-Aldrich).
Gel extraction purification kit (Zymo Research).
Restriction enzymes NdeI, KpnI-HF, and XhoI with supplied buffers and 100× BSA solution (New England Biolabs).
PCR purification kit (Qiagen).
2.2 Cloning of DNA Constructs
pETCON yeast surface expression vector (with NdeI and XhoI cloning sites) (Addgene reference).
DH5 alpha bacteria (either electro- or chemically competent, depending on method of bacterial transformation).
0.1 cm electroporation cuvettes (if using electroporation) (Bio-Rad).
Electroporator (Bio-Rad).
Luria broth (LB) for bacterial culture.
Carbenicillin antibiotic (Bioline).
LB-carbenicillin bacterial agar plates (1:1,000 antibiotic).
Mini- or maxi-prep plasmid isolation kit (Qiagen).
Restriction enzymes NdeI, KpnI-HF, and XhoI with supplied buffers and 100× BSA solution (New England Biolabs).
Agarose, molecular grade (Bioline).
1× Tris–Acetic acid–EDTA buffer (TAE) (Fisher Scientific).
Ethidium bromide solution (10 mg/mL) (Sigma-Aldrich).
Gel electrophoresis equipment.
Scalpel (for gel extraction).
Gel extraction DNA purification kit (Zymo Research).
UV gel illumination box with low intensity setting (for extraction of gel bands without damaging the DNA).
PCR purification kit (Qiagen).
Spectrophotometer.
T4 DNA Ligase with supplied buffer (New England Biolabs).
BigDye Terminator v3.1 sequencing mix (Applied Biosystems).
5× sequencing buffer: 400 mM Tris, pH 9.0, 10 mM MgCl2
-
pETCON sequencing primers:
Forward primer: 5′-GTTCCAGACTACGCTCTGCAGG-3′
Reverse primer: 5′-GATTTTGTTACATCTACACTGTTG-3′
Thermal cycler.
2.3 Dual-Labeled Double-Stranded DNA Substrates
Platinum® Taq High Fidelity DNA Polymerase, with 10× buffer and 50 mM MgSO4 (Invitrogen).
Target site oligonucleotide template with flanking universal primer sites, standard desalting purification (Integrated DNA Technologies [IDT]) (Fig. 5).
Biotin-labeled universal forward primer (IDT) (Fig. 5) (see Note 1).
A647-labeled universal reverse primer (IDT) (Fig. 5) (see Note 1).
10 mM dNTPs.
0.2 mL PCR strip tubes and/or 96-well PCR plates.
Thermal cycler.
Exonuclease I (New England Biolabs).
MultiScreen HTS-HV filter plate (Fisher Scientific).
illustra Sephadex G-100 (GE Healthcare): Prepare sephadex solution at 1 g/20 mL in water; allow at least 24 h for bead hydration; store at room temperature for a maximum of 4 months.
Tabletop centrifuge fit for spinning plates.
Odyssey infrared imaging system (Li-Cor Biosciences) or UV transilluminator.
NanoDrop or similar microvolume spectrophotometer (Thermo Scientific).
Fig. 5.

Reagents for PCR production of fluorescently labeled DNA target oligonucleotide substrate. Determination of the chimeric target recognition sequence requires the simple fusion of two parental half-sites (divided at the center of the native targets). If orientation of the parent enzyme on its DNA target site is NOT known, then one must test both the forward and reverse-complement sequences for that parent. To create the target site PCR template, the desired target sequence should be flanked by forward and reverse “universal primer” sequences (sample sequences are those currently used in our laboratories; alternate primer sequences may be substituted). Primers are ordered with A647 or biotin modifications for the PCR production of dual-labeled double-stranded DNA substrates
2.4 Polyacrylamide Gel
30 % acrylamide–bis acrylamide (19:1) solution.
10× Tris–borate–EDTA (TBE) buffer (see Note 2).
N,N,N′,N′-tetramethylethylenediamine (TEMED).
Ammonium persulfate (Acros Organics): 10 % w/v solution in water.
Plastic gel cassette, 1.0 mm.
Vertical gel electrophoresis apparatus.
6× Ficoll loading buffer: 18 % (w/v) Ficoll-400 (Sigma-Aldrich) in 6× TBE, with NO added dyes (see Note 3).
2.5 Yeast Transformation
EBY100 yeast (Invitrogen).
2× YPAD non-selective yeast media: 20 g Bacto yeast extract (BD), 40 g Bacto peptone (BD), 100 mg Adenine hemisulfate (Sigma-Aldrich), 50 g D-Glucose (Fisher Scientific), water to 1 l total volume, pH to 6.0. Filter-sterilize or autoclave and store at 4 °C (see Note 4).
Salmon sperm DNA (Sigma-Aldrich), 2 mg/mL solution in 1× TE buffer.
1 M Lithium acetate solution in water.
50 % w/v Polyethylene glycol in water, MW 3350 (PEG 3350) (Amaxa).
Plasmid DNA encoding a chimeric endonuclease in the pET-CON yeast surface display vector.
42 °C water bath.
Yeast selective growth media “SC–Ura–Trp”: 6.7 g Yeast nitrogen base without amino acids (Sigma-Aldrich), 1.4 g yeast synthetic drop-out media supplement without Trp, Ura, His, Leu (Sigma-Aldrich), 76 mg Histidine, 380 mg Leucine, 4.34 g MES, and water to 900 mL. Adjust pH to 5.25 with HCl. Sterilize by autoclaving 20 min. Prior to use, add penicillin (100 i.u./mL), streptomycin (100 μg/mL), and kanamycin (25 μg/mL). Store at 4 °C.
20 % w/v D-glucose (Fisher Scientific) solution, filter-sterilized. Store at 4 °C.
Selective growth media agar plates: add 20 g of bacteriological agar (BD) to 900 mL of SC–Ura–Trp selective growth media and autoclave for 20 min. Add 100 mL pre-warmed (55 °C) 20 % w/v D-glucose and penicillin (100 i.u./mL), streptomycin (100 μg/mL), and kanamycin (25 μg/mL). Pour into petri dishes and let solidify at room temperature. Store plates at 4 °C.
Water-jacketed incubator.
2.6 Yeast Growth and Induction
EBY100 yeast transformed with surface-expression vector containing chimeric homing endonuclease.
SC–Ura–Trp selective growth media (for recipe, see Subheading 2.5).
20 % w/v D-glucose (Fisher Scientific) solution, filter-sterilized. Store at 4 °C.
20 % w/v D-(+)-raffinose pentahydrate (Sigma-Aldrich)+ 0.1 % w/v glucose solution, filter-sterilized. Store at room temperature.
20 % w/v D-(+)-galactose (Sigma-Aldrich) solution, filter-sterilized. Store at 4 °C.
Baffled Erlenmeyer flask(s).
Disposable 15-mL culture tubes.
Deep-well 96-well plate (flat bottom).
Shaking incubator.
Spectrophotometer.
2.7 Yeast Surface Display Flow-Cytometric DNA Binding and Cleavage Assays
Induced EBY100 yeast with surface expressed chimeric homing endonuclease (see Subheading 3.9).
10× Yeast Staining Buffer (YSB): 1.8 M KCl, 0.1 M NaCl, 0.1 M HEPES, 2 % BSA, 1 % w/v D-(+)-Galactose, adjust pH to 7.5 with KOH (see Note 5). Filter-sterilize and store at 4 °C in a light-protected or foil-wrapped container.
1× high-salt Yeast Staining Buffer (YSB + KCl): Dilute 10× YSB to 1× and add an additional 400 mM KCl for a final KCl concentration of 580 mM. Store at 4 °C in light-protected or foil-wrapped container.
10× In-vitro Oligonucleotide Cleavage Buffer (IOCB): 1.5 M KCl, 0.1 M NaCl, 0.1 M HEPES, 0.05 M K-Glu (L-Glutamic Acid Potassium Salt Monohydrate), 0.5 % BSA, adjusted to pH 8.25 with KOH (see Note 5). Filter-sterilize solution and store at 4 °C in a light-protected or foil-wrapped container.
1 M CaCl2 solution. Filter-sterilize and store at room temperature.
1 M MgCl2 solution. Filter-sterilize and store at room temperature.
Biotin-labeled anti-HA antibody (Covance).
Streptavidin-PE (BD Biosciences).
FITC-conjugated chicken anti-cMyc antibody (Immunology Consultants Laboratory Inc.).
Costar 96-well V-bottom plate (Sigma-Aldrich).
BD FACScalibur or LSRII™ cytometer (BD Biosciences) or other cytometer with equivalent optics.
FloJo software (Tree Star Inc).
2.8 Non-tethered In Vitro Cleavage Assay and Gel
Induced EBY100 yeast with surface expressed chimeric homing endonuclease (see Subheading 3.9).
1× IOCB (for recipe, see Subheading 2.7).
1 M solution CaCl2 (to be diluted to 5 mM).
1 M solution MgCl2 (to be diluted to 5 mM).
0.2 M Dithiothreitol (DTT).
Polyacrylamide gel (for components, see Subheading 2.4).
Vertical gel electrophoresis apparatus.
3 Methods
Carry out all procedures on ice, unless otherwise specified.
3.1 Analysis and Preparation of Parent Homing Endonuclease DNA Coding Sequences
Codon optimize the DNA coding sequences of interest for expression in yeast (or dual optimize for expression in both yeast and bacteria if planning downstream assays in bacteria).
Search the codon-optimized DNA sequences for the following restriction sites: KpnI, NdeI, and XhoI. Replace any instances of these sites with alternate codons to remove the sites.
Align the amino acid sequences of interest with the I-OnuI sequence (or other reference sequence) to identify the positions of the two LAGLIDADG (Fig. 6).
-
Number backwards from the start of the second LAGLIDADG helix (in the I-OnuI reference structure, numbering begins with Asn167) (Fig. 6 inset).
0=Asn167
1=Pro166
2=Ile165
3=Asn164
4=Lys163
5=Asn162
Substitute a serine at position 5, a glycine at position 4, and a threonine at position 3. Use the sequence “GGTACC” for the Gly-Thr to introduce a KpnI restriction site. (Alternately, if structures are available, align the structure of interest to I-OnuI and replace the equivalent N–K–N residues with S–G–T.) (see Note 6).
Add an NdeI restriction site (CATATG) to the 5′ end of the coding sequence and an XhoI restriction site (CTCGAG) to the 3′ end (for cloning into the pETCON vector).
Order the resulting sequence (we used synthesized genes from Genscript) or generate by assembly PCR (see Subheading 3.2). Clone into the pETCON yeast surface expression vector between the NdeI and XhoI restriction sites (see Subheading 3.3 for preparation of open vector).
Fig. 6.

Alignment of native endonuclease sequences. Alignment of a well-characterized reference sequence like I-OnuI is used to define the N- and C-terminal domains of the I-PanMI enzyme. The location of the second LAGLIDADG helix is used to guide the placement of the -SGT-tripeptide linker. Counting backwards from the residue at the top of the second LAGLIDADG helix (marked residue “0” in the magnified inset ), the -SGT-linker is substituted at positions 3 through 5 (marked with a bracket). This location was chosen as a region where there are minimal interactions between the native linker and the adjacent protein domain
3.2 Assembly PCR
Derive your desired homing endonuclease sequence, as described in Subheading 3.1 (codon-optimized for yeast expression and including the -SGT-tri-residue bridging linker between the N- and C-terminal halves). Position the sequence as it would appear inside the pETCON vector in order to see the sequence directly preceding and following the endonucle-ase reading frame.
Using Primer3 [7] (or similar primer design program), find forward and reverse primers for the N-terminal domain (see Note 7), including six or more base pairs preceding the NdeI site and six or more base pairs following the KpnI site (see Note 8). This is most easily accomplished by inputting a limited region including potential forward sites, and determining only the forward primer (uncheck “pick right primer or use right primer below”). Repeat for reverse primer.
Repeat step 2 for the C-terminal domain (using the KpnI and XhoI restriction sites).
-
Input the coding sequences for each half of the chimera, including sequence adjacent to the restriction sites as described above, into the DNAWorks online assembly PCR design tool [8]. We used the following settings:
Annealing temperature: 61–63 °C.
-
Oligo length: 40–60 bp (see Note 9).
Be sure to check the “Random” box, which allows the program to use oligonucleotides of varying lengths to find optimal overlaps. If left unchecked, the program will return a set of oligonucleotides of identical length (see Note 10).
The remaining options were left as default.
Paste your DNA sequence in the last field, and select “nucleotide” as the type of sequence.
Be sure to input your email address and a title for your job (do NOT include spaces in your title).
Once DNAWorks returns a completed set of solutions, scroll to the very bottom of the resulting logfile to find a table of statistics. The best set of oligonucleotides will have the LOWEST overall score. A low score will correspond to the smallest number of misprimes. Note the number of the design run with the lowest score, and scroll back up the logfile to find that set of oligonucleotides.
Order the set of oligonucleotides. DNA synthesis on the 25 nM scale with standard desalting purification is adequate.
Resuspend the oligonucleotides at 100 μM concentration in ultrapure water.
Create a “pool” of all the oligonucleotides for each domain by combining 1 μL of each assembly primer. Add water to bring the final concentration of each primer in the pool to 1 μM. (Example: add 1 μL from each of 30 primers, each at a stock concentration of 100 μM. Then add 70 μL water to bring the total volume of the pool to 100 μL, and the final concentration of each primer to 1 μM) (see Note 11).
The assembly procedure consists of two separate PCR reactions. The first reaction allows the set of oligonucleotide primers to properly assemble. The second reaction amplifies the full-length assembly products. See Table 1 for PCR recipes and thermal cycler programs. Perform the primary assembly reaction for each domain. No purification of the reaction is necessary.
Perform the secondary assembly reaction for each domain (Table 1), using the forward and reverse primers you designed in steps 2 and 3 above.
Run 1–5 μL of the secondary assembly reaction on a 1 % aga-rose gel with ethidium bromide. If there is a single, clean band at approximately 400 bp (representing one full-length domain), proceed directly to digestion. If there is significant contamination by products of other sizes, run the entire reaction on a 1 % agarose gel and purify by gel extraction of the correct band.
Digest the full-length assembly product with NdeI and KpnI-HF (for N-terminal domain), or KpnI-HF and XhoI (for C-terminal domain) to prepare for ligation into the pETCON vector (see Note 12).
Purify the digestion reaction with a PCR purification kit.
Clone into the pETCON yeast surface expression vector between the NdeI and XhoI restriction sites (see Subheading 3.3 for preparation of open vector).
Table 1.
Assembly PCR recipes and thermal cycler programs
| Primary assembly reaction (50 μL) | Secondary assembly reaction (50 μL) |
|---|---|
| 10 μL 5× Phusion buffer | 10 μL 5× Phusion buffer |
| 2 μL oligo pool | 5 μL primary assembly reaction |
| 2.5 μL dNTPs (10 mM) | 2.5 μL dNTPs (10 mM) |
| 1 μL Phusion enzyme | 2.5 μL forward primer (10 μM) |
| 34.5 μL water | 2.5 μL reverse primer (10 μM) 1 μL Phusion enzyme 26.5 μL water |
| Thermal Cycler Program | |
| 1. 98 °C for 1 min | 1. 98 °C for 1 min |
| 2. 98 °C for 10 s | 2. 98 °C for 10 s |
| 3. 58 °C for l5 sa | 3. 63°Cfor 15 sb |
| 4. 72 °C for 1 min | 4. 72 °C for 1 min |
| 5. Go to step 2, 29 more times | 5. Go to step 2, 29 more times |
| 6. 72 °C for 5 min | 6. 72 °C for 5 min |
| 7. 4 °C for ever | 7. 4 °C for ever |
| 8. End | 8. End |
Adjust annealing temperature according to Tm of assembly oligonucleotides
Adjust annealing temperature according to Tm of designed primers
3.3 Preparation of Open pETCON Vector DNA
Transform the pETCON vector DNA into DH5 alpha bacteria by standard electroporation (see Note 13) or chemical transformation protocols and plate on an LB + carbenicillin (carb) agar plate. Incubate overnight at 37 °C.
Culture a single colony from the transformation plate in LB+carb media.
Use a plasmid isolation kit (mini or maxi) to obtain a large quantity of the intact vector (a minimum of 10 μg of DNA should be enough for downstream reactions). Elute with sterile water.
Digest 5–10 μg of the intact vector with the restriction enzymes NdeI and XhoI. Digest at least 5–10 μg of the vector DNA (see Note 14).
Determine the efficiency of the double digest by running 1 μL of the digest products on a 0.5 % agarose gel (with ethidium bromide) in 1× TAE buffer for 30–60 min at 120–130 V. The correct size of the open vector is approximately 6,200 bp.
If more than one product size is visible (suggesting incomplete digestion), run the entire digestion reaction on a 0.5 % agarose gel (in multiple lanes or a single large lane). Excise the appropriate band for the linearized vector, and proceed with gel extraction purification (see Note 15). If only a single band of the correct size is visible, the reaction can be purified with a PCR purification kit. Elute with sterile water.
Quantify the purified open pETCON vector using a spectrophotometer.
3.4 Preparation of N- and C-Terminal Domain DNA for Ligation
This protocol is designed to prepare chimeras from several parent enzymes simultaneously.
Transform DH5 alpha bacterial cells (using standard electro-poration or chemical transformation) with the parent homing endonuclease DNA coding sequence in the pETCON yeast surface expression vector, and plate on an LB + carb agar plate. Incubate overnight at 37 °C.
Culture a single colony from the transformation plate in LB+carb media.
Use a plasmid isolation kit (mini or maxi) to obtain a large quantity of the intact vector (a minimum of ten micrograms of DNA should be enough for downstream reactions). Elute with sterile water.
Set up double digest reactions with 5–10 μg of the intact vector DNA using the following restriction enzyme combinations: (a) NdeI and KpnI-HF (to cut out the N-terminal domain) (see Notes 12 and 14) and (b) KpnI-HF and XhoI (to obtain the C-terminal domain fragment).
Run the digested DNA in a large lane on a 0.8 % agarose gel (with 2 μL/100 mL ethidium bromide) in 1× TAE buffer at 120–130 V for 45–60 min.
Excise the correct-sized DNA fragments from the gel, using a low-intensity UV light box and minimizing the length of time that the product is exposed to UV light. For I-OnuI, the NTD fragment is 477 bp and the CTD fragment is 419 bp. Purify using a gel extraction purification kit, and elute with sterile water.
3.5 Ligation of N- and C-Terminal Domains into the pETCON Yeast Surface Expression Vector
Determine the volume of open pETCON vector you will use for each ligation reaction (use 100–200 ng vector).
Based on the size (in bp) and concentration (in ng/μL) of your purified N- and C-terminal half-inserts, calculate the nanograms of each half you will need to create a 3:1 molar ratio of insert to vector. The following online “Ligation Calculator” works well: http://www.insilico.uni-duesseldorf.de/Lig_Input.html.
Combine the N-terminal half-insert, C-terminal half-insert, and open pETCON vector with 1 μL 10× T4 ligase buffer and 0.5 μL T4 DNA ligase enzyme. Add water to a final volume of 10 μL.
Incubate the ligation reaction overnight at 16 °C, followed by heat inactivation at 65 °C for 10 min.
3.6 Transformation and Screening of Chimeric Homing Endonuclease Constructs
Transform the completed ligation reaction into bacterial cells (using either chemical transformation or electroporation). Use up to 4 μL of the ligation reaction for chemical transformation and up to 2 μL for electroporation.
Plate the transformed cells. In most cases, chemical transformation is sufficient to produce many colonies for screening. Electroporation should be used if fewer than ten colonies are obtained.
Screen the resulting colonies by either: (a) colony sequencing (see Note 16) or (b) culture single colonies, isolate plasmids using a miniprep kit, and sequence the plasmid DNA (see Note 17).
3.7 Preparation of Dual-Labeled Double-Stranded DNA Substrates
In 0.2 mL PCR tubes, mix 0.08 μL Platinum High Fidelity Taq, 2 μL 10× Taq buffer, 1.3 μL 50 mM MgSO4, 0.4 μL 10 mM dNTPs, 2 nM (final concentration) target site template oligonucleotide, and 0.55 nM (final concentration) each of the A647 universal FP and biotin universal RP. Add H2O to a final volume of 20 μL (see Note 18).
-
Thermal cycler program (for PCR amplification of target and incorporation of labels):
90°C×1 min.
40× (86 °C × 15 s, 48 °C × 15 s, 60 °C × 30 s).
60°C×15 min.
40× (70 °C × 30 s, decrease by 1 °C every cycle) (see Note 19).
Hold at 4 °C.
Digest excess single-stranded DNA with Exonuclease I: Add 2 U of ExoI to each 20 μL PCR reaction in 2 μL total volume of water (see Note 20). Digest 4 h at 37 °C. This reaction can be stored at 4 °C overnight or at −20 °C for extended periods.
Load the hydrated sephadex G-100 suspension into the filter plate. For each 20 μL PCR reaction to be purified, add 500 μL total volume of suspension to a filter plate well. This is best accomplished by loading 320 μL sephadex suspension (using wide bore tips) into each necessary well of the filter plate, centrifuging briefly up to a speed of 500 × g, discarding water, and adding the remaining 180 μL sephadex (see Note 21). The plate should then be dehydrated by centrifugation at 2,000 × g for 7 min. Do not use any sephadex columns that contain cracks.
Load the 22 μL PCR + ExoI reaction directly to the center of each sephadex column. Secure a 96-well PCR plate below the filter plate to catch the purified flow-through, using tape if necessary. Centrifuge for 5 min at 2,000 × g. Approximately 12–14 μL of flow-through should be present in each recipient well.
Determine the concentration of purified target site substrates using a NanoDrop spectrophotometer. Target concentration should be approximately 10–25 ng/μL. A concentration greater than 25 ng/μL suggests inadequate ExoI digestion or sephadex purification. Sephadex purification can be repeated, but this will not help purify inadequately ExoI-digested samples.
Run a sample of the purified target oligonucleotides on a 15 % polyacrylamide gel. For a 7.5-mL gel, combine 0.75 mL 10×TBE, 3.75 mL 30 % acrylamide–bis acrylamide (19:1), and 2.9 mL water. Add 100 μL 10 % APS and 10 μL TEMED, then immediately mix and pipette into the prepared gel cassette. Once the gel is set, load 0.5–1.0 μL purified target, diluted with 1.0 μL 6× Ficoll loading buffer and 4 μL water (see Note 22). Use 0.1 μL of the A647-labeled primer as a size standard. Run the gel for 90 min at 120 V.
Visualize the gel on a Licor Odyssey infrared imager, using the 700 nM laser. The gel should show a prominent single PCR product and minimal contamination by other bands or leftover primers. Alternatively, the gel can be stained with 1× SybrGold in 1× TBE for 20 min, washed in 1× TBE or water, and visualized on a UV transilluminator.
3.8 Yeast Transformation
(This transformation procedure is based on published protocols by Gietz and Scheizl) [9].
Thaw and spin down a frozen aliquot of EBY100 competent yeast cells (see Note 23).
Resuspend the pellet in the following transformation mixture: 50 μL denatured 2 mg/mL salmon sperm DNA (denature by heating at 95 °C for 5 min, then transfer immediately to ice), 36 μL 1 M LiAc, 260 μL 50 % PEG 3350, and 14 μL water plus plasmid DNA (up to 1 μg) (see Notes 24 and 25).
Incubate the yeast and transformation mixture at 42 °C for 40 min (see Note 26).
Fill the tube with SC–Ura–Trp + 2 % glucose media and spin down the cells. Remove supernatant.
Resuspend the yeast pellet in 1 mL SC–Ura–Trp + 2 % glucose media.
Plate 1–10 μL transformed yeast on selective growth media agar plates (SC–Ura–Trp + 2 % glucose) and incubate in a 30 °C water-jacketed incubator. Colonies of an appropriate size for picking should appear by 48–72 h.
3.9 Growth and Induction of Yeast
Transfer a single colony of transformed yeast into 1.5 mL SC–Ura–Trp+2 % raffinose + 0.1 % glucose media (see Note 27).
Incubate overnight in a 15-mL culture tube at 30 °C with 250 RPM shaking (see Note 28) until the cells reach a density of 90–120 million/mL (see Note 29) and place on ice for up to 24 h.
Wash 30 million cells twice with water and transfer to 1.5 mL of SC–Ura–Trp + 2 % galactose media (see Note 30).
Incubate the galactose culture on the benchtop (room temperature with no shaking) for up to 16 h (see Note 31).
3.10 Yeast Surface Display Flow-Cytometric DNA Cleavage Assay
All components should be kept on ice throughout the assay, unless otherwise specified, including YSB and IOCB buffers. If possible, the centrifuge should also be kept at 4 °C.
Determine the density of induced yeast in the galactose culture. This can be accomplished using a hemocytometer and microscope or estimated by spectrophotometer (see Note 29). Aliquot 500,000 yeast per sample into a 96-well, V-bottom plate (see Notes 32 and 33).
Wash cells twice with 200 μL 1× YSB, centrifuging the V-bottom plate at 3,000 × g for 3 min and discarding the supernatant. Yeast cells often not form a visible pellet until washed with YSB.
Gently resuspend cells at a concentration of 50 million/mL in 1× YSB with 1:250 dilution of anti-HA-biotin antibody (i.e., consider the anti-HA-biotin to be a 250× stock). Incubate at 4 °C for 30–60 min, mixing gently every 10–15 min (see Note 34).
During the anti-HA stain, prepare target substrates for conjugation in either 1.5-mL microcentrifuge tubes or plate format. For 500,000 cells (final cell density of 50 million/mL), use 25 μL total volume per well. SAV-PE should be diluted to 5 nM in the high salt 1× YSB + KCl buffer. Add the labeled ds-oligonucleotide target substrate to a final concentration of 40 nM (see Notes 35 and 36). Aliquot to plates (if necessary) and incubate in the dark and on ice for 30 min.
Following the anti-HA incubation, centrifuge cells for 3 min at 3,000×g and wash twice with 200 μL ice-cold high-salt 1× YSB+KCl.
Following the second wash, resuspend yeast cells with target site conjugates. Gently vortex or pipette to resuspend.
Incubate 30 min at 4 °C, in the dark or in a light-protected container, mixing briefly every 5–10 min.
During this incubation, make 5 mM MgCl2 and CaCl2 solutions in 1× IOCB, and pre-warm to 37 °C.
Following incubation with conjugated target substrate, wash yeast with 200 μL ice-cold high-salt 1× YSB + KCl, and centrifuge for 2 min at 3,000 × g. Resuspend conjugated yeast in 200 μL ice-cold 1× IOCB (containing no divalent ions).
Create duplicate wells (one will contain calcium for no cleavage, and one will contain magnesium to allow cleavage) by transferring half of the resuspended volume into an adjacent set of wells. Add an additional 100 μL ice-cold 1× IOCB to each well (giving a final wash volume of 200 μL). Centrifuge for 3 min at 3,000 × g. Discard the supernatant and blot/tap vigorously on paper towels to remove as much buffer as possible.
Add 30 μL 1× IOCB containing either Ca2+ or Mg2+ (pre-warmed to 37 °C) to each set of duplicate wells. Add Ca2+ IOCB first to minimize background cleavage events in the negative control, and work as quickly as possible.
Incubate 20–30 min at 37 °C.
Fill wells with ice cold 1× YSB to stop the reaction, centrifuge 3 min at 3,000 × g, and discard the supernatant.
Resuspend in 25 μL 1× YSB containing 1:100 dilution of anti-Myc FITC to give a final cell density of 100 million/mL.
Incubate 1–4 h at 4 °C, with foil wrap or in a refrigerator to protect from light, gently vortexing occasionally to keep the cells in suspension (see Note 37).
Wash cells twice with 1× YSB, and resuspend in a final volume of 60 μL. This will lead to an acquisition rate of 1,000–2,000 events/s depending on the acquisition settings for the cytometer.
Acquire data on a BD Biosciences LSRII with HTS. Record data for FSC-A, FSC-H, SSC-A, SSC-H, APC, PE, and FITC parameters.
Analyze the flow cytometry data using FloJo. Gate live cells (FSC-A by SSC-A), then singlets (FSC-H by FSC-A), then cells staining for both FITC and PE (representing full length expression of both the C′ and N′ termini) (Fig. 7). Visualize this final subset as APC (y-axis) versus PE (x-axis). Superimpose the Ca2+ and Mg2+ samples to observe any cleavage-induced shift in APC signal. Quantitative measurements of cleavage efficiency can be obtained by determining the median APC signal within a small gated subset of live, singlet, expressing cells. A greater Ca2+-to-Mg2+ median APC ratio represents increased cleavage efficiency (Fig. 7d).
Fig. 7.

FlowJo analysis of flow cytometry data from the tethered DNA cleavage assay. (a ) Live cells are gated first, using FSC-A and SSC-A parameters, followed by (b) singlet cells using FSC-A and FSC-H. With proper induction conditions, approximately 50 % of cells will display the enzyme, and (c) the high-expressing population is gated using the PE signal (bound via a biotin–streptavidin bridge to the N-terminal HA tag) and FITC signal (bound directly by a fluorescence-conjugated antibody to the C-terminal Myc tag). Cleavage activity is visualized within this expressing population using a plot of (d) A647 fluorescence (observed in the APC channel, conjugated to the free end of the cleaved DNA) versus PE fluo-rescence (N-terminal HA tag). Cleavage activity can be quantified by gating on a PE-normalized subset of the expressing cells and calculating the ratio of median APC fluorescence for the uncleaved (Ca2+) versus cleaved (Mg2+) conditions
3.11 Yeast Surface Display Flow-Cytometric DNA Binding Assay
All components should be kept on ice throughout the assay, unless otherwise specified. If possible, the centrifuge should also be kept at 4 °C.
Determine the density of induced yeast. This can be done using a hemocytometer or by spectrophotometry (see Note 29).
Aliquot 100,000 yeast to appropriate wells in a 96-well plate (for 384-well format, see Note 38) and wash twice with 200 μL 1× IOCB containing 5 mM CaCl2. Centrifuge at 3,000 × g for 3 min and discard the supernatant.
Resuspend cells in 30 μL 1× IOCB containing 5 mM CaCl2, 1:100 anti-Myc FITC, and the desired concentration of target site oligonucleotide (see Note 39). Incubate at 4 °C for 2 h, vortexing gently every 30 min.
Wash twice with 1× IOCB + 5 mM CaCl2.
Resuspend in 60 μL 1× IOCB + 5 mM Ca2+ for flow cytometry analysis. A final volume of 60 μL will lead to 500–1,000 events/s on the flow cytometer.
Acquire data on a BD Biosciences LSRII with HTS. Record data for FSC-A, FSC-H, SSC-A, SSC-H, APC, and FITC parameters. If using HTS for collecting 96- or 384-well plate samples, set the machine to mix samples at minimum 3×.
Analyze the flow cytometry data using FloJo. Gate first the live cells, then singlets, then expressing cells, as described above. APC signal represents binding of the A647-labeled oligonucleotide to the surface-expressed homing endonucle-ase. Quantitative measurements of binding can be obtained by determining the ratio of median APC signal of the FITC-positive cells (which are expressing full-length enzyme) to the median APC signal of the FITC-negative cells (no enzyme expression).
3.12 Non-tethered in Vitro Cleavage Assay and Gel
All components should be kept on ice throughout the assay, unless otherwise specified.
Induce expression of the desired enzyme on the surface of yeast, as described above in Subheading 3.9 (see Note 40).
Determine the density of induced yeast (in galactose media) by spectrophotometer, as described above in 3.10 (see Notes 29 and 41).
Wash 5 million yeast cells with cleavage buffer (1× IOCB), spinning for 1 min at 10,000 × g or 3 min at 4,000 × g in a tabletop centrifuge. Resuspend in 200 μL IOCB (in plate) or 1 mL IOCB (in microcentrifuge tubes).
Divide the washed cells evenly into two separate tubes/wells. One sample will be run in the presence of calcium (control) and one in the presence of magnesium (cleavage).
Spin down the divided cells, and resuspend in the following mixtures: 50 μL total volume of 1× IOCB with 10 mM DTT, 5 mM CaCl2 (control) or 5 mM MgCl2 (cleavage), and 20–50 nM A647-labeled DNA substrate (see Subheading 3.7).
Mix thoroughly, and incubate at 37 °C for 5–60 min (see Note 42). Mix every 5–10 min throughout the incubation.
Transfer tubes/plates to ice to stop the reaction.
Spin the yeast at 4,000 × g for 3 min, or 10,000 × g for 1 min. Carefully pipette supernatant away from the cell pellets and transfer to a separate tube for storage (see Note 43).
Mix 2 μL of the 6× Ficoll loading buffer with 10 μL of the supernatant. Do not include any colored dyes. No DNA ladder is necessary. The calcium control lane(s) will provide an uncut control for size-comparison.
Carefully load the 12-uL samples into a 15 % acrylamide gel (see Subheading 3.7 for gel recipe) (see Note 22).
Run the gel for 60–90 min at 120 V.
Visualize the gel on a Licor Odyssey infrared imager, using the 700 nM laser. The Odyssey analysis software can be used for crude quantification of cleavage activity by manually gating cleaved and uncleaved bands for each sample, and determining the ratio of signal intensities.
3.13 Optimization of Partially Active Chimeras
In our experience, up to 50 % of I-OnuI family chimeras show significant cleavage activity with this direct domain fusion method [4]. However, certain enzyme combinations will be incompatible and require further engineering to achieve DNA cleavage activity. There are many different options for optimization of these enzymes and subsequent selection of the desired final outcome. Though we do not present step-by-step instructions for each option (as that is beyond the scope of this chapter), the following list can provide some initial guidance for beginning the optimization process. For chimeras with low cleavage activity, we have been able to improve stability and/or activity using the following strategies:
Variation of residues at the DNA-distal end of the LAGLIDADG helices. The LAGLIDADG helices form a major portion of the chimeric interface, and they show remarkable structural conservation between family members. However, this conservation breaks down at the DNA-distal end of the helices (Fig. 8a). We have found that manipulation of these distal helical residues can greatly increase the overall stability and resulting activity of a low-performing chimera [4]. Assembly PCR (see Subheading 3.2) is a great method to use for introduction of variation at one or more positions in a chimeric enzyme. Degenerate codons can be used to allow more than one residue at a given position (Fig. 9 inset), and the resulting library can be screened for the desired cleavage activity.
Error-prone PCR. Introduction of random variation across the entire coding sequence can often produce an enzyme with improved stability and/or activity.
Parent interface substitution. Another way to potentially improve an incompatible chimeric interface is to engineer the entire interface to match one of the two parent enzymes (Fig. 8b) [4]. This strategy can help overcome problems associated with steric clashes between interacting residues, but only if the surrounding core residues can support the new set of residues.
Library of interface residues. To find a functional solution for an important, but poorly functioning chimera, variation can be introduced along the entire chimeric interface. Again, assembly PCR with degenerate codons can be used to construct this type of interface library, which can then be screened for active enzymes. Care must be taken to limit the size of the overall library to that which can be reasonably screened by available yeast or bacterial cleavage selection methods.
Fig. 8.

Structural representation of areas for optimization of partially active chimeras. (a) The I-OnuI structure (gray) is shown with superimposed LAGLIDADG helices from five different enzymes: I-OnuI helices are colored purple, I-LtrI are green, I-MpeMI (homology model) are cyan, I-PanMI are magenta, and I-GzeII are yellow. Side chains are depicted as “sticks” at the DNA-distal end of the LAGLIDADG helices. A magnified view highlights the lack of conservation at these positions. (b) A significant portion of the chimeric interface is formed between the two adjacent LAGLIDADG helices. Residues from a single parent enzyme (blue) can be substituted onto BOTH sides of the LAGLIDADG interface to help overcome problems with an otherwise incompatible chimeric interface
Fig. 9.

Example of assembly PCR primers and introduction of variation via degenerate codons. Each colored selection represents a single assembly primer; the sum of all primers is designed to produce the entire coding sequence shown. An NdeI restriction site (CATATG) has been added to the N-terminal end of the sequence, and an XhoI restriction site (CTCGAG) has been added to the C-terminal end. The magnified inset towards the C-terminal end of the sequence gives an example of introducing variation using the degenerate codon “RAA.” The “R” base designates the introduction of either a guanine (G) or adenine (A) base at that position, resulting in a translated protein sequence with either glutamic acid (E) or lysine (K)
Acknowledgments
This work was supported by NIH grants RO1CA133832, RL1GM133833, 5RL1GM84433-04, and U19AI096111.
Footnotes
The biotin and A647 labels can be affixed to either the forward or the reverse primer. Maximum cleavage signal is achieved empirically by testing each endonuclease with both labeling schemes. For reasons that remain speculative, a more pronounced cleavage shift may occur with the A647 label on one end of the oligonucleotide versus the other.
10× TAE buffer can be used in place of 10× TBE buffer. If this substitution is made, be sure to use 1× TAE as the gel running buffer in place of 1× TBE.
Double-stranded oligonucleotide targets are visualized by the incorporated fluorescent A647 tag, and single-stranded, unincorporated A647 primer is used as a control to determine approximate purity of the double-stranded target. Dyes used to track DNA location on the gel also fluoresce, and should therefore be avoided. Loading of the colorless DNA solution into the gel lanes is difficult, so we suggest using a marker to trace each lane on the outside of the gel cassette and number the wells for reference. Watch the gel loading tip carefully to make sure it is correctly inserted into the desired well, and load the DNA slowly.
If autoclaving, add the glucose after autoclaving. To increase the shelf life of this media, make a 50× stock of the adenine hemisulfate and add it at 1× concentration just prior to use. Store the 50× stock at −20 °C (upon thawing, there will be a small amount of precipitation which will not go back into solution).
Solution should be adjusted to pH 7.5 using KOH. This limits introduction of additional sodium ions.
The position of this flexible S–G–T tripeptide was chosen to replace a short portion of the linker sequence making minimal to no interactions with the adjacent structure (to minimize detrimental effects on overall protein stability or enzyme activity).
These primers will be used to amplify full-length gene assembly PCR product for the N-terminal domain, and should have reasonably well-matched melting temperatures (Tm).
The sequence flanking the restriction sites can, theoretically, be set to any sequence. However, matching the sequence to the actual vector allows for downstream use of recombination-based cloning techniques, such as InFusion (Clontech), if standard sticky-end ligation methods are not yielding adequate product. For this purpose, we prefer to include 15 or more vector-matched base pairs on either side of the NdeI and XhoI restriction sites.
Using oligonucleotides over 70 bp in length greatly increases the error rate associated with synthesis of longer oligonucle-otides. We kept our lengths at 60 bp or less to allow ordering of 25 nM-scale standard oligonucleotides from IDT. If there are large sections of the gene in which no subsequent alterations or variation is desired, ultramers may be considered in place of oligonucleotides.
If too many variables are included in a design run, the program will time-out and return NO results. This is especially true when you have the “Random” box selected. If you are NOT getting successful design runs, decrease the number of variations possible. One way to do this is to significantly decrease the range of oligo lengths.
Save an aliquot of this pool in the event that any subsequent steps fail or need to be repeated, to avoid having to re-mix the full set of assembly primers.
The high fidelity (HF) version of the KpnI restriction enzyme is used for its compatibility with NdeI or XhoI enzymes in NEB Buffer #4.
Transformation of intact, supercoiled plasmid DNA is a high efficiency reaction. If using electroporation for this step, a single aliquot of electrocompetent cells can be used for up to ten transformations. To do this, we dilute an aliquot of thawed competent cells (commonly 50 μL) to the appropriate volume for ten individual transformation reactions (500 μL total volume), using sterile, ice-cold 10 % glycerol in water. Also, only a small amount of plasmid DNA is needed. Dilute the plasmid DNA 1:10 with water, and use 1 μL diluted DNA for the transformation.
The restriction enzyme NdeI (from New England Biolabs) is not as efficient as the other enzymes at cleaving DNA purified by plasmid isolation kits (as described in the technical details of the NEB product catalog). Compensate for this by using a larger volume of this enzyme in the reaction or by letting the digest run for a longer duration.
Undigested vector will significantly contaminate subsequent transformations, and this step should be performed carefully. If a considerable number of clones are identified as undigested vector in subsequent sequencing reactions, a new vector digestion and purification should be performed.
Colony sequencing: lightly touch a single colony with a small pipette tip, and transfer the tip to a PCR tube containing 6 μL of sterile, PCR-grade water. Heat to 96 °C for 6 min to lyse the cells. If the water was cloudy after adding the bacterial colony, it should now be clear. If the mixture is still cloudy after heating/lysis, this suggests that too many bacteria have been transferred, and the subsequent sequencing steps may yield poor data and possibly clog the sequencing machine. To the lysed cells, add 0.25 μL BigDye mix, 0.25 μL 10 mM primer, 2 μL 5× sequencing buffer, and water to a final volume of 10 μL. Proceed with PCR sequencing reaction. If using an external sequencing service, permission should be requested before submitting colony sequencing reactions.
After sequencing your clones, if contamination by undigested vector is a problem, use the following trick: Start with pET-CON vector containing a stuffer sequence which contains a unique restriction site NOT found in the pETCON vector sequence. It is best to use an enzyme which is compatible with NEB Buffer #1, because this buffer composition most closely matches that of the DNA ligation buffer. In our case, we started with pETCON vector containing a homing endonucle-ase ORF with a PacI restriction site. After the ligation step and PRIOR to transformation into bacteria, we added 10 μL of 1× NEB buffer #1 and 2× BSA to the ligation reaction (giving a new reaction volume of 20 μL), and 0.3 μL of the PacI restriction enzyme. Digesting for 1 h (followed by heat inactivation of the restriction enzyme) eliminates any undigested original vector from the ligation reaction, and the DESIRED ligation product remains intact. For the subsequent transformation step, we used DNA from the 20 μL PacI-treated ligation reaction (no purification necessary).
Target oligonucleotide substrate can be made in large batches and stored at −80 °C in a light-protected container. One 20 μL PCR reaction should yield approximately 12–14 μL of purified 300–500 nM substrate. When scaling up production of these substrates, maintain 20 μL PCR reaction volumes, and increase the number of reactions simultaneously run.
This gradual decrease in final temperature allows for high effi-ciency annealing into double-stranded DNA target oligonucle-otide (leaving minimal single-stranded or mis-annealed target).
ExoI is diluted with water to a total volume of 2 μL per sample for ease and accuracy of transfer to the PCR reaction. Do not add any of the supplied ExoI buffer.
Special care should be taken to avoid any bubbles in the sephadex suspension when mixing or aliquoting to the filter plate. Bubbles will lead to cracks within the final, centrifuged sephadex columns, and cracked columns should be discarded. We find that careful pipetting, using wide bore tips or standard p200 tips cut at approximately the 50 μL gradation, reduces frequency of cracking. We also find that allowing 30 min between pipetting and centrifugation can significantly reduce column cracking.
While it is more difficult to load samples without dye in the loading buffer, this allows for clear visualization of the target oligonucleotide and A647-labeled primer. Colored dyes will fluoresce under the Licor excitation wavelength and confound the image.
Frozen competent cells are prepared according to the published protocol by Gietz and Schiestl [10]. Add 2.5 × 109 EBY100 cells from an overnight 2× YPAD culture to 500 mL fresh 2× YPAD media. Grow at 30 °C to a density of at least 20 million/ mL. Pellet the cells and wash with sterile water. Resuspend the washed cell pellet in 5 mL of 5 % v/v glycerol + 10 % v/v DMSO in water. Aliquot 50-μL volumes to microcentrifuge tubes. Pack the tubes into a styrofoam rack with lid (or similar form of insulation) and place at −80 °C. (The insulation allows for gradual freezing of the cells.)
When transforming a library of variant homing endonucleases, increase the number of yeast and volume of the transformation mixture according to Gietz and Schiestl [11].
When transforming high quality plasmid DNA, a single frozen aliquot of competent yeast cells in the described volume of transformation mixture can be used for multiple reactions. In this case, divide the resuspended cells (prior to addition of DNA) into up to 15 equal volumes and add up to 1 μL total volume of plasmid DNA to each aliquot. Proceed to the incubation step.
We have found that an incubation time of 40–42 min at 42 °C provides the highest transformation efficiency with lowest cell death. Longer incubation times can lead to significant cell death. If using a high-quality plasmid, a shorter incubation time of 20 min will suffice for the generation of transformed clones.
Raffinose cultures can be successfully started using a single colony from a selective media + glucose plate. Alternatively, we have found that an initial overnight incubation in YPAD media (at 30 °C with 250 RPM shaking) can substantially increase induction efficiency, and the absence of selective media at this stage does not result in significant plasmid loss.
When using vertical tube racks inside a shaking incubator, position the 15-mL culture tubes at a slant to allow for maximum aeration, and do not use more than 1.5 mL of media.
On our spectrophotometer, the density of a yeast culture can be estimated by mixing a 1:10 dilution of yeast in water and measuring the resulting OD600. A simple calculation of OD600 × 300 provides an estimated value for density of the culture in millions of cells per mL. The validity of this estimate should be checked when using a different instrument.
Care should be taken to wash yeast from the raffinose culture at least twice before transferring to the galactose media. This limits carry-over of raffinose and/or glucose.
Induced galactose cultures should be kept on ice or at 4 °C. Well-folded homing endonucleases will be stably expressed on the yeast surface for several days, although the total expression levels and catalytic activity may decrease slightly over time, depending on the endonuclease.
If running a large number of samples, the assay can be performed in a 384-well conical-bottom plate, with 50,000 cells/ well. Volumes for staining and wash steps for a 384 well plate are: 8 μL anti-HA stain, 8 μL conjugated DNA-SAV-PE stain, and 10 μL anti-Myc-FITC stain. All washes should use a minimum 100 μL buffer.
If running a small number of samples, the assay can be performed using 1.5 mL microcentrifuge tubes. In this format, cells can be spun down in a tabletop centrifuge at speeds up to 10,000×g for 1 min. Perform 4 °C incubation steps on slow rotator, if possible.
A rotator at 4 °C can be used to assure continued suspension for thorough staining.
Include the volume of DNA target substrate in calculations for total conjugation reaction, as the dilution volume should be around 1:8 to 1:14, and is therefore substantial. The resulting slight decrease in IOCB salt concentration is not problematic at this point, and can be disregarded.
If using 1.5 mL microcentrifuge tubes, pipette the DNA target substrate onto the side of the tube not contacting SAV-PE, then gently vortex to mix the SAV-PE and DNA quickly. Likewise, in plate format the DNA target substrate should be pipetted onto the side of plate wells not contacting the SAV-PE mixture, if possible, and quickly mixed by vortex or multi-channel pipette.
Yeast can be left in anti-Myc FITC stain overnight, if necessary. Due to relatively low affinity of this antibody, FITC-stained cells should not be washed or diluted greater than 2× if cells are to sit for more than 4 h prior to acquisition.
25,000 yeast can be assayed in a 384-well plate format. Use a total volume of 20 μL. Low cell number is important to ensure that the effective concentration of DNA target substrate is not altered in affinity titration experiments.
For the I-OnuI family of homing endonucleases, specific binding can be detected from 100 pM to 50 nM. Higher concentrations of DNA target substrate may be non-specifically bound, and lower concentrations can be difficult to detect. This protocol can also be used to determine binding along a titration of various DNA concentrations.
Yeast can be used for up to 3 days following induction with preservation of most cleavage activity (when stored at 4 °C). However, because enzyme activity degrades over time, one should collect data from fresh samples for optimal activity.
The total concentration of enzyme used in this assay cannot be reliably standardized between populations, as efficiency of surface expression depends on the enzyme and varies considerably. Standard induction of a stable LHE should yield up to 105 individual enzymes per yeast cell [12]. Therefore this assay is used to approximate relative activities. In order to determine more exact values for Km or Kcat, or compare enzymes with a high degree of sensitivity, recombinant enzyme should be produced in and purified from bacteria.
Highly active, stable enzymes should show considerable cleavage after only 5 min. Cleavage of the DNA substrate should be completed by 1 h, and further incubation rarely yields any detectable increase in cleavage product. Shorter incubation times should be used when observing subtle differences between enzymes with comparable activities.
Supernatant can be stored at −20 °C in a light-protected container.
References
- 1.Takeuchi R, Lambert AR, Mak AN-S, et al. Tapping natural reservoirs of homing endonucleases for targeted gene modification. Proc Natl Acad Sci U S A. 2011;108:13077–13082. doi: 10.1073/pnas.1107719108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chevalier BS, Kortemme T, Chadsey MS, et al. Design, activity, and structure of a highly specific artificial endonuclease. Mol Cell. 2002;10:895–905. doi: 10.1016/s1097-2765(02)00690-1. [DOI] [PubMed] [Google Scholar]
- 3.Epinat JC, Arnould S, Chames P, et al. A novel engineered meganuclease induces homologous recombination in yeast and mammalian cells. Nucleic Acids Res. 2003;31:2952–2962. doi: 10.1093/nar/gkg375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baxter S, Lambert AR, Kuhar R, et al. Engineering domain fusion chimeras from I-OnuI family LAGLIDADG homing endo-nucleases. Nucleic Acids Res. 2012;40:7985–8000. doi: 10.1093/nar/gks502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Volná P, Jarjour J, Baxter S, et al. Flow cytometric analysis of DNA binding and cleavage by cell surface-displayed homing endonu-cleases. Nucleic Acids Res. 2007;35:2748–2758. doi: 10.1093/nar/gkm182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jarjour J, West-Foyle H, Certo MT, et al. High-resolution profiling of homing endonuclease binding and catalytic specificity using yeast surface display. Nucleic Acids Res. 2009;37:6871–6880. doi: 10.1093/nar/gkp726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rozen S, Skaletsky HJ. Primer3 on the WWW for general users and for biologist programmers. In: Krawetz S, Misener S, editors. Bioinformatics methods and protocols: methods in molecular biology. Humana; Totowa, NJ: 2000. pp. 365–386. [DOI] [PubMed] [Google Scholar]
- 8.Hoover DM, Lubkowski J. DNAWorks: an automated method for designing oligonu-cleotides for PCR-based gene synthesis. Nucleic Acids Res. 2002;30:e43. doi: 10.1093/nar/30.10.e43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gietz RD, Schiestl RH. High-efficiency yeast transformation using the LiAc/SS carrier DNA/PEG method. Nat Protoc. 2007;2:31–34. doi: 10.1038/nprot.2007.13. [DOI] [PubMed] [Google Scholar]
- 10.Gietz RD, Schiestl RH. Frozen competent yeast cells that can be transformed with high efficiency using the LiAc/SS carrier DNA/PEG method. Nat Protoc. 2007;2:1–4. doi: 10.1038/nprot.2007.17. [DOI] [PubMed] [Google Scholar]
- 11.Gietz RD, Schiestl RH. Large-scale high-efficiency yeast transformation using the LiAc/SS carrier DNA/PEG method. Nat Protoc. 2007;2:38–41. doi: 10.1038/nprot.2007.15. [DOI] [PubMed] [Google Scholar]
- 12.Boder ET, Wittrup KD. Yeast surface display for screening combinatorial polypeptide libraries. Nat Biotechnol. 1997;15:553–557. doi: 10.1038/nbt0697-553. [DOI] [PubMed] [Google Scholar]