

Abstract
Autoimmune and autoinflammatory diseases arise as a consequence of complex interactions of environmental factors with genetic traits. Although specific allelic variations cluster in predisposed individuals and promote the generation and/or expansion of autoreactive T and B lymphocytes, auto-immunity appears in various disease phenotypes and localizes to diverging tissues. Furthermore, the discovery that allelic variations within genes encoding components of the innate immune system drive self-reactive immune responses as well, led to the distinction of immune responses against host tissues into auto-immune and autoinflammatory diseases. In both categories of disorders, different pathogenic mechanisms and/or subsequent orders of tissue assaults may underlie the target cell specificity of the respective autoimmune attack. Furthermore, the transition from the initial tissue assault to the development of full-blown disease is likely driven by several factors. Thus, the development of specific forms of autoimmunity and autoinflammation reflects a multi-factorial process. The delineation of the specific factors involved in the pathogenic process is hampered by the fact that certain symptoms are assembled under the umbrella of a specific disease, although they might originate from diverging pathogenic pathways. These multi-factorial triggers and pathogenic pathways may also explain the inter-individual divergent courses and outcomes of diseases among humans. Here, we will discuss the impact of different environmental factors in general and microbial pathogens in particular on the regulation/ expression of genes encoded within susceptibility alleles, and its consequences on subsequent autoimmune and/or autoinflammatory tissue damage utilizing primarily the chronic cholestatic liver disease primary biliary cirrhosis as model.
Keywords: Autoimmunity, Autoinflammation, Genetic susceptibility, Infection, Host–pathogen interactions
Pathogenic Mechanisms Underlying the Induction of Self-Reactive Immune Responses
It is well established that defects in the selection of T and B cells lead to aberrant lymphocyte activation and subsequent autoimmune tissue damage (Davidson and Diamond 2001). The recent elucidation of mechanisms underlying self-directed tissue inflammation mediated through the innate immune response independent of abnormalities of the adaptive immune system led to the distinction of autoimmune from autoinflammatory disorders (McGonagle and McDermott 2006). Although several diseases have been labeled as autoimmune, not all of them have been firmly associated with the major histocompatibility complex and/ or specific autoantibodies (Rose and Bona 1993). Thus, the categorization and definition of several disease entities might need to be revised.
The complex interactions between multiple genetic traits underlying the generation of autoreactive and tissue-destructive immune responses in autoimmune diseases have been difficult to elucidate (Bjorses et al. 1998; Rioux and Abbas 2005; Walker and Abbas 2002). The complexity of these polygenic interactions and the fact that the incidence and prevalence of some of these autoimmune and/or autoinflammatory diseases are very rare complicate the definition of the interplay between gene variations encoding disease-promoting allelic polymorphisms and the consequences for the immune response.
Genetic susceptibility alone might not be sufficient to drive autoimmunity and/or autoinflammation. Nowadays, the induction of immune-mediated disease is considered to be the result of complex interactions between environmental factors and genetic traits (He et al. 2006) (Fig. 1). Although common tolerance mechanisms appear to be affected in several autoimmune diseases (Gregersen and Olsson 2009), the mechanisms leading to a tissue-specific autoimmune damage in the respective disorder have remained unknown. The fact that the autoimmune and/or autoinflammatory attack targets distinct organs and defined tissue sites becomes even more puzzling, as many of the autoantigens detected in autoimmune diseases are ubiquitously expressed (Rosen and Casciola-Rosen 2009).
Fig. 1.
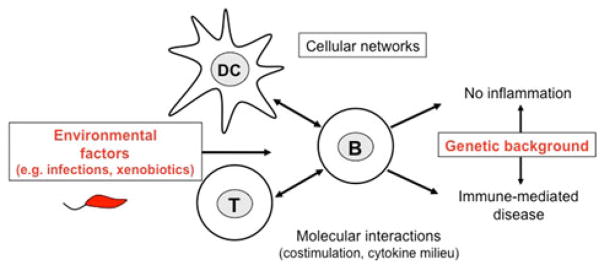
PBC is driven by complex interactions between environmental factors and genetic traits. Environmental factors trigger complex cellular networks and molecular circuits during the immune response of the host. Naturally occurring genetic polymorphisms of the host determine the immune response to these environmental triggers and subsequently the susceptibility of an individual to immune-mediated diseases such as PBC. Thus, the genetic background defines whether the interaction of the host with the environmental factor results in the tolerogenic resolution of the immune response and/or the elimination of the trigger in resistant individuals or contrarily leads to tissue damage and inflammation and/or persistence of the trigger in susceptible individuals
Infection Triggered Autoimmunity and/or Autoinflammation
The immune system has the difficult duty to distinguish between self- and foreign antigen patterns in order to fight infections without harming host cells. Autoimmune diseases arise as a consequence of impaired tolerance against self-antigens and the subsequent immune-mediated destruction of host tissues. Although inflammatory responses are primarily initiated to prevent or suspend tissue damage caused by invading pathogens, an exaggerated inflammation in response to infection can also cause tissue damage in genetically susceptible individuals (Janssen and Henson 2012).
Infections have been suggested as one of the factors that elicit immune responses against self-antigens. Two mechanisms promoting tissue damage by pathogens have been discussed in this context (Munz et al. 2009):
First, microbial pathogens create an inflammatory environment resulting in cellular damage and tissue destruction. Subsequently, increased numbers of self-antigens are presented to autoreactive lymphocytes that become activated in a pathogen-induced inflammatory environment.
Second, molecular mimicry has been implicated in the pathogenesis of several diseases that have been associated with specific bacteria including Guillain–Barre syndrome (Shahrizaila and Yuki 2011), rheumatic fever (Cunningham 2000), autoimmune gastritis (Amedei et al. 2003) and primary biliary cirrhosis (PBC) (Bogdanos et al. 2003, 2004a, b; Fussey et al. 1990; Mattner et al. 2008; Selmi et al. 2003). Structural homology between microbial and self-antigens is suspected to drive host tissue damage under these conditions.
Next to the growing body of evidence, that autoimmune diseases might be triggered by infections, both genetic susceptibility (Suzuki et al. 2011) and female predisposition (Fairweather and Rose 2004) are also commonly found in multiple autoimmune diseases. Although some distinct risk factors have been associated with defined autoimmune disorders, they may not be sufficient to drive on their own the complete pathogenesis of the respective disease. Thus, full-blown autoimmunity likely evolves in a multi-factorial process. Nonetheless, the initial tissue damage is likely the trigger that initiates the detrimental sequelae of pathogenic events in susceptible individuals. However, the identification of these factors has remained a challenge.
Primary Biliary Cirrhosis
PBC is a chronic cholestatic liver disease (Kaplan 1996) characterized by the immune-mediated destruction of small intrahepatic bile ducts and T and B cells that recognize the E2 subunit of the mitochondrial pyruvate dehydrogenase complex (PDC-E2) (Coppel et al. 1988; Gershwin et al. 1987; Shimoda et al. 1995; Van de Water et al. 1995; Yeaman et al. 1988). These E2 subunits are also found within other mitochondrial enzymes like the branched chain 2-oxo acid dehydrogenase complex, and the 2-oxoglutarate dehydrogenase complex and form the dominant antigenic epitope for T and B cells in PBC patients (Fussey et al. 1988).
Although PBC is referred to as autoimmune disease, PBC patients poorly respond to immune suppressants. Furthermore, the involvement of multiple components belonging to the innate immune system in the pathogenic processes suggests an important autoinflammatory disease component.
Histopathologic lesions in PBC present with a mixed lymphoid/mononuclear infiltrate around the bile ducts and an enhanced inflammatory cytokine profile while interleukin (IL)-10 levels are reduced (Baba et al. 2006; Honda et al. 2005; Shackel et al. 2001). Although B cells represent the cellular source for the signature anti-PDC-E2 autoantibodies, the clinical hallmark of PBC and are abundantly found within the periportal infiltrates, their role in the pathogenic process has remained unclear.
In contrast, T cells have been implicated in the pathogenesis of PBC (Invernizzi et al. 2010; Yeaman et al. 2000). Both CD4- and CD8-positive T cells have been shown to contribute to biliary damage. The pathogenicity of T cells that promote or inhibit disease is strongly influenced by the cytokines they secrete and requires antigen presentation by activated antigen-presenting cells (APC) and costimulatory interactions. From the three polarized disease-promoting CD4-positive T cell subsets, T helper (Th)1 and Th17 cells have been implicated in the pathogenesis of PBC (Lan et al. 2009; Mohammed et al. 2011). Thus, the interaction of the APC with the respective T cell needs to be tightly controlled.
Another T cell subset that has been implicated in the pathogenesis of PBC is the natural killer T (NKT) cells. NKT cells recognize glycosphingolipid (GSL) antigens presented by CD1d on dendritic cells (DCs), macrophages and B cells and release immediately various copies of cytokines upon T cell receptor (TCR) engagement. The numbers of NKT cells are increased in the livers of PBC patients (Harada et al. 2003; Kita et al. 2002). Furthermore, an increased expression of CD1d was observed in the livers of PBC patients (Tsuneyama et al. 1998). As the deletion of NKT cells in mouse models of PBC ameliorated the development of liver lesions (Chuang et al. 2008; Mattner et al. 2008), NKT cells appear to play a dominant role in the promotion of inflammatory processes leading to biliary bile duct destruction.
PBC as Infection Triggered Disease
The pathogenic mechanisms of biliary injury in PBC are poorly defined. The specific stimuli that trigger autoreactivity are unknown but include both genetic and environmental factors (He et al. 2006). Some bacteria and viruses have been suggested to trigger PBC (Abdulkarim et al. 2004; Burroughs et al. 1984; Hopf et al. 1989; Nilsson et al. 2000; Xu et al. 2003). In addition, patients with repeated urinary tract infections bear an increased risk to develop PBC (Bogdanos et al. 2003; Butler et al. 1993; Gershwin et al. 2005). These observations and the fact that PBC patients exhibit frequently a polyclonal hyper-immunoglobulin (Ig)M syndrome (Daniels et al. 2009; Feizi 1968; Moreira et al. 2010) that can also be observed during chronic infections support the hypothesis of a microbe being a likely causative agent. On the other hand, the antigen specificity of these antibody responses needs to be carefully examined as these polyclonal IgMs might react with many unrelated epitopes only by chance.
Molecular mimicry has been proposed as mechanism for the development of autoimmunity in PBC (Rieger and Gershwin 2007). Serum antibodies of PBC patients cross-react to conserved bacterial PDC-E2 homologs of Escherichia coli, Mycobacteria, and Lactobacilli species (Bogdanos et al. 2004a, b, 2005; Fussey et al. 1990). Among the infectious agents reported, lipoproteins of the ubiquitous alpha proteobacterium Novosphingobium aromaticivorans had the highest homology to the mammalian mitochondrial PDC-E2 epitopes, the signature antigens in PBC (Padgett et al. 2005; Selmi et al. 2003).
Novosphingobium aromaticivorans is a Gram-negative alpha proteobacterium (Takeuchi et al. 2001) that is ubiquitously found in the environment like other Novosphingobium/ Sphingomonas spp. Novosphingobium/Sphingomonas spp. have also been detected on human mucosal surfaces (Selmi et al. 2003), and several reports described opportunistic infections in immunocompromised patients (summarized in Ryan and Adley 2010). Novosphingobium/Sphingomonas spp. utilize GSLs instead of lipopolysaccharides to build their cell wall (Kawahara et al. 2000; Takeuchi et al. 2001). NKT cells recognize these GSL antigens and form the major innate recognition pathway for these bacteria in the absence of Toll-like receptor 4 signaling (Kinjo et al. 2005; Mattner et al. 2005). This observation and the fact that PBC patients exhibit a striking redistribution of NKT cells from the blood to the liver (Kita et al. 2002) support the idea that PBC patients may have experienced an infection with GSL-antigen bearing bacteria in the past.
Novosphingobium/Sphingomonas spp. also exhibit a capability to metabolize xenobiotics and to degrade poly-cyclic aromatics by utilizing them as the sole carbon source (Fredrickson et al. 1991; Habe and Omori 2003; Shi et al. 2001; Shuttleworth et al. 2000). Therefore, they could also interfere with the human hormone metabolism. Novo-sphingobium/Sphingomonas spp. are regularly isolated from terrestrial sites which are polluted with polycyclic aromatic hydrocarbons (Gupta et al. 2009; Suzuki and Hiraishi 2007). Thus, Novosphingobium/Sphingomonas spp. have even been used to detoxify industrial polluted soils (Notomista et al. 2011; Tiirola et al. 2002).
Based on these associations, we speculate that anti-PDC-E2 responses might reflect a chronic immune reaction against a latent bacterial infection rather than a detrimental autoimmune response. Reports suggesting that antimitochondrial antibodies (AMA)-negative PBC patients exhibit greater biliary pathology than AMA-positive patients (Jin et al. 2012) and that there is no correlation between antibody titers and disease activity (Benson et al. 2004; Van Norstrand et al. 1997) support the notion that B cell responses per se may not be pathogenic and/or not primarily target mammalian autoantigens. As anti-PDC-E2 antibody responses proceed the induction of liver pathology (Metcalf et al. 1996), the period between the detection of antibody responses and the onset of biliary pathology may mark a time frame in which the application of antibiotics may halt the development of full-blown PBC, assuming that the underlying pathogenic mechanisms are triggered by a bacterial infection.
Although bacteria expressing conserved homologous epitopes to the signature antigens of PBC have been associated with the initiation of biliary inflammation and the propagation of autoreactive T and B cells in this particular disease, additional trigger(s) and/or interactions with genetic susceptibility alleles contribute to the progression to liver fibrosis/cirrhosis, while other individuals expressing protective alleles may cope with the bacterial pathogen and/or autoreactive immune cells without causing tissue damage. Thus, our study not only suggests a critical role of infection in the initiation of autoimmune tissue damage, but also addresses the complex network of interactions that has to follow this initial assault in order to trigger full-blown disease.
Despite being the diagnostic hallmark of PBC, anti-PDC-E2 antibodies can occur in patients that do not have a florid liver disease (Mattalia et al. 1998; Metcalf et al. 1996). However, it remains unclear what triggers the development of biliary pathology as patients can remain asymptomatic besides being positive for AMAs. Unfortunately, a systematic analysis of these individuals has not been performed so far. Thus, one might speculate that these individuals may carry genetic resistance factors that allow the control of infection and/or prevent infection-induced liver damage, or they may not have experienced additional assaults that drive the subsequent development of full-blown disease and/or the transition to liver fibrosis/cirrhosis. In addition, it is likely that several steps are critical from the initial tissue assault to the induction of biliary inflammation, fibrosis, cirrhosis, and liver failure. Thus, it needs to be determined if a single tissue assault is sufficient to drive pathology or if a sequence of different events is required.
Genetic Susceptibility to Autoimmune Diseases
Allelic variations within defined genetic regions have been associated with increased susceptibility or resistance to autoimmunity (Gregersen and Olsson 2009). Single nucleotide polymorphisms (SNPs) often underlie these allelic variations and affect the function and/or expression of the respective encoded proteins. The defect of a single gene and/or altered interactions between several defective alleles are considered as one(s) of the most important underlying mechanisms for driving irregular immune responses and subsequent autoimmune tissue damage. As immune responses need to be tightly regulated, these allelic variations may affect the control over invading pathogens on the one hand and/or the limitation of exaggerated inflammatory processes on the other hand. Both are essential processes for the integrity of the host.
The mechanisms underlying the evolution of allelic variations are unknown, but it is suspected that environmental pressure elicited, for example, by microbial pathogens drives the development of SNPs. The best described example for a genetic mutation conferring protection from a pathogen is the HBB gene in β-thalassemia (summarized in Weatherall 2008): while the subsequent reduced or abolished synthesis of the β-globin chain of hemoglobin causes severe anemia and hepatosplenomegaly (Galanello and Origa 2010), the characteristic clinical symptoms of thalassemia, it enhances the resistance to malaria in endemic areas when expressed in heterozygous carriers (Hill et al. 1988; Willcox et al. 1983).
Nowadays, mechanisms that initiate, regulate, and terminate the immune response are distinguished. Particularly, an aberrant regulation of lymphocyte activation is considered as critical for the susceptibility to autoimmune disease and the initiation of autoimmune tissue damage. Here, we describe the regulation of lymphocyte activation by costimulatory molecules and cytokine signaling, but also address the impact of the hormone metabolism on different functions of the immune system and the predominance of autoimmune diseases in women.
Genetic Susceptibility to PBC
Several candidate genes have been described as genetic susceptibility factors for PBC. Among those there are susceptibility genes encoding costimulatory molecules like Cd80 (Mells et al. 2011) or Ctla-4 (Agarwal et al. 2000; Fan et al. 2004; Joshita et al. 2010; Juran et al. 2008a, b; Oertelt et al. 2005; Poupon et al. 2008; Walker et al. 2009); another group is represented by genes that have been implicated in the initiation of the innate immune response like the Il-12 (Hirschfield et al. 2009) pathway or Irf-5 (Hirschfield et al. 2010). While the two latter candidate pathways support the hypothesis that an inappropriate innate immune response toward infection may contribute to the initiation of PBC, the first ones hint to dysregulated adaptive immune responses.
Costimulatory Molecules
Costimulatory molecules are expressed on various (sub-) populations of APCs and lymphocytes (Leitner et al. 2010). The interaction of costimulatory molecules with their respective ligands enhances or suppresses the antigen-driven activation of T and B cells through their TCR and B cell receptor. Thus, positive and negative costimulatory molecules can be distinguished.
One potential candidate gene that may underlie this inadequate accumulation of serum IgM is the cytotoxic T lymphocyte antigen (CTLA)-4. SNP analyses identified Ctla-4 as a susceptibility gene for PBC (Agarwal et al. 2000; Fan et al. 2004; Joshita et al. 2010; Juran et al. 2008a, b; Oertelt et al. 2005; Poupon et al. 2008; Walker et al. 2009). CTLA-4 protein competes with CD28 for binding CD80 and CD86 (Masteller et al. 2000; Rudd 2008). While CTLA-4 is a negative costimulatory molecule that limits T cell activation (Tivol et al. 1995; Walunas et al. 1994; Waterhouse et al. 1995) and is a key regulator of self-tolerance (Takahashi et al. 2000; Wing et al. 2008), CD28 is a positive costimulatory molecule that augments signals transmitted through the TCR (Bluestone 1995; Jenkins et al. 1991; Linsley et al. 1991; Thompson et al. 1989). Most T cells, with the exception of FoxP3-positive regulatory T cells (Takahashi et al. 2000), express CTLA-4 only upon activation (Linsley et al. 1992) in contrast to CD28 that is constitutively expressed on naive T cells (summarized in Lenschow et al. 1996). As one of the mechanisms described for the suppressive action of CTLA-4, engagement of CTLA-4 on T cells with CD80 and CD86 on APCs results in the reduced expression of the latter ones, making them less available for the interaction with CD28 (Qureshi et al. 2011).
Ctla-4 polymorphisms belong to the leading examples of genetic variants that confer strong associations with multiple autoimmune diseases (summarized in Gough et al. 2005; Holmberg et al. 2005; Scalapino and Daikh 2008). Two SNPs within Ctla-4 associated with PBC correlate with enhanced T cell activation and autoantibody production (Agarwal et al. 2000; Fan et al. 2004; Juran et al. 2008b; Maurer et al. 2002; Oertelt et al. 2005; Zaletel et al. 2010), although this appears to be restricted to defined populations of PBC patients (Bittencourt et al. 2003). Furthermore, it needs to be elucidated, if polymorphisms within Ctla-4 are also involved in the promotion of these polyclonal IgM responses. Nonetheless, microbial pathogens may alter the functions of CTLA-4, either by inhibiting or enhancing its expression on T cells or by modulating the expression of its interaction partners, the B7 molecules (CD80/CD86) on APCs. Thus, one could speculate that an enhanced activation of CTLA-4 may disrupt the interaction of T with B cells subsequently impairing a proper isotype switch to IgG on the one hand.
As SNPs within Cd80 have been identified as genetic susceptibility factors for PBC as well (Mells et al. 2011), one could speculate that the interaction of CTLA-4 and CD80 might be disturbed resulting in augmented T cell activation and subsequent T cell-driven tissue damage on the other hand. Accordingly, aberrant T cell activation might result from a decreased function and/or expression of CTLA-4, while CD80 expression/function could be even enhanced despite interacting with CTLA-4. Both mechanisms are suitable for promoting the interaction of CD80/ CD86 with CD28 on T cells resulting in subsequent T cell activation. Thus, disease-associated SNPs have to be carefully examined in the respective disease circumstances and the critical cell populations involved in the pathological process.
Both types of costimulatory molecules are tightly controlled as they are critical for an appropriate response to different antigens. If a self-antigen, due to the uptake of materials released from apoptotic cells, is presented to the TCR (Bellone et al. 1997), the APC presenting that antigen likely creates a tolerizing environment by upregulating negative costimulatory molecules (Getts et al. 2011) and releasing immune suppressive cytokines such as IL-10 (Gray et al. 2007; Voll et al. 1997) or transforming growth factor β (Fadok et al. 1998). In order to mount a protective immune response to foreign antigenic material, in example during a microbial infection, an inflammatory response has to be created. Thus, T cells require positive costimulatory signals (Leitner et al. 2010) and inflammatory cytokines for their activation (Curtsinger et al. 1999). Both the expression of positive costimulatory signals and the release of inflammatory cytokines are induced by APCs after engagement of innate immune receptors through defined microbial antigen patterns (Banchereau and Steinman 1998; Iwasaki and Medzhitov 2004; Jakob et al. 1998; Sparwasser et al. 1998; Verhasselt et al. 1997). Thus, these additional signals likely evolved together with T cells in order to help the immune system to distinguish between self- and foreign antigens. SNPs within these signal-promoting molecules might be the consequence of microbial pressure increasing the survival of populations under infectious thread, but lowering the threshold for T cell activation in inflammatory responses not related to fighting microbial infection.
Cytokine Signaling
Similar to costimulatory molecules, which positively or negatively regulate interactions between APCs and T cells, cytokines are important to activate or inhibit the immune response. Not surprisingly, several cytokines or mediators of their downstream signaling cascades have been associated with a number of autoimmune diseases. Recently, the association of a SNP within the tyrosine kinase 2 (Tyk2) encoding gene with PBC was reported (Liu et al. 2012). Previously, this locus has also been associated with other autoimmune diseases such as multiple sclerosis (MS) (Ban et al. 2009), type 1 diabetes (Wallace et al. 2010), and systemic lupus erythematosus (SLE) (Suarez-Gestal et al. 2009). As a member of the Janus kinase family, Tyk2 transmits signals of activated cytokine receptors via phosphorylation of signal transducer and activator of transcription (STATs) (Ghoreschi et al. 2009; Krolewski et al. 1990). One important cytokine that activates Tyk2 is IL-12 (Bacon et al. 1995a), which is produced by APCs upon pathogen encounter (Del Vecchio et al. 2007). The IL-12 signal is mainly transmitted by STAT-4 (Bacon et al. 1995b; Jacobson et al. 1995) and drives the Th1 differentiation of naïve CD4-positive T cells and the interferon c production by T cells and NK cells (Chan et al. 1991; Manetti et al. 1993; Shimoda et al. 2000). Accordingly, Tyk2-deficient mice are more susceptible to viral and bacterial infections (Aizu et al. 2006; Karaghiosoff et al. 2000). Due to the reduced inhibitory effects of Th1 signals on Th2 immunity, Tyk2-deficient mice show increased antigen-induced allergic inflammation (Seto et al. 2003). With respect to autoimmune diseases, Tyk2-mutant mice are resistant to collagen-induced arthritis and experimental autoimmune encephalomyelitis (Ortmann et al. 2001; Oyamada et al. 2009; Spach et al. 2009). The risk allele of the PBC-associated SNP rs34536443 was shown to confer an enhanced enzyme activity in comparison with the protective allele (Couturier et al. 2011). Further analysis revealed that the reduced activity of Tyk2 expressed by the protective C allele enforces a shift toward Th2 immune responses (Couturier et al. 2011). Hence, an elevated Tyk2 activity in susceptible PBC patients might be responsible for an increased Th1 cytokine milieu that drives biliary tissue damage. Considering PBC as a microbe-driven disease, acute or chronic infections might even further amplify this Th1 prone status.
Hormone Metabolism and Female Predisposition
Females appear to benefit from some differences in their immune response compared to men: (a) female newborns are less vulnerable to some microbial pathogens; (b) during later stages of life, women also reveal improved clinical outcome and survival rates from infectious diseases, trauma or injury (reviewed in Libert et al. 2010). Females exhibit higher levels of serum IgM (Butterworth et al. 1967; Stoop et al. 1969), increased numbers of CD4-positive T cells (Amadori et al. 1995; Lee et al. 1996; Tollerud et al. 1989), and enhanced cytokine production in response to infections (Nalbandian and Kovats 2005; Rubtsov et al. 2010). On the other hand, these differences in the immune response may contribute to the elevated incidence rates of autoimmune diseases like Sjogren’s syndrome, SLE, PBC, autoimmune thyroid diseases, RA and MS among women (Beeson 1994).
Sex hormones likely contribute to these different clinical outcomes, as they have been reported to impact different immunological functions (Bouman et al. 2005; Cunningham and Gilkeson 2011): Both innate and adaptive immune cells express estrogen receptors, including B cells, CD8-positive T cells, monocytes, and neutrophils (Cohen et al. 1983; Molero et al. 2002; Stimson 1988; Suenaga et al. 1998). Estrogen also contributes to the differentiation of DCs (Paharkova-Vatchkova et al. 2004) and increases IgG production by B cells (Kanda and Tamaki 1999; Kanda et al. 1999) as well as cytokine secretion by CD4-positive T cells (Gilmore et al. 1997). As sex hormones are also involved in the modulation of the hypothalamic–pituitary–adrenal axis, they can affect the immune system indirectly through the regulation of other hormones (Wilder 1995). Another indication for the important role of sex hormones in autoimmune diseases emerges from observations obtained during pregnancy: during the third trimester, when estrogen and progesterone levels are massively increased, an amelioration of the disease phenotypes in MS and RA patients is monitored (Confavreux et al. 1998; Nelson and Ostensen 1997). The hormones presumably suppress Th1 signaling by peripheral T lymphocytes (Forger et al. 2008; Munoz-Suano et al. 2012). In contrast, pregnancy tends to worsen clinical symptoms in patients with SLE, a Th2-related disease, in which autoantibodies and B cells are responsible for the disease phenotype, which are also aberrantly activated under Th2 conditions (Rubtsov et al. 2010; Wegmann et al. 1993).
Similar in many other autoimmune diseases, there exists a strong female predominance (gender ratio up to 10:1) in PBC. However, it remains speculative if this is due to an X-chromosome-linked locus of susceptibility (Invernizzi et al. 2004; Miozzo et al. 2007; Mitchell et al. 2011). Furthermore, it has been suggested that estrogen is involved in the homeostatic proliferation of cholangiocytes in the early stages of PBC (Alvaro et al. 2004; Fouassier et al. 2009) and that polymorphisms in estrogen receptor genes are associated with disease (Lakatos et al. 2002). In addition, it has been shown that bacteria can interfere with the hormone metabolism as outlined below.
Interference of Sphingomonas/Novosphingobium spp. with Xenobiotic Materials and the Hormone Metabolism
As described before, Sphingomonas/Novosphingobium spp. have the unique capability to interfere with xenobiotics and the hormone metabolism (Stolz 2009). Although our animal studies provided evidence that Sphingomonas/ Novosphingobium spp. have the potential to initiate liver damage and to promote severe biliary inflammation dependent on the genetic background of the mice, none of the infected mouse strains progressed to liver fibrosis or cirrhosis (Mattner et al. 2008; Mohammed et al. 2011). Despite the technical difficulties to induce liver fibrosis/ cirrhosis in mouse models, a second signal might be required to initiate the progression from inflammation to fibrosis/cirrhosis. As the incidence of PBC is higher in polluted areas, chemicals might be involved in the pathologic process (Ala et al. 2006). Chemical xenobiotics like 6-bromohexanoate have been reported to induce anti-PDC-E2 responses and liver pathology as well (Leung et al. 2003, 2007). Thus, a combination of a bacterium and a chemical substance might be critical for the induction of a complete histopathologic PBC picture in the mouse model. As Sphingomonas/Novosphingobium spp. can metabolize xenobiotic compounds, the interference with the bacterial metabolism may create a biological toxic substance for the promotion of liver damage with subsequent fibrosis.
Among the xenobiotic compounds degraded by Novosphingobium/Sphingomonas strains biphenyl, carbazole (chlorinated) dibenzo-p-dioxins, (chlorinated) furans, fluorene, (substituted) naphthalenes, (substituted) phenanthrenes, chlorinated phenols and pyrene as well as different herbicides and pesticides can be found (Basta et al. 2004; Fredrickson et al. 1991, 1995; Habe et al. 2002; Sohn et al. 2004; Stolz 2009; Suzuki and Hiraishi 2007; Tiirola et al. 2002; Yan et al. 2007; Yuan et al. 2009). Of particular, interest is the capacity of Novosphingobium/Sphingomonas spp. to interfere with the estradiol metabolism (Fujii et al. 2002), especially with respect to the fact that PBC is characterized by a striking female predominance, similarly as many other autoimmune diseases (Beeson 1994). Studies evaluating the presence of putative endocrine disruptors in natural sources described the isolation of Novosphingobium/Sphingomonas strains from various environmental samples (Corvini et al. 2006; Fujii et al. 2002, 2003; Gabriel et al. 2005; Kang et al. 2007; Porter and Hay 2007; Tanghe et al. 1999; Ternes et al. 1999).
Summary and Outlook
The etiology of PBC has remained largely unknown. Only one single drug, ursodeoxycholic acid, has been approved to date for treating this devastating liver disease (Heathcote 2000; Ishibashi et al. 2007; Poupon et al. 1987), although its first description dates back to 1851 (Heathcote 2003). While animal models provided valuable insights into the genetic susceptibility to PBC (Leung et al. 2012; Tsuneyama et al. 2012), the trigger(s) driving the inflammatory and tissue-destructive cellular network have not been identified. Although recent reports suggested that bacteria initiate biliary damage and promote the expansion of autoreactive T and B lymphocytes, the inflammatory liver lesions initiated in those models do not progress to liver fibrosis and/or cirrhosis. Thus, at least a second environmental and/or genetic factor might be required for the induction of full-blown disease. Complementary to these studies, it needs to be addressed if appropriate antibiotic treatment is sufficient to prevent the progression to biliary liver disease in AMA-positive individuals.
Acknowledgments
J. Mattner is supported by the Interdisciplinary Center for Clinical Research of the Universitätsklinikum Erlangen (IZKF_JB10_A48), the German Research Foundation DFG (MA 2621/2-1) and by a grant (award number R01DK084054) from the National Institute of Diabetes and Digestive and Kidney Diseases.
Abbreviations
- AMA
Antimitochondrial antibodies
- APC
Antigen-presenting cell
- CTLA-4
Cytotoxic T lymphocyte antigen 4
- DC
Dendritic cell
- GSL
Glycosphingolipid
- IgM
Immunoglobulin M
- IL-10
Interleukin 10
- MS
Multiple sclerosis
- NKT
Natural killer T cell
- PBC
Primary biliary cirrhosis
- PDC-E2
Pyruvate dehydrogenase complex E2 subunit
- RA
Rheumatoid arthritis
- SLE
Systemic lupus erythematosus
- SNP
Single nucleotide polymorphism
- TCR
T cell receptor
- Th cells
T helper cells
- Tyk2
Tyrosine kinase 2
Contributor Information
Claudia Danzer, Mikrobiologisches Institut, Klinische Mikrobiologie, Immunologie Und Hygiene, Universitätsklinikum Erlangen and Friedrich-Alexander Universität Erlangen-Nürnberg, Wasserturmstr. 3/5, 91054 Erlangen, Germany.
Jochen Mattner, Email: Jochen.Mattner@uk-erlangen.de, Mikrobiologisches Institut, Klinische Mikrobiologie, Immunologie Und Hygiene, Universitätsklinikum Erlangen and Friedrich-Alexander Universität Erlangen-Nürnberg, Wasserturmstr. 3/5, 91054 Erlangen, Germany. Division of Cellular and Molecular Immunology, Cincinnati Children’s Hospital, Cincinnati, OH 45229, USA.
References
- Abdulkarim AS, Petrovic LM, Kim WR, et al. Primary biliary cirrhosis: an infectious disease caused by Chlamydia pneumoniae? J Hepatol. 2004;40:380–384. doi: 10.1016/j.jhep.2003.11.033. [DOI] [PubMed] [Google Scholar]
- Agarwal K, Jones DE, Daly AK, et al. CTLA-4 gene polymorphism confers susceptibility to primary biliary cirrhosis. J Hepatol. 2000;32:538–541. doi: 10.1016/s0168-8278(00)80213-5. [DOI] [PubMed] [Google Scholar]
- Aizu K, Li W, Yajima T, et al. An important role of Tyk2 in APC function of dendritic cells for priming CD8+ T cells producing IFN-gamma. Eur J Immunol. 2006;36:3060–3070. doi: 10.1002/eji.200636173. [DOI] [PubMed] [Google Scholar]
- Ala A, Stanca CM, Bu-Ghanim M, et al. Increased prevalence of primary biliary cirrhosis near Superfund toxic waste sites. Hepatology. 2006;43:525–531. doi: 10.1002/hep.21076. [DOI] [PubMed] [Google Scholar]
- Alvaro D, Invernizzi P, Onori P, et al. Estrogen receptors in cholangiocytes and the progression of primary biliary cirrhosis. J Hepatol. 2004;41:905–912. doi: 10.1016/j.jhep.2004.08.022. [DOI] [PubMed] [Google Scholar]
- Amadori A, Zamarchi R, De Silvestro G, et al. Genetic control of the CD4/CD8 T-cell ratio in humans. Nat Med. 1995;1:1279–1283. doi: 10.1038/nm1295-1279. [DOI] [PubMed] [Google Scholar]
- Amedei A, Bergman MP, Appelmelk BJ, et al. Molecular mimicry between Helicobacter pylori antigens and H+, K+— adenosine triphosphatase in human gastric autoimmunity. J Exp Med. 2003;198:1147–1156. doi: 10.1084/jem.20030530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baba N, Kobashi H, Yamamoto K, et al. Gene expression profiling in biliary epithelial cells of primary biliary cirrhosis using laser capture microdissection and cDNA microarray. Transl Res. 2006;148:103–113. doi: 10.1016/j.trsl.2006.04.007. [DOI] [PubMed] [Google Scholar]
- Bacon CM, McVicar DW, Ortaldo JR, et al. Interleukin 12 (IL-12) induces tyrosine phosphorylation of JAK2 and TYK2: differential use of Janus family tyrosine kinases by IL-2 and IL-12. J Exp Med. 1995a;181:399–404. doi: 10.1084/jem.181.1.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacon CM, Petricoin EF, 3rd, Ortaldo JR, et al. Interleukin 12 induces tyrosine phosphorylation and activation of STAT4 in human lymphocytes. Proc Natl Acad Sci USA. 1995b;92:7307–7311. doi: 10.1073/pnas.92.16.7307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ban M, Goris A, Lorentzen AR, et al. Replication analysis identifies TYK2 as a multiple sclerosis susceptibility factor. Eur J Hum Genet. 2009;17:1309–1313. doi: 10.1038/ejhg.2009.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature. 1998;392:245–252. doi: 10.1038/32588. [DOI] [PubMed] [Google Scholar]
- Basta T, Keck A, Klein J, et al. Detection and characterization of conjugative degradative plasmids in xenobiotic-degrading Sphingomonas strains. J Bacteriol. 2004;186:3862–3872. doi: 10.1128/JB.186.12.3862-3872.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beeson PB. Age and sex associations of 40 autoimmune diseases. Am J Med. 1994;96:457–462. doi: 10.1016/0002-9343(94)90173-2. [DOI] [PubMed] [Google Scholar]
- Bellone M, Iezzi G, Rovere P, et al. Processing of engulfed apoptotic bodies yields T cell epitopes. J Immunol. 1997;159:5391–5399. [PubMed] [Google Scholar]
- Benson GD, Kikuchi K, Miyakawa H, et al. Serial analysis of antimitochondrial antibody in patients with primary biliary cirrhosis. Clin Dev Immunol. 2004;11:129–133. doi: 10.1080/10446670410001722113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bittencourt PL, Palacios SA, Farias AQ, et al. Analysis of major histocompatibility complex and CTLA-4 alleles in Brazilian patients with primary biliary cirrhosis. J Gastroenterol Hepatol. 2003;18:1061–1066. doi: 10.1046/j.1440-1746.2003.03091.x. [DOI] [PubMed] [Google Scholar]
- Bjorses P, Aaltonen J, Horelli-Kuitunen N, et al. Gene defect behind APECED: a new clue to autoimmunity. Hum Mol Genet. 1998;7:1547–1553. doi: 10.1093/hmg/7.10.1547. [DOI] [PubMed] [Google Scholar]
- Bluestone JA. New perspectives of CD28-B7-mediated T cell costimulation. Immunity. 1995;2:555–559. doi: 10.1016/1074-7613(95)90000-4. [DOI] [PubMed] [Google Scholar]
- Bogdanos DP, Baum H, Butler P, et al. Association between the primary biliary cirrhosis specific anti-sp100 antibodies and recurrent urinary tract infection. Dig Liver Dis. 2003;35:801–805. doi: 10.1016/s1590-8658(03)00466-3. [DOI] [PubMed] [Google Scholar]
- Bogdanos DP, Baum H, Grasso A, et al. Microbial mimics are major targets of crossreactivity with human pyruvate dehydro-genase in primary biliary cirrhosis. J Hepatol. 2004a;40:31–39. doi: 10.1016/s0168-8278(03)00501-4. [DOI] [PubMed] [Google Scholar]
- Bogdanos DP, Pares A, Baum H, et al. Disease-specific cross-reactivity between mimicking peptides of heat shock protein of Mycobacterium gordonae and dominant epitope of E2 subunit of pyruvate dehydrogenase is common in Spanish but not British patients with primary biliary cirrhosis. J Autoimmun. 2004b;22:353–362. doi: 10.1016/j.jaut.2004.03.002. [DOI] [PubMed] [Google Scholar]
- Bogdanos DP, Baum H, Okamoto M, et al. Primary biliary cirrhosis is characterized by IgG3 antibodies cross-reactive with the major mitochondrial autoepitope and its Lactobacillus mimic. Hepatology. 2005;42:458–465. doi: 10.1002/hep.20788. [DOI] [PubMed] [Google Scholar]
- Bouman A, Heineman MJ, Faas MM. Sex hormones and the immune response in humans. Hum Reprod Update. 2005;11:411–423. doi: 10.1093/humupd/dmi008. [DOI] [PubMed] [Google Scholar]
- Burroughs AK, Rosenstein IJ, Epstein O, et al. Bacteriuria and primary biliary cirrhosis. Gut. 1984;25:133–137. doi: 10.1136/gut.25.2.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler P, Valle F, Hamilton-Miller JM, et al. M2 mitochondrial antibodies and urinary rough mutant bacteria in patients with primary biliary cirrhosis and in patients with recurrent bacteriuria. J Hepatol. 1993;17:408–414. doi: 10.1016/s0168-8278(05)80225-9. [DOI] [PubMed] [Google Scholar]
- Butterworth M, McClellan B, Allansmith M. Influence of sex in immunoglobulin levels. Nature. 1967;214:1224–1225. doi: 10.1038/2141224a0. [DOI] [PubMed] [Google Scholar]
- Chan SH, Perussia B, Gupta JW, et al. Induction of interferon gamma production by natural killer cell stimulatory factor: characterization of the responder cells and synergy with other inducers. J Exp Med. 1991;173:869–879. doi: 10.1084/jem.173.4.869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang YH, Lian ZX, Yang GX, et al. Natural killer T cells exacerbate liver injury in a transforming growth factor beta receptor II dominant-negative mouse model of primary biliary cirrhosis. Hepatology. 2008;47:571–580. doi: 10.1002/hep.22052. [DOI] [PubMed] [Google Scholar]
- Cohen JH, Danel L, Cordier G, et al. Sex steroid receptors in peripheral T cells: absence of androgen receptors and restriction of estrogen receptors to OKT8-positive cells. J Immunol. 1983;131:2767–2771. [PubMed] [Google Scholar]
- Confavreux C, Hutchinson M, Hours MM, et al. Rate of pregnancy-related relapse in multiple sclerosis. Pregnancy in multiple sclerosis group. N Engl J Med. 1998;339:285–291. doi: 10.1056/NEJM199807303390501. [DOI] [PubMed] [Google Scholar]
- Coppel RL, McNeilage LJ, Surh CD, et al. Primary structure of the human M2 mitochondrial autoantigen of primary biliary cirrhosis: dihydrolipoamide acetyltransferase. Proc Natl Acad Sci USA. 1988;85:7317–7321. doi: 10.1073/pnas.85.19.7317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corvini PF, Hollender J, Ji R, et al. The degradation of alpha-quaternary nonylphenol isomers by Sphingomonas sp. strain TTNP3 involves a type II ipso-substitution mechanism. Appl Microbiol Biotechnol. 2006;70:114–122. doi: 10.1007/s00253-005-0080-0. [DOI] [PubMed] [Google Scholar]
- Couturier N, Bucciarelli F, Nurtdinov RN, et al. Tyrosine kinase 2 variant influences T lymphocyte polarization and multiple sclerosis susceptibility. Brain. 2011;134(Pt 3):693–703. doi: 10.1093/brain/awr010. [DOI] [PubMed] [Google Scholar]
- Cunningham MW. Pathogenesis of group A streptococcal infections. Clin Microbiol Rev. 2000;13:470–511. doi: 10.1128/cmr.13.3.470-511.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham M, Gilkeson G. Estrogen receptors in immunity and autoimmunity. Clin Rev Allergy Immunol. 2011;40:66–73. doi: 10.1007/s12016-010-8203-5. [DOI] [PubMed] [Google Scholar]
- Curtsinger JM, Schmidt CS, et al. Inflammatory cytokines provide a third signal for activation of naive CD4+ and CD8+ T cells. J Immunol. 1999;162:3256–3262. [PubMed] [Google Scholar]
- Daniels JA, Torbenson M, Anders RA, et al. Immunostaining of plasma cells in primary biliary cirrhosis. Am J Clin Pathol. 2009;131:243–249. doi: 10.1309/AJCP8WHR0IEVUUOJ. [DOI] [PubMed] [Google Scholar]
- Davidson A, Diamond B. Autoimmune diseases. N Engl J Med. 2001;345:340–350. doi: 10.1056/NEJM200108023450506. [DOI] [PubMed] [Google Scholar]
- Del Vecchio M, Bajetta E, Canova S, et al. Interleukin-12: biological properties and clinical application. Clin Cancer Res. 2007;13:4677–4685. doi: 10.1158/1078-0432.CCR-07-0776. [DOI] [PubMed] [Google Scholar]
- Fadok VA, Bratton DL, Konowal A, et al. Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-beta, PGE2, and PAF. J Clin Invest. 1998;101:890–898. doi: 10.1172/JCI1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fairweather D, Rose NR. Women and autoimmune diseases. Emerg Infect Dis. 2004;10:2005–2011. doi: 10.3201/eid1011.040367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan LY, Tu XQ, Cheng QB, et al. Cytotoxic T lymphocyte associated antigen-4 gene polymorphisms confer susceptibility to primary biliary cirrhosis and autoimmune hepatitis in Chinese population. World J Gastroenterol. 2004;10:3056–3059. doi: 10.3748/wjg.v10.i20.3056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feizi T. Immunoglobulins in chronic liver disease. Gut. 1968;9:193–198. doi: 10.1136/gut.9.2.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forger F, Marcoli N, Gadola S, et al. Pregnancy induces numerical and functional changes of CD4 + CD25 high regulatory T cells in patients with rheumatoid arthritis. Ann Rheum Dis. 2008;67:984–990. doi: 10.1136/ard.2007.075283. [DOI] [PubMed] [Google Scholar]
- Fouassier L, Rosenberg P, Mergey M, et al. Ezrin-radixin-moesin-binding phosphoprotein (EBP50), an estrogen-inducible scaffold protein, contributes to biliary epithelial cell proliferation. Am J Pathol. 2009;174:869–880. doi: 10.2353/ajpath.2009.080079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredrickson JK, Brockman FJ, Workman DJ, et al. Isolation and characterization of a subsurface bacterium capable of growth on toluene, naphthalene, and other aromatic compounds. Appl Environ Microbiol. 1991;57:796–803. doi: 10.1128/aem.57.3.796-803.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredrickson JK, Balkwill DL, Drake GR, et al. Aromatic-degrading Sphingomonas isolates from the deep subsurface. Appl Environ Microbiol. 1995;61:1917–1922. doi: 10.1128/aem.61.5.1917-1922.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujii K, Kikuchi S, Satomi M, et al. Degradation of 17beta-estradiol by a gram-negative bacterium isolated from activated sludge in a sewage treatment plant in Tokyo, Japan. Appl Environ Microbiol. 2002;68:2057–2060. doi: 10.1128/AEM.68.4.2057-2060.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujii K, Yamamoto R, Tanaka T, et al. Potential of a new biotreatment: sphingomonas cloacae S-3T degrades nonylphenol in industrial wastewater. J Ind Microbiol Biotechnol. 2003;30:531–535. doi: 10.1007/s10295-003-0072-y. [DOI] [PubMed] [Google Scholar]
- Fussey SP, Guest JR, James OF, et al. Identification and analysis of the major M2 autoantigens in primary biliary cirrhosis. Proc Natl Acad Sci USA. 1988;85:8654–8658. doi: 10.1073/pnas.85.22.8654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fussey SP, Ali ST, Guest JR, et al. Reactivity of primary biliary cirrhosis sera with Escherichia coli dihydrolipoamide acetyl-transferase (E2p): characterization of the main immunogenic region. Proc Natl Acad Sci USA. 1990;87:3987–3991. doi: 10.1073/pnas.87.10.3987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabriel FL, Giger W, Guenther K, et al. Differential degradation of nonylphenol isomers by Sphingomonas xenophaga Bayram. Appl Environ Microbiol. 2005;71:1123–1129. doi: 10.1128/AEM.71.3.1123-1129.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galanello R, Origa R. Beta-thalassemia. Orphanet J Rare Dis. 2010;5:11. doi: 10.1186/1750-1172-5-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gershwin ME, Mackay IR, Sturgess A, et al. Identification and specificity of a cDNA encoding the 70 kd mitochondrial antigen recognized in primary biliary cirrhosis. J Immunol. 1987;138:3525–3531. [PubMed] [Google Scholar]
- Gershwin ME, Selmi C, Worman HJ, et al. Risk factors and comorbidities in primary biliary cirrhosis: a controlled interview-based study of 1032 patients. Hepatology. 2005;42:1194–1202. doi: 10.1002/hep.20907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Getts DR, Turley DM, Smith CE, et al. Tolerance induced by apoptotic antigen-coupled leukocytes is induced by PD-L1+ and IL-10-producing splenic macrophages and maintained by T regulatory cells. J Immunol. 2011;187:2405–2417. doi: 10.4049/jimmunol.1004175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghoreschi K, Laurence A, O’Shea JJ. Janus kinases in immune cell signaling. Immunol Rev. 2009;228:273–287. doi: 10.1111/j.1600-065X.2008.00754.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilmore W, Weiner LP, Correale J. Effect of estradiol on cytokine secretion by proteolipid protein-specific T cell clones isolated from multiple sclerosis patients and normal control subjects. J Immunol. 1997;158:446–451. [PubMed] [Google Scholar]
- Gough SC, Walker LS, Sansom DM. CTLA4 gene polymorphism and autoimmunity. Immunol Rev. 2005;204:102–115. doi: 10.1111/j.0105-2896.2005.00249.x. [DOI] [PubMed] [Google Scholar]
- Gray M, Miles K, Salter D, et al. Apoptotic cells protect mice from autoimmune inflammation by the induction of regulatory B cells. Proc Natl Acad Sci USA. 2007;104:14080–14085. doi: 10.1073/pnas.0700326104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregersen PK, Olsson LM. Recent advances in the genetics of autoimmune disease. Annu Rev Immunol. 2009;27:363–391. doi: 10.1146/annurev.immunol.021908.132653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta SK, Lal D, Lal R. Novosphingobium panipatense sp. nov. and Novosphingobium mathurense sp. nov., from oil-contaminated soil. Int J Syst Evol Microbiol. 2009;59(Pt 1):156–161. doi: 10.1099/ijs.0.65743-0. [DOI] [PubMed] [Google Scholar]
- Habe H, Omori T. Genetics of polycyclic aromatic hydrocarbon metabolism in diverse aerobic bacteria. Biosci Biotechnol Biochem. 2003;67:225–243. doi: 10.1271/bbb.67.225. [DOI] [PubMed] [Google Scholar]
- Habe H, Ashikawa Y, Saiki Y, et al. Sphingomonas sp. strain KA1, carrying a carbazole dioxygenase gene homologue, degrades chlorinated dibenzo-p-dioxins in soil. FEMS Microbiol Lett. 2002;211:43–49. doi: 10.1111/j.1574-6968.2002.tb11201.x. [DOI] [PubMed] [Google Scholar]
- Harada K, Isse K, Tsuneyama K, et al. Accumulating CD57 + CD3 + natural killer T cells are related to intrahepatic bile duct lesions in primary biliary cirrhosis. Liver Int. 2003;23:94–100. doi: 10.1034/j.1600-0676.2003.00807.x. [DOI] [PubMed] [Google Scholar]
- He XS, Ansari AA, Ridgway WM, et al. New insights to the immunopathology and autoimmune responses in primary biliary cirrhosis. Cell Immunol. 2006;239:1–13. doi: 10.1016/j.cellimm.2006.04.006. [DOI] [PubMed] [Google Scholar]
- Heathcote EJ. Management of primary biliary cirrhosis. The American Association for the Study of Liver Diseases practice guidelines. Hepatology. 2000;31:1005–1013. doi: 10.1053/he.2000.5984. [DOI] [PubMed] [Google Scholar]
- Heathcote EJ. Primary biliary cirrhosis: historical perspective. Clin Liver Dis. 2003;7:735–740. doi: 10.1016/s1089-3261(03)00098-9. [DOI] [PubMed] [Google Scholar]
- Hill AV, Bowden DK, O’Shaughnessy DF, et al. Beta thalassemia in Melanesia: association with malaria and characterization of a common variant (IVS-1 nt 5 G—C) Blood. 1988;72:9–14. [PubMed] [Google Scholar]
- Hirschfield GM, Liu X, Xu C, et al. Primary biliary cirrhosis associated with HLA, IL12A, and IL12RB2 variants. N Engl J Med. 2009;360:2544–2555. doi: 10.1056/NEJMoa0810440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirschfield GM, Liu X, Han Y, et al. Variants at IRF5-TNPO3, 17q12-21 and MMEL1 are associated with primary biliary cirrhosis. Nat Genet. 2010;42:655–657. doi: 10.1038/ng.631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmberg D, Cilio CM, Lundholm M, et al. CTLA-4 (CD152) and its involvement in autoimmune disease. Autoimmunity. 2005;38:225–233. doi: 10.1080/08916930500050210. [DOI] [PubMed] [Google Scholar]
- Honda M, Kawai H, Shirota Y, et al. Differential gene expression profiles in stage I primary biliary cirrhosis. Am J Gastroenterol. 2005;100:2019–2030. doi: 10.1111/j.1572-0241.2005.41662.x. [DOI] [PubMed] [Google Scholar]
- Hopf U, Moller B, Stemerowicz R, et al. Relation between Escherichia coli R(rough)-forms in gut, lipid A in liver, and primary biliary cirrhosis. Lancet. 1989;2:1419–1422. doi: 10.1016/s0140-6736(89)92034-5. [DOI] [PubMed] [Google Scholar]
- Invernizzi P, Miozzo M, Battezzati PM, et al. Frequency of monosomy X in women with primary biliary cirrhosis. Lancet. 2004;363:533–535. doi: 10.1016/S0140-6736(04)15541-4. [DOI] [PubMed] [Google Scholar]
- Invernizzi P, Selmi C, Gershwin ME. Update on primary biliary cirrhosis. Dig Liver Dis. 2010;42:401–408. doi: 10.1016/j.dld.2010.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishibashi H, Komori A, Shimoda S, et al. Guidelines for therapy of autoimmune liver disease. Semin Liver Dis. 2007;27:214–226. doi: 10.1055/s-2007-979472. [DOI] [PubMed] [Google Scholar]
- Iwasaki A, Medzhitov R. Toll-like receptor control of the adaptive immune responses. Nat Immunol. 2004;5:987–995. doi: 10.1038/ni1112. [DOI] [PubMed] [Google Scholar]
- Jacobson NG, Szabo SJ, Weber-Nordt RM, et al. Interleukin 12 signaling in T helper type 1 (Th1) cells involves tyrosine phosphorylation of signal transducer and activator of transcription (Stat)3 and Stat4. J Exp Med. 1995;181:1755–1762. doi: 10.1084/jem.181.5.1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakob T, Walker PS, Krieg AM, et al. Activation of cutaneous dendritic cells by CpG-containing oligodeoxynucleotides: a role for dendritic cells in the augmentation of Th1 responses by immunostimulatory DNA. J Immunol. 1998;161:3042–3049. [PubMed] [Google Scholar]
- Janssen WJ, Henson PM. Cellular regulation of the inflammatory response. Toxicol Pathol. 2012;40:166–173. doi: 10.1177/0192623311428477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins MK, Taylor PS, Norton SD, et al. CD28 delivers a costimulatory signal involved in antigen-specific IL-2 production by human T cells. J Immunol. 1991;147:2461–2466. [PubMed] [Google Scholar]
- Jin Q, Moritoki Y, Lleo A, et al. Comparative analysis of portal cell infiltrates in AMA positive versus AMA negative PBC. Hepatology. 2012;55:1495–1506. doi: 10.1002/hep.25511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshita S, Umemura T, Yoshizawa K, et al. Association analysis of cytotoxic T-lymphocyte antigen 4 gene polymorphisms with primary biliary cirrhosis in Japanese patients. J Hepatol. 2010;53:537–541. doi: 10.1016/j.jhep.2010.03.017. [DOI] [PubMed] [Google Scholar]
- Juran BD, Atkinson EJ, Schlicht EM, et al. Primary biliary cirrhosis is associated with a genetic variant in the 3′ flanking region of the CTLA4 gene. Gastroenterology. 2008a;135:1200–1206. doi: 10.1053/j.gastro.2008.06.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juran BD, Atkinson EJ, Schlicht EM, et al. Interacting alleles of the coinhibitory immunoreceptor genes cytotoxic T-lymphocyte antigen 4 and programmed cell-death 1 influence risk and features of primary biliary cirrhosis. Hepatology. 2008b;47:563–570. doi: 10.1002/hep.22048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanda N, Tamaki K. Estrogen enhances immunoglobulin production by human PBMCs. J Allergy Clin Immunol. 1999;103(2 Pt 1):282–288. doi: 10.1016/s0091-6749(99)70503-8. [DOI] [PubMed] [Google Scholar]
- Kanda N, Tsuchida T, Tamaki K. Estrogen enhancement of anti-double-stranded DNA antibody and immunoglobulin G production in peripheral blood mononuclear cells from patients with systemic lupus erythematosus. Arthritis Rheum. 1999;42:328–337. doi: 10.1002/1529-0131(199902)42:2<328::AID-ANR16>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- Kang JH, Asai D, Katayama Y. Bisphenol A in the aquatic environment and its endocrine-disruptive effects on aquatic organisms. Crit Rev Toxicol. 2007;37:607–625. doi: 10.1080/10408440701493103. [DOI] [PubMed] [Google Scholar]
- Kaplan MM. Primary biliary cirrhosis. N Engl J Med. 1996;335:1570–1580. doi: 10.1056/NEJM199611213352107. [DOI] [PubMed] [Google Scholar]
- Karaghiosoff M, Neubauer H, Lassnig C, et al. Partial impairment of cytokine responses in Tyk2-deficient mice. Immunity. 2000;13:549–560. doi: 10.1016/s1074-7613(00)00054-6. [DOI] [PubMed] [Google Scholar]
- Kawahara K, Moll H, Knirel YA, et al. Structural analysis of two glycosphingolipids from the lipopolysaccharide-lacking bacterium Sphingomonas capsulata. Eur J Biochem. 2000;267:1837–1846. doi: 10.1046/j.1432-1327.2000.01189.x. [DOI] [PubMed] [Google Scholar]
- Kinjo Y, Wu D, Kim G, et al. Recognition of bacterial glycosphingolipids by natural killer T cells. Nature. 2005;434:520–525. doi: 10.1038/nature03407. [DOI] [PubMed] [Google Scholar]
- Kita H, Naidenko OV, Kronenberg M, et al. Quantitation and phenotypic analysis of natural killer T cells in primary biliary cirrhosis using a human CD1d tetramer. Gastroenterology. 2002;123:1031–1043. doi: 10.1053/gast.2002.36020. [DOI] [PubMed] [Google Scholar]
- Krolewski JJ, Lee R, Eddy R, et al. Identification and chromosomal mapping of new human tyrosine kinase genes. Oncogene. 1990;5:277–282. [PubMed] [Google Scholar]
- Lakatos LP, Bajnok E, Hegedus D, et al. Vitamin D receptor, oestrogen receptor-alpha gene and interleukin-1 receptor antagonist gene polymorphisms in Hungarian patients with primary biliary cirrhosis. Eur J Gastroenterol Hepatol. 2002;14:733–740. doi: 10.1097/00042737-200207000-00004. [DOI] [PubMed] [Google Scholar]
- Lan RY, Salunga TL, Tsuneyama K, et al. Hepatic IL-17 responses in human and murine primary biliary cirrhosis. J Autoimmun. 2009;32:43–51. doi: 10.1016/j.jaut.2008.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee BW, Yap HK, Chew FT, et al. Age- and sex-related changes in lymphocyte subpopulations of healthy Asian subjects: from birth to adulthood. Cytometry. 1996;26:8–15. doi: 10.1002/(SICI)1097-0320(19960315)26:1<8::AID-CYTO2>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- Leitner J, Grabmeier-Pfistershammer K, Steinberger P. Receptors and ligands implicated in human T cell costimulatory processes. Immunol Lett. 2010;128:89–97. doi: 10.1016/j.imlet.2009.11.009. [DOI] [PubMed] [Google Scholar]
- Lenschow DJ, Walunas TL, Bluestone JA. CD28/B7 system of T cell costimulation. Annu Rev Immunol. 1996;14:233–258. doi: 10.1146/annurev.immunol.14.1.233. [DOI] [PubMed] [Google Scholar]
- Leung PS, Quan C, Park O. Immunization with a xenobiotic 6-bromohexanoate bovine serum albumin conjugate induces antimitochondrial antibodies. J Immunol. 2003;170:5326–5332. doi: 10.4049/jimmunol.170.10.5326. [DOI] [PubMed] [Google Scholar]
- Leung PS, Park O, Tsuneyama K. Induction of primary biliary cirrhosis in guinea pigs following chemical xenobiotic immunization. J Immunol. 2007;179:2651–2657. doi: 10.4049/jimmunol.179.4.2651. [DOI] [PubMed] [Google Scholar]
- Leung PS, Yang GX, Dhirapong A, et al. Animal models of primary biliary cirrhosis: materials and methods. Methods Mol Biol. 2012;900:291–316. doi: 10.1007/978-1-60761-720-4_14. [DOI] [PubMed] [Google Scholar]
- Libert C, Dejager L, Pinheiro I. The X chromosome in immune functions: when a chromosome makes the difference. Nat Rev Immunol. 2010;10:594–604. doi: 10.1038/nri2815. [DOI] [PubMed] [Google Scholar]
- Linsley PS, Brady W, Grosmaire L, et al. Binding of the B cell activation antigen B7 to CD28 costimulates T cell proliferation and interleukin 2 mRNA accumulation. J Exp Med. 1991;173:721–730. doi: 10.1084/jem.173.3.721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linsley PS, Greene JL, Tan P, et al. Coexpression and functional cooperation of CTLA-4 and CD28 on activated T lymphocytes. J Exp Med. 1992;176:1595–1604. doi: 10.1084/jem.176.6.1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu JZ, Almarri MA, Gaffney DJ, et al. Dense fine-mapping study identifies new susceptibility loci for primary biliary cirrhosis. Nat Genet. 2012;44:1137–1141. doi: 10.1038/ng.2395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manetti R, Parronchi P, Giudizi MG, et al. Natural killer cell stimulatory factor (interleukin 12 [IL-12]) induces T helper type 1 (Th1)-specific immune responses and inhibits the development of IL-4-producing Th cells. J Exp Med. 1993;177:1199–1204. doi: 10.1084/jem.177.4.1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masteller EL, Chuang E, Mullen AC, et al. Structural analysis of CTLA-4 function in vivo. J Immunol. 2000;164:5319–5327. doi: 10.4049/jimmunol.164.10.5319. [DOI] [PubMed] [Google Scholar]
- Mattalia A, Quaranta S, Leung PS, et al. Characterization of antimitochondrial antibodies in health adults. Hepatology. 1998;27:656–661. doi: 10.1002/hep.510270303. [DOI] [PubMed] [Google Scholar]
- Mattner J, Debord KL, Ismail N, et al. Exogenous and endogenous glycolipid antigens activate NKT cells during microbial infections. Nature. 2005;434:525–529. doi: 10.1038/nature03408. [DOI] [PubMed] [Google Scholar]
- Mattner J, Savage PB, Leung P, et al. Liver autoimmunity triggered by microbial activation of natural killer T cells. Cell Host Microbe. 2008;3:304–315. doi: 10.1016/j.chom.2008.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurer M, Loserth S, Kolb-Maurer A, et al. A polymorphism in the human cytotoxic T-lymphocyte antigen 4 (CTLA4) gene (exon 1 +49) alters T-cell activation. Immunogenetics. 2002;54:1–8. doi: 10.1007/s00251-002-0429-9. [DOI] [PubMed] [Google Scholar]
- McGonagle D, McDermott MF. A proposed classification of the immunological diseases. PLoS Med. 2006;3:e297. doi: 10.1371/journal.pmed.0030297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mells GF, Floyd JA, Morley KI, et al. Genome-wide association study identifies 12 new susceptibility loci for primary biliary cirrhosis. Nat Genet. 2011;43:329–332. doi: 10.1038/ng.789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metcalf JV, Mitchison HC, Palmer JM, et al. Natural history of early primary biliary cirrhosis. Lancet. 1996;348:1399–1402. doi: 10.1016/S0140-6736(96)04410-8. [DOI] [PubMed] [Google Scholar]
- Miozzo M, Selmi C, Gentilin B, et al. Preferential X chromosome loss but random inactivation characterize primary biliary cirrhosis. Hepatology. 2007;46:456–462. doi: 10.1002/hep.21696. [DOI] [PubMed] [Google Scholar]
- Mitchell MM, Lleo A, Zammataro L, et al. Epigenetic investigation of variably X chromosome inactivated genes in monozygotic female twins discordant for primary biliary cirrhosis. Epigenetics. 2011;6:95–102. doi: 10.4161/epi.6.1.13405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohammed JP, Fusakio ME, Rainbow DB, et al. Identification of Cd101 as a susceptibility gene for Novosphingobium aromaticivorans-induced liver autoimmunity. J Immunol. 2011;187:337–349. doi: 10.4049/jimmunol.1003525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molero L, Garcia-Duran M, Diaz-Recasens J, et al. Expression of estrogen receptor subtypes and neuronal nitric oxide synthase in neutrophils from women and men: regulation by estrogen. Cardiovasc Res. 2002;56:43–51. doi: 10.1016/s0008-6363(02)00505-9. [DOI] [PubMed] [Google Scholar]
- Moreira RK, Revetta F, Koehler E. Diagnostic utility of IgG and IgM immunohistochemistry in autoimmune liver disease. World J Gastroenterol. 2010;16:453–457. doi: 10.3748/wjg.v16.i4.453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munoz-Suano A, Kallikourdis M, Sarris M, et al. Regulatory T cells protect from autoimmune arthritis during pregnancy. J Autoimmun. 2012;38:J103–J108. doi: 10.1016/j.jaut.2011.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munz C, Lunemann JD, Getts MT, et al. Antiviral immune responses: triggers of or triggered by autoimmunity? Nat Rev Immunol. 2009;9:246–258. doi: 10.1038/nri2527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nalbandian G, Kovats S. Understanding sex biases in immunity: effects of estrogen on the differentiation and function of antigen-presenting cells. Immunol Res. 2005;31:91–106. doi: 10.1385/IR:31:2:091. [DOI] [PubMed] [Google Scholar]
- Nelson JL, Ostensen M. Pregnancy and rheumatoid arthritis. Rheum Dis Clin North Am. 1997;23:195–212. doi: 10.1016/s0889-857x(05)70323-9. [DOI] [PubMed] [Google Scholar]
- Nilsson HO, Taneera J, Castedal M, et al. Identification of Helicobacter pylori and other Helicobacter species by PCR, hybridization, and partial DNA sequencing in human liver samples from patients with primary sclerosing cholangitis or primary biliary cirrhosis. J Clin Microbiol. 2000;38:1072–1076. doi: 10.1128/jcm.38.3.1072-1076.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Notomista E, Pennacchio F, Cafaro V, et al. The marine isolate Novosphingobium sp. PP1Y shows specific adaptation to use the aromatic fraction of fuels as the sole carbon and energy source. Microb Ecol. 2011;61:582–594. doi: 10.1007/s00248-010-9786-3. [DOI] [PubMed] [Google Scholar]
- Oertelt S, Kenny TP, Selmi C, et al. SNP analysis of genes implicated in T cell proliferation in primary biliary cirrhosis. Clin Dev Immunol. 2005;12:259–263. doi: 10.1080/17402520500317859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortmann R, Smeltz R, Yap G, et al. A heritable defect in IL-12 signaling in B10.Q/J mice. I. In vitro analysis. J Immunol. 2001;166:5712–5719. doi: 10.4049/jimmunol.166.9.5712. [DOI] [PubMed] [Google Scholar]
- Oyamada A, Ikebe H, Itsumi M, et al. Tyrosine kinase 2 plays critical roles in the pathogenic CD4 T cell responses for the development of experimental autoimmune encephalomyelitis. J Immunol. 2009;183:7539–7546. doi: 10.4049/jimmunol.0902740. [DOI] [PubMed] [Google Scholar]
- Padgett KA, Selmi C, Kenny TP, et al. Phylogenetic and immunological definition of four lipoylated proteins from Novosphingobium aromaticivorans, implications for primary biliary cirrhosis. J Autoimmun. 2005;24:209–219. doi: 10.1016/j.jaut.2005.01.012. [DOI] [PubMed] [Google Scholar]
- Paharkova-Vatchkova V, Maldonado R, et al. Estrogen preferentially promotes the differentiation of CD11c + CD11b (intermediate) dendritic cells from bone marrow precursors. J Immunol. 2004;172:1426–1436. doi: 10.4049/jimmunol.172.3.1426. [DOI] [PubMed] [Google Scholar]
- Porter AW, Hay AG. Identification of opdA, a gene involved in biodegradation of the endocrine disrupter octylphenol. Appl Environ Microbiol. 2007;73:7373–7379. doi: 10.1128/AEM.01478-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poupon R, Chretien Y, Poupon RE, et al. Is ursodeoxycholic acid an effective treatment for primary biliary cirrhosis? Lancet. 1987;1:834–836. doi: 10.1016/s0140-6736(87)91610-2. [DOI] [PubMed] [Google Scholar]
- Poupon R, Ping C, Chretien Y, et al. Genetic factors of susceptibility and of severity in primary biliary cirrhosis. J Hepatol. 2008;49:1038–1045. doi: 10.1016/j.jhep.2008.07.027. [DOI] [PubMed] [Google Scholar]
- Qureshi OS, Zheng Y, Nakamura K, et al. Trans-endocytosis of CD80 and CD86: a molecular basis for the cell-extrinsic function of CTLA-4. Science. 2011;332:600–603. doi: 10.1126/science.1202947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieger R, Gershwin ME. The X and why of xenobiotics in primary biliary cirrhosis. J Autoimmun. 2007;28:76–84. doi: 10.1016/j.jaut.2007.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rioux JD, Abbas AK. Paths to understanding the genetic basis of autoimmune disease. Nature. 2005;435:584–589. doi: 10.1038/nature03723. [DOI] [PubMed] [Google Scholar]
- Rose NR, Bona C. Defining criteria for autoimmune diseases (Witebsky’s postulates revisited) Immunol Today. 1993;14:426–430. doi: 10.1016/0167-5699(93)90244-F. [DOI] [PubMed] [Google Scholar]
- Rosen A, Casciola-Rosen L. Autoantigens in systemic autoimmunity: critical partner in pathogenesis. J Intern Med. 2009;265:625–631. doi: 10.1111/j.1365-2796.2009.02102.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubtsov AV, Rubtsova K, Kappler JW, et al. Genetic and hormonal factors in female-biased autoimmunity. Autoimmun Rev. 2010;9:494–498. doi: 10.1016/j.autrev.2010.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudd CE. The reverse stop-signal model for CTLA4 function. Nat Rev Immunol. 2008;8:153–160. doi: 10.1038/nri2253. [DOI] [PubMed] [Google Scholar]
- Ryan MP, Adley CC. Sphingomonas paucimobilis: a persistent Gram-negative nosocomial infectious organism. J Hosp Infect. 2010;75:153–157. doi: 10.1016/j.jhin.2010.03.007. [DOI] [PubMed] [Google Scholar]
- Scalapino KJ, Daikh DI. CTLA-4: a key regulatory point in the control of autoimmune disease. Immunol Rev. 2008;223:143–155. doi: 10.1111/j.1600-065X.2008.00639.x. [DOI] [PubMed] [Google Scholar]
- Selmi C, Balkwill DL, Invernizzi P, et al. Patients with primary biliary cirrhosis react against a ubiquitous xenobiotic-metabolizing bacterium. Hepatology. 2003;38:1250–1257. doi: 10.1053/jhep.2003.50446. [DOI] [PubMed] [Google Scholar]
- Seto Y, Nakajima H, Suto A, et al. Enhanced Th2 cell-mediated allergic inflammation in Tyk2-deficient mice. J Immunol. 2003;170:1077–1083. doi: 10.4049/jimmunol.170.2.1077. [DOI] [PubMed] [Google Scholar]
- Shackel NA, McGuinness PH, Abbott CA, et al. Identification of novel molecules and pathogenic pathways in primary biliary cirrhosis: cDNA array analysis of intrahepatic differential gene expression. Gut. 2001;49:565–576. doi: 10.1136/gut.49.4.565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahrizaila N, Yuki N. Guillain-barre syndrome animal model: the first proof of molecular mimicry in human autoimmune disorder. J Biomed Biotechnol. 2011;2011:829129. doi: 10.1155/2011/829129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi T, Fredrickson JK, Balkwill DL. Biodegradation of polycyclic aromatic hydrocarbons by Sphingomonas strains isolated from the terrestrial subsurface. J Ind Microbiol Biotechnol. 2001;26:283–289. doi: 10.1038/sj.jim.7000130. [DOI] [PubMed] [Google Scholar]
- Shimoda S, Nakamura M, Ishibashi H, et al. HLA DRB4 0101-restricted immunodominant T cell autoepitope of pyruvate dehydrogenase complex in primary biliary cirrhosis: evidence of molecular mimicry in human autoimmune diseases. J Exp Med. 1995;181:1835–1845. doi: 10.1084/jem.181.5.1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimoda K, Kato K, Aoki K, et al. Tyk2 plays a restricted role in IFN alpha signaling, although it is required for IL-12-mediated T cell function. Immunity. 2000;13:561–571. doi: 10.1016/s1074-7613(00)00055-8. [DOI] [PubMed] [Google Scholar]
- Shuttleworth KL, Sung J, Kim E, et al. Physiological and genetic comparison of two aromatic hydrocarbon-degrading Sphingomonas strains. Mol Cells. 2000;10:199–205. doi: 10.1007/s10059-000-0199-x. [DOI] [PubMed] [Google Scholar]
- Sohn JH, Kwon KK, Kang JH, et al. Novosphingobium pentaromativorans sp. nov., a high-molecular-mass polycyclic aromatic hydrocarbon-degrading bacterium isolated from estu-arine sediment. Int J Syst Evol Microbiol. 2004;54(Pt 5):1483–1487. doi: 10.1099/ijs.0.02945-0. [DOI] [PubMed] [Google Scholar]
- Spach KM, Noubade R, McElvany B, et al. A single nucleotide polymorphism in Tyk2 controls susceptibility to experimental allergic encephalomyelitis. J Immunol. 2009;182:7776–7783. doi: 10.4049/jimmunol.0900142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparwasser T, Koch ES, Vabulas RM, et al. Bacterial DNA and immunostimulatory CpG oligonucleotides trigger maturation and activation of murine dendritic cells. Eur J Immunol. 1998;28:2045–2054. doi: 10.1002/(SICI)1521-4141(199806)28:06<2045::AID-IMMU2045>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- Stimson WH. Oestrogen and human T lymphocytes: presence of specific receptors in the T-suppressor/cytotoxic subset. Scand J Immunol. 1988;28:345–350. doi: 10.1111/j.1365-3083.1988.tb01459.x. [DOI] [PubMed] [Google Scholar]
- Stolz A. Molecular characteristics of xenobiotic-degrading sphingomonads. Appl Microbiol Biotechnol. 2009;81:793–811. doi: 10.1007/s00253-008-1752-3. [DOI] [PubMed] [Google Scholar]
- Stoop JW, Zegers BJ, Sander PC, et al. Serum immunoglobulin levels in healthy children and adults. Clin Exp Immunol. 1969;4:101–112. [PMC free article] [PubMed] [Google Scholar]
- Suarez-Gestal M, Calaza M, Endreffy E, et al. Replication of recently identified systemic lupus erythematosus genetic associations: a case-control study. Arthritis Res Ther. 2009;11:R69. doi: 10.1186/ar2698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suenaga R, Evans MJ, Mitamura K, et al. Peripheral blood T cells and monocytes and B cell lines derived from patients with lupus express estrogen receptor transcripts similar to those of normal cells. J Rheumatol. 1998;25:1305–1312. [PubMed] [Google Scholar]
- Suzuki S, Hiraishi A. Novosphingobium naphthalenivorans sp. nov., a naphthalene-degrading bacterium isolated from poly-chlorinated-dioxin-contaminated environments. J Gen Appl Microbiol. 2007;53:221–228. doi: 10.2323/jgam.53.221. [DOI] [PubMed] [Google Scholar]
- Suzuki A, Kochi Y, Okada Y, et al. Insight from genome-wide association studies in rheumatoid arthritis and multiple sclerosis. FEBS Lett. 2011;585:3627–3632. doi: 10.1016/j.febslet.2011.05.025. [DOI] [PubMed] [Google Scholar]
- Takahashi T, Tagami T, Yamazaki S, et al. Immunologic self-tolerance maintained by CD25(+)CD4(+) regulatory T cells constitutively expressing cytotoxic T lymphocyte-associated antigen 4. J Exp Med. 2000;192:303–310. doi: 10.1084/jem.192.2.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi M, Hamana K, Hiraishi A. Proposal of the genus Sphingomonas sensu stricto and three new genera, Sphingobium, Novosphingobium and Sphingopyxis, on the basis of phyloge-netic and chemotaxonomic analyses. Int J Syst Evol Microbiol. 2001;51(Pt 4):1405–1417. doi: 10.1099/00207713-51-4-1405. [DOI] [PubMed] [Google Scholar]
- Tanghe T, Dhooge W, Verstraete W. Isolation of a bacterial strain able to degrade branched nonylphenol. Appl Environ Microbiol. 1999;65:746–751. doi: 10.1128/aem.65.2.746-751.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ternes TA, Stumpf M, Mueller J, et al. Behavior and occurrence of estrogens in municipal sewage treatment plants–I. Investigations in Germany, Canada and Brazil. Sci Total Environ. 1999;225:81–90. doi: 10.1016/s0048-9697(98)00334-9. [DOI] [PubMed] [Google Scholar]
- Thompson CB, Lindsten T, Ledbetter JA, et al. CD28 activation pathway regulates the production of multiple T-cell-derived lymphokines/cytokines. Proc Natl Acad Sci USA. 1989;86:1333–1337. doi: 10.1073/pnas.86.4.1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiirola MA, Mannisto MK, Puhakka JA, et al. Isolation and characterization of Novosphingobium sp. strain MT1, a dominant polychlorophenol-degrading strain in a groundwater bioremediation system. Appl Environ Microbiol. 2002;68:173–180. doi: 10.1128/AEM.68.1.173-180.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tivol EA, Borriello F, Schweitzer AN, et al. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity. 1995;3:541–547. doi: 10.1016/1074-7613(95)90125-6. [DOI] [PubMed] [Google Scholar]
- Tollerud DJ, Clark JW, Brown LM, et al. The influence of age, race, and gender on peripheral blood mononuclear-cell subsets in healthy nonsmokers. J Clin Immunol. 1989;9:214–222. doi: 10.1007/BF00916817. [DOI] [PubMed] [Google Scholar]
- Tsuneyama K, Yasoshima M, Harada K, et al. Increased CD1d expression on small bile duct epithelium and epithelioid granuloma in livers in primary biliary cirrhosis. Hepatology. 1998;28:620–623. doi: 10.1002/hep.510280303. [DOI] [PubMed] [Google Scholar]
- Tsuneyama K, Moritoki Y, Kikuchi K, et al. Pathological features of new animal models for primary biliary cirrhosis. Int J Hepatol. 2012;2012:403954. doi: 10.1155/2012/403954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van de Water J, Ansari A, Prindiville T, et al. Heterogeneity of autoreactive T cell clones specific for the E2 component of the pyruvate dehydrogenase complex in primary biliary cirrhosis. J Exp Med. 1995;181:723–733. doi: 10.1084/jem.181.2.723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Norstrand MD, Malinchoc M, Lindor KD, et al. Quantitative measurement of autoantibodies to recombinant mitochondrial antigens in patients with primary biliary cirrhosis: relationship of levels of autoantibodies to disease progression. Hepatology. 1997;25:6–11. doi: 10.1002/hep.510250103. [DOI] [PubMed] [Google Scholar]
- Verhasselt V, Buelens C, Willems F, et al. Bacterial lipopoly-saccharide stimulates the production of cytokines and the expression of costimulatory molecules by human peripheral blood dendritic cells: evidence for a soluble CD14-dependent pathway. J Immunol. 1997;158:2919–2925. [PubMed] [Google Scholar]
- Voll RE, Herrmann M, Roth EA, et al. Immunosuppressive effects of apoptotic cells. Nature. 1997;390:350–351. doi: 10.1038/37022. [DOI] [PubMed] [Google Scholar]
- Walker LS, Abbas AK. The enemy within: keeping self-reactive T cells at bay in the periphery. Nat Rev Immunol. 2002;2:11–19. doi: 10.1038/nri701. [DOI] [PubMed] [Google Scholar]
- Walker EJ, Hirschfield GM, Xu C, et al. CTLA4/ICOS gene variants and haplotypes are associated with rheumatoid arthritis and primary biliary cirrhosis in the Canadian population. Arthritis Rheum. 2009;60:931–937. doi: 10.1002/art.24412. [DOI] [PubMed] [Google Scholar]
- Wallace C, Smyth DJ, Maisuria-Armer M, et al. The imprinted DLK1-MEG3 gene region on chromosome 14q32.2 alters susceptibility to type 1 diabetes. Nat Genet. 2010;42:68–71. doi: 10.1038/ng.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walunas TL, Lenschow DJ, Bakker CY, et al. CTLA-4 can function as a negative regulator of T cell activation. Immunity. 1994;1:405–413. doi: 10.1016/1074-7613(94)90071-x. [DOI] [PubMed] [Google Scholar]
- Waterhouse P, Penninger JM, Timms E, et al. Lymphoproliferative disorders with early lethality in mice deficient in Ctla-4. Science. 1995;270:985–988. doi: 10.1126/science.270.5238.985. [DOI] [PubMed] [Google Scholar]
- Weatherall DJ. Genetic variation and susceptibility to infection: the red cell and malaria. Br J Haematol. 2008;141:276–286. doi: 10.1111/j.1365-2141.2008.07085.x. [DOI] [PubMed] [Google Scholar]
- Wegmann TG, Lin H, Guilbert L, et al. Bidirectional cytokine interactions in the maternal-fetal relationship: is successful pregnancy a TH2 phenomenon? Immunol Today. 1993;14:353–356. doi: 10.1016/0167-5699(93)90235-D. [DOI] [PubMed] [Google Scholar]
- Wilder RL. Neuroendocrine-immune system interactions and autoimmunity. Annu Rev Immunol. 1995;13:307–338. doi: 10.1146/annurev.iy.13.040195.001515. [DOI] [PubMed] [Google Scholar]
- Willcox M, Bjorkman A, Brohult J, et al. A case-control study in northern Liberia of Plasmodium falciparum malaria in haemoglobin S and beta-thalassaemia traits. Ann Trop Med Parasitol. 1983;77:239–246. doi: 10.1080/00034983.1983.11811704. [DOI] [PubMed] [Google Scholar]
- Wing K, Onishi Y, Prieto-Martin P, et al. CTLA-4 control over Foxp3+ regulatory T cell function. Science. 2008;322:271–275. doi: 10.1126/science.1160062. [DOI] [PubMed] [Google Scholar]
- Xu L, Shen Z, Guo L, et al. Does a betaretrovirus infection trigger primary biliary cirrhosis? Proc Natl Acad Sci USA. 2003;100:8454–8459. doi: 10.1073/pnas.1433063100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan QX, Hong Q, Han P, et al. Isolation and characterization of a carbofuran-degrading strain Novosphingobium sp. FND-3. FEMS Microbiol Lett. 2007;271:207–213. doi: 10.1111/j.1574-6968.2007.00718.x. [DOI] [PubMed] [Google Scholar]
- Yeaman SJ, Fussey SP, Danner DJ, et al. Primary biliary cirrhosis: identification of two major M2 mitochondrial autoantigens. Lancet. 1988;1:1067–1070. doi: 10.1016/s0140-6736(88)91894-6. [DOI] [PubMed] [Google Scholar]
- Yeaman SJ, Kirby JA, Jones DE. Autoreactive responses to pyruvate dehydrogenase complex in the pathogenesis of primary biliary cirrhosis. Immunol Rev. 2000;174:238–249. doi: 10.1034/j.1600-0528.2002.00021h.x. [DOI] [PubMed] [Google Scholar]
- Yuan J, Lai Q, Zheng T, et al. Novosphingobium indicum sp. nov., a polycyclic aromatic hydrocarbon-degrading bacterium isolated from a deep-sea environment. Int J Syst Evol Microbiol. 2009;59(8):2084–2088. doi: 10.1099/ijs.0.002873-0. [DOI] [PubMed] [Google Scholar]
- Zaletel K, Krhin B, Gaberscek S, et al. Association of CT60 cytotoxic T lymphocyte antigen-4 gene polymorphism with thyroid autoantibody production in patients with Hashimoto’s and postpartum thyroiditis. Clin Exp Immunol. 2010;161:41–47. doi: 10.1111/j.1365-2249.2010.04113.x. [DOI] [PMC free article] [PubMed] [Google Scholar]