

Abstract
Chemotherapy in prostate cancer (CaP) even as an adjunct has not been a success. In this communication, we report the pre-clinical efficacy of a nitroacridine derivative, C-1748 (9[2′-hydroxyethylamino]-4-methyl-1-nitroacridine) in CaP cell culture and human xenograft animal models. C-1748, a DNA intercalating agent has been derived from its precursor C-857 that was a potent anti-cancer drug, but failed clinical development “methyl” group imparted novel properties, the most interesting of which is the difference in the IC50 values between LnCaP (22.5 nM), a CaP cell line and HL-60, a leukemia cell line (>100 nM). Using γH2AX as an intervention marker of DNA double strand breaks, we concluded that C-1748 is more efficacious in CaP cells than in HL-60 cells. In hormone dependent cells, the androgen receptor (AR) was identified as an additional target of C-1748. In xenograft studies, administration of C-1748 intra-peritoneally inhibited tumor growth by 80–90% with minimal toxicity. These studies identify C-1748 as a novel acridine drug that has a high therapeutic index and low cytotoxicity on myelocytic cells with potential for clinical development.
Keywords: chemotherapy, xenografts, 1-nitroacridine derivative, systemic treatment, DNA double strand breaks, γH2AX
INTRODUCTION
The role of chemotherapy in the treatment of prostate cancer (CaP) is mostly restricted to palliative use in metastatic or hormone refractory disease.1–6 Since other coexisting diseases may occur in men with advanced age, coupled with the lack of CaP specificity of current regimens7,8 makes chemotherapy a less attractive option in management of CaP. In general, the slow growth of CaP cells implies an inherent resistance to most chemotherapeutic agents that target the DNA synthetic machinery of cancer cells.9,10 Another serious problem with many chemotherapeutic agents is the side effects observed with aggressive high dose chemotherapy11–15 that in severe cases extend to fatal myelo-suppression16 and neuropathy.17 Among several chemotherapeutic regimens being explored are docetaxel, in phase II trials, with response rates between 20–40%;8,18–20 estramustine plus taxane in phase III Southwest Oncology Group (SWOG) trial with objective responses in less than 20% of patients;8,20,21 ketoconazole in Eastern Cooperative Oncology Group (ECOG)-sponsored phase III trial8,22–24 and carboplatin in phase II trials, showed significant prostate specific antigen (PSA) reductions, while less than 10% of patients exhibited a complete response.25 Only a subpopulation of patients benefit from chemotherapy and since none of the agents are specific for CaP alongwith unacceptable toxicity profiles30–38 makes it imperative to search for alternative chemotherapeutic agents.
The 1-nitroacridines are potent DNA binding agents26,27 inhibiting growth of hypoxic cells. In vitro studies on the parent compound ledakrin (Fig. 1), showed significant differential cytotoxicity against non-leukemia human cancer cell lines.28–32 Ledakrin, a prodrug, is activated by cellular enzymes followed by covalent crosslinking to DNA26–28 leading to cell cycle perturbation and initiation of apoptosis.29 However, it produced side effects such as intense nausea and vomiting and clinical development was stopped. Analogs of ledakrin retained the anti-cancer activity with lower toxicity. One such derivative with an amino-alkyl substitution in the C9 position was designated as 9-(2′-hydroxyethylamino)-1-nitroacridine (C-857) and found to have high toxicity33 necessitating modifications in order to find substituted derivatives that retained both specificity and anti-tumor potency for CaP resulting in the generation of C-1748, a 9-hydroxy-alkylamino-4-methyl-1-nitro-acridine derivative.
Figure 1.

Chemical structures of ledakrin, C-1748, (9(2′-hydroxy-ethylamino)-4-methyl-1-nitroacridine) and C-1748 analogs are represented as C-1860 (9(2′-propionoxyethylamino)-4-methyl-1-nitroacridine), C-1872 (9(3′-acetoxypropylamino)-4-methyl-1-nitroacridine), C-1873 (9(3′-propionoxypropylamino)-4-methyl-1-nitroacridine).
Structure-activity relationship studies indicated that the 1-nitro group is crucial for biological activity of 1-nitroacridines, but is extremely conducive to reduction leading to formation of very reactive and unstable products resulting in high systemic toxicity of 1-nitroacridines. The introduction of methyl-electron donating group- in position 4, i.e., para to the 1-nitro group, leading to 4-methyl derivatives that exhibit lower toxicity30 (Fig. 1). Our pre-clinical toxicology studies in rodents33 and dogs34 showed C-1748 to have very low systemic toxicity along with a lowered mutagenic potential.35 The 4-methyl-1-nitroacridines retain the differential cytotoxic capacity of ledakrin, while demonstrating a striking specificity for CaP and lowered systemic toxicities.36 The present study uses cell culture models and human cancer cell xeno-grafts to demonstrate that C-1748 has specific anti-cancer activity towards CaP and excellent host tolerance with potential for further clinical development as a chemotherapeutic drug for CaP.
MATERIALS AND METHODS
Cell culture
Cell lines used in this study LnCaP (human androgen dependent prostate cancer), TSU (derived from bladder carcinoma cells)37 and HL-60 (human leukemia) were grown in RPMI 1640 (Mediatech) supplemented with 10% fetal bovine serum (FBS) (Gemini), penicillin 50 IU/ml, streptomycin 50 μg/ml (Mediatech) and 2 mM L-glutamine (Mediatech). The methanesulfonate derivative of C-1748 is water-soluble and was used for the in vivo and in vitro experiments.
XTT assay
XTT assay was performed as described by Ashok et al.38 Cells (2 × 103) were plated into 96 well plates and incubated overnight to allow cell adherence. The media was removed and C-1748 was added at concentrations from 10 nM to 1 μM and incubated for 24, 48, 72 and 96 hours. The medium was replaced with fresh medium to which 50 μl of XTT solution (1 mg/ml in serum free RPMI + Phenazine methosulfate (25 nM) of XTT before use). Plates were read after 3–4 hr in a microplate reader at 450 nm and 630 nm. The mean OD values were calculated and percent survival in the treated cells was calculated compared to untreated control and plotted as a function of the time and dose. XTT assays were used to assess the response of LnCaP to the synthetic androgen R1881 (0.1 nM) and anti-androgen flutamide (1 μM) and performed as described in combination with different doses of C-1748 at the end of 24 or 48 hr.
Xenograft experiments to test efficacy of drugs
Male nude mice (Balb/c/nu/nu) (Charles River, Wilmington, MA), weighing about 15–20 g; 8–10 weeks age were fed standard chow and water ad libitum and allowed one week of acclimatization before start of experiment. Human cancer cells-LnCaP (5 × 106 cells/mouse); TSU (2×106 cells/mouse) were used as tumor xenografts (six to ten mice/group) and efficacy of C-1748 was evaluated. The cells were harvested using Trypsin-EDTA, washed with PBS, counted using Trypan Blue and cell suspension injected intra-dermally (0.2 ml) on the left flank of nude mice. Drug treatment was started when tumors were palpable and were injected intra-peritoneally (i.p.) at doses of 0.8 and 1.0 mg/kg. The therapeutic dose concentrations were over 12 fold lower than the calculated LD50 doses. Treatment schedule was once weekly for 6–7 weeks as mentioned for each set of xenografts. The animals were monitored for activity, physical condition, body weight, and rate of tumor growth. Tumor size was determined by caliper measurement in two perpendicular diameters of the implant and tumor volumes were calculated as (described earlier Ref. 39): Tumor volume (cm3) = 0.4 X Long diameter X (Short diameter)2
Detection of phosphorylated histone H2AX (γH2AX)
The detection of γH2AX was performed as (described in refs. 40 and 41). LnCaP and HL-60 cells were used for these experiments. At the onset of the experiments, there was less than 5 × 105 cells per ml in culture and the cells were at an exponential and asynchronous phase of growth. The cells were treated with C-1748 (25 nM or 50 nM) and/or the DNA polymerase α inhibitor, aphidicolin (2 μM). In some experiments, cells were pretreated with 2 μM aphidicolin for 20 min prior adding C-1748. The cells were washed twice in PBS and suspended in 0.2% Triton X-100 (Sigma) in a 1% (w/v) solution of bovine serum albumin (BSA; Sigma) in PBS for 30 min followed by centrifugation (200 G, 5 min) and the cell pellet was suspended in 100 μl of 1% BSA containing 1:100 diluted phospho-histone H2AX (Ser139) antibody (Cell Signaling, MA). Cells were incubated for 2 h at room temperature, washed twice with PBS and resuspended in 100 μl of 1:30 diluted FITC-conjugated F(ab′)2 fragment of goat anti-mouse immunoglobulin (DAKO, Carpinteria, CA) for 30 min at room temperature in the dark, counterstained with 5 μg/ml of propidium iodide (PI; Molecular Probes, Eugene, OR) dissolved in PBS containing 100 μg/ml of DNase-free RNase A (Sigma), for 20 min at room temperature. Cellular green (histone γH2AX) and red (PI) fluorescence was measured using a FACScan cytometer (Becton Dickinson, San Jose, CA) with the standard emission filters for green (FL1) and red (FL3) fluorescence.
Quantification of γH2AX IF
To compare the changes in γH2AX IF intensity e.g., in relation to cell cycle phase or C-1748 treatment, the mean γH2AX IF positivity was calculated in each phase of the cycle by gating G1, S and G2/M cell subpopulations, based on differences in DNA content.40,41 Further details on methods are provided in figure legends.
Western blot analysis of the steady-state level of cellular proteins
Cells were treated with C-1748 (25 and 50 nM) for 24 hr. At the end of the incubation period, cells were harvested, washed with PBS and lysed (1 × 106 cells/100 μl of lysis buffer) using RIPA buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 0.2% sodium deoxycholate, 0.1% SDS, 0.5% NP-40, 1 μM Pefabloc) and lysates were subjected to Western blotting using antibodies to androgen receptor and estrogen receptor-β (Santa Cruz).
RESULTS
Anti-tumor activity of C-1748 compared to its analogs in CaP
The anti-tumor efficacy of different derivatives was tested in TSU xenograft model at a dose of 0.8 mg/kg body weight and once weekly and efficacy compared to C-1748 (Fig. 2). Untreated control animals exhibited steady tumor growth, however, the rate of inhibition of tumor growth varied considerably between different drug treated groups. C-1860 had significantly reduced tumor volumes with greater than 50% reduction compared to controls at the end of six weeks. C-1872 and C-1873 were comparable with slower tumor growth rates and tumor volume reduction of ~60%. Final tumor volumes in the C-1748 treated group were significantly lowered by 80% compared to controls (Fig. 2). A paired Student’s t test shows a ‘p’ value of 0.019 for C-1748 treated group compared to controls (p < 0.05). A marked inhibition in the rate of tumor growth was observed in the “log phase” of the growth curve for 3 weeks. In the same time period, C-1748 treated xenografts showed an 80% decrease in rate of tumor growth (p < 0.016). C-1748 was well tolerated by the host without adverse effects and no loss of body weight. The results single out C-1748 as the most efficacious compound compared to other derivatives. The total cumulative dose of C-1748 used was 4.8 mg/kg, which when extrapolated is an achievable human dose.
Figure 2.
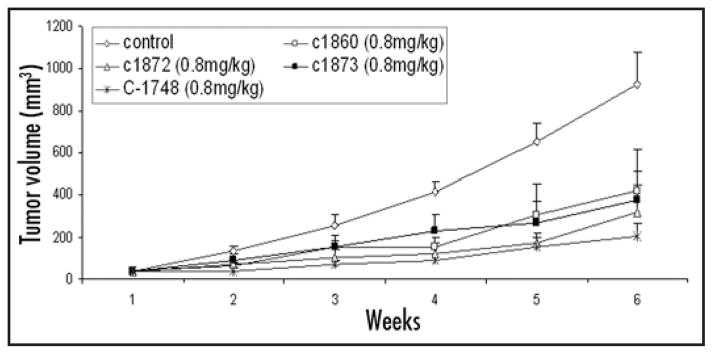
Anti-tumor efficacy of C-1748 and C-1860, C-1872, C-1873 in human TSU xenografts in nude mice. Animals were randomized into treatment and control groups (six mice/group). Treatment was started when tumors were palpable (40 mm3) and continued once a week for six weeks. Tumor growth was measured using Vernier calipers and tumor volume calculated.
C-1748 inhibits the growth of human LnCaP xenografts in nude mice
The efficacy of C-1748 was studied on androgen dependent LnCaP xenografts transplanted in nude mice (Fig. 3). Untreated control exhibited steady tumor growth in seven weeks. Two different treatment groups of mice were treated with 0.8 mg/kg and 1 mg/kg concentrations of C-1748 (Fig. 3). The lower dose of C-1748 consistently inhibited the rate of tumor growth resulting in 80% decrease in final tumor volumes compared to control. This statistically significant effect (p < 0.05) correlated with the efficacy observed in TSU xenografts. During the log phase of tumor growth, growth of C-1748 treated LnCaP decreased by 75% per day (p < 0.02). The cumulative dose of C-1748 was 7 mg/kg was well tolerated and sustained anti-tumor activity without adverse effects on physical activity, appetite or weight loss (Fig. 3).
Figure 3.

Anti-tumor efficacy of C-1748 in human LnCaP CaP xenografts in nude mice. Animals were randomized into treatment and control groups (10 mice/group). Cells were injected intra-dermally and treatment started when tumors were palpable at 40 mm3. C-1748 (0.8 mg/kg, 1 mg/kg) or saline (control) were administered i.p. once a week for seven weeks. Tumor growth was measured using Vernier calipers and tumor volume calculated.
C-1748 has preferential cytotoxicity towards LnCaP cells compared to human leukemia HL-60 cells
We then investigated in vitro effects of C-1748 by XTT assay to correlate in vivo results using LnCaP cells and human leukemia cells (HL-60) representing cells of hematopoietic lineage. IC50 values clearly indicate that LnCaP is highly sensitive to C-1748 (22.5 nM) in contrast to HL-60 with IC50 values atleast four fold higher than CaP (~100 nM) (Fig. 4). Similar results were observed with hormone independent DU145 CaP (2.5–5 nM) and the results strongly support our finding that C-1748 had a unique sensitivity towards CaP.42
Figure 4.

Dose and time dependent cytotoxic effects of C-1748 on LnCaP and HL-60 cells determined by XTT assay. Cells were plated at a density of 2000 cells/well and after overnight incubation varying concentrations of C-1748 (5–1000 nM) were added to each well for 96 h. Percent cell survival was determined by XTT assay and plotted as a function of time and dose of C-1748.
Induction of γH2AX is more pronounced in C-1748 treated LnCaP than HL-60
In order to assess extent of DNA DSBs induced by C-1748 in cells, LnCaP and HL-60 cells were treated with 25 and 50 nM of C-1748 for 3 h and extent of γH2AX induction was analyzed by the increase in immunofluorescence over untreated controls (Fig. 5). Induction of γH2AX was 9 fold and 3.5 fold higher in LnCaP than HL-60 with 25 and 50 nM C-1748 respectively (Fig. 5A). Based on cellular DNA content, LnCaP and HL-60 cells were gated into G1, S and G2 phases of the cell cycle and the difference in γH2AX immunofluorescence was assessed in each phase (Fig. 5A). γH2AX induction in C-1748 treated LnCaP was greater compared to HL-60 cells in all phases of the cell cycle (4.7 fold higher in G1; 7.8 fold in S phase and 23 fold in G2 phase with 25 nM C-1748; 4.8 fold higher in G1, 3 fold in S and G2 phases with 50 nM C-1748) (Fig. 5A).
Figure 5.

(A) C-1748 induces γH2AX in LnCaP and HL-60. Exponentially growing LnCaP (upper panel) and HL-60 (lower panel) cells, untreated (Control) or exposed to 25 and 50 nM C-1748 for 3 h and fixed with formaldehyde. Expression of γH2AX was measured concurrently with cellular DNA content by flow cytometry and data shown as bivariate γH2AX IF versus DNA content distributions. Over 93% cells (LnCaP) and 99% (HL-60) from the untreated (Control) culture had γH2AX IF below the threshold marked by the solid line. Inset in each panel shows cellular DNA content frequency histogram of untreated and treated cells. The dashed horizontal line indicates intrinsic H2A.X IF associated with each cell line. The dashed vertical lines in the left panel show boundaries separating cells in G1 vs S vs G2M phases of the cell cycle. (B) Aphidicolin inhibits induction of γH2AX in S phase LnCaP cells treated with C-1748. Exponentially growing LnCaP cells, untreated (control) or exposed to 2 μM aphidicolin for 20 min or 25 and 50 nM C-1748 in the presence of aphidicolin for 3 h. Expression of γH2AX IF was measured by FITC-tagged secondary antibody to γH2AX concurrently with cellular DNA content by flow cytometry and data shown as bivariate γH2AX IF versus DNA content distributions.
While all phases of the cell cycle are affected by agents that induce DSBs, topoisomerase I inhibitors like topotecan induce γH2AX at higher levels in S phase. This is because the non-cleavable topoisomerase complexes on DNA can convert single strand DNA breaks to DSBs during the collision of the replication fork with the complexes during DNA synthesis.41 To assess the mechanism of DSB induction by C-1748, LnCaP cells were first pretreated with the DNA polymerase inhibitor aphidicolin that inhibits replication, and subsequently treated with C-1748 (Fig. 5B). A decrease in the mean γH2AX fluorescence by approximately 50% in treated LnCaP was seen compared to that without aphidicolin pretreatment (Fig. 5B).
C-1748 downregulates androgen receptor (AR) in LnCaP cells
AR levels showed a four fold downregulation with 25 and 50 nM C-1748 in LnCaP cells at 48 h (Fig. 6A). Simultaneously, levels of ERβ, an anti-proliferative marker, showed a two fold increase with C-1748 treatment (Fig. 6A). The synthetic AR agonist, R1881 was used to assess the effect of C-1748 on functionality of AR. R1881 stimulated LnCaP has a markedly higher proliferative index with two fold increased cell survival compared to controls (Fig. 6B). However, in LnCaP cells simultaneously exposed to R1881 and C-1748, no such increase in cell viability was seen. Studies to evaluate the functional consequence of C-1748 mediated AR downregulation were performed using the anti-androgen flutamide and C-1748. It was found that untreated LnCaP was susceptible to flutamide with a greater than 50% inhibition in cell viability after 24 h and statistically significant (p < 0.02), while simultaneous treatment with 25 nM C-1748 conferred resistance to flutamide (Fig. 6C) (statistically significant p < 0.02), which could be partly due to downregulation of AR in C-1748 treated LnCaP.
Figure 6.

(A) Effect of C-1748 on AR and ERβ in LnCaP cells. LnCaP cells were treated with 25 and 50 nM C-1748 for 48 h and expression levels of AR and ERβ were assessed by western blotting. Densitometric analysis of western blots using actin as loading control is shown in the right panel for AR and ERβ. The standard deviation is the mean of three independent experiments. (B) Dose dependent effect of R1881 0.1 nM, 1 nM and 1 μM flutamide and (Fig. 6C) in combination with 25 nM C-1748 on survival of LnCaP cells determined by XTT assay.
DISCUSSION
Chemotherapy has not achieved the success in solid tumors that is observed in hematopoietic cancers.43 Therefore, the primary therapy for CaP at present is often surgery in combination with hormonal therapy or local irradiation.44 The anti-cancer potential of 1-nitroacridines has been studied for decades in order to develop a viable chemotherapeutic agent.31,45 The original drug, ledakrin exhibited differential cytotoxicity for many cancers, but revealed a lack of specificity and poor tolerance in animal models.46 It is a prodrug and most metabolites proved biologically unstable. However, a metabolite of ledakrin was persistently stable30,31 and derivatives of this group led to the development of 4-methyl-1-nitroacridines that showed anti-CaP efficacy. The introduction of a methyl electron donating group at C4 lowered the ability of 1-nitroacridines to undergo reduction and exhibited lower toxicity and mutagenicity than parent 1-nitroacridines33–35 along with excellent host tolerance and survival even after 7 weeks of treatment. Interestingly, increasing the size of the amino alkyl side chain on C9 did not improve efficacy or drug toxicity profiles.
C-1748 derivative exhibited highest efficacy on TSU xenografts. A maximum growth rate was observed during the log phase of growth. A dose of 0.8 mg/kg of C-1748 once weekly, resulted in a decrease of over 70% compared to the untreated group. A similar 70% decrease in growth rate was observed in the log linear phase of growth curve of LnCaP xenografts in the C-1748 treated group. This is highly significant since the dose of C-1748 required to bring this striking reduction was 0.8 mg/Kg, which corresponds to 31 mg/m2 in humans. More importantly, the mice treated with C-1748 remained healthy and did not exhibit any signs of systemic toxicity or weight loss.
These in vivo efficacy studies showed an excellent correlation with the in vitro assessment of the cytotoxic potential of C-1748. Our observations indicated that C-1748 is highly specific for CaP (IC50 22.5 nM) and potentially non-toxic to hematopoietic cells (IC50 > 100 nM) (over four fold higher) in HL-60 leukemia cell line. These effects of C-1748 on a leukemia cell line translate into similar activity on normal hematopoietic cells as observed in our pre-clinical toxicity studies33–35 with profound implications for patient therapy.
DNA damage involving double strand breaks in DNA (DSB) is a common mechanism induced by chemotherapeutic agents in cancer including mitoxantrone and acridines.41 Induction of DNA DSBs triggers phosphorylation of the histone, histone H2AX40,41 leading its phosphorylation on Ser139 at the site of the DSB. The phosphorylated H2AX (γH2AX) recruits DNA repair machinery and is essential for recognition and repair of damaged DNA.
We investigated whether C-1748, a nitroacridine and a DNA damaging agent, has DNA damaging potential by induction of DNA DSBs using γH2AX as a specific and sensitive marker and observed a 9-fold higher induction of DNA DSBs in LnCaP cells compared to HL-60 cells. These results correlate with the difference in IC50 values between LnCaP and HL-60 since LnCaP cells with a much greater larger number of DSBs per cell would be more likely to undergo apoptosis in response to C-1748. Further, there was a lack of G1 arrest in C-1748 treated HL-60 cells in contrast to the marked G1 arrest in C-1748 treated LnCaP cells. This reinforces the observation that the molecular targets conferring specificity to C-1748 in CaP are distinct from the DNA damage potential of nitroacridines and hence we investigated additional targets of C-1748.
In hormone sensitive CaP androgen receptor (AR) plays a key role and has been the basis for androgen ablation therapy used as a primary modality in the treatment of CaP.43 Several studies have documented a link between DNA DSB repair genes and steroid hormone receptors.46,47 Our studies indicate that in LnCaP that express the native AR, C-1748 treatment markedly downregulates AR while upregulating another ERβ a key steroid receptor that has anti-proliferative action in the prostate. The downregulation we observed in the steady state protein levels of AR is also a functional downregulation as C-1748 treated LnCaP is resistant to the effects of androgen agonist R1881 and antagonist flutamide. The upregulation in expression of ERβ is a further validation of the cytotoxicity of C-1748 in CaP.48,49
We have identified C-1748 as a putative anti-CaP chemotherapeutic agent that overcomes the systemic toxicities and limitations of its precursors as the therapeutic efficacy doses are well tolerated in animal models. The steroid hormone receptors have also been identified as its additional anti-CaP targets while maintaining its DNA strand breaks characteristics. The differential activity on DNA strand breaks may explain its lowered myelosuppressive activity as observed in the toxicology analysis.33–35 Coupled with the observed low mutagenecity, C-1748 is a putative anti-CaP agent that needs further clinical investigation.
Acknowledgments
This research was funded by Prostagenics LLC. RKT and DB have significant financial interest.
References
- 1.Dawson NA, Slovin SF. Novel approaches to treat asymptomatic, hormone-naive patients with rising prostate-specific antigen after primary treatment for prostate cancer. Urology. 2003;62:102–18. doi: 10.1016/j.urology.2003.10.027. [DOI] [PubMed] [Google Scholar]
- 2.Kelly WK, Slovin SF. Chemotherapy for androgen-independent prostate cancer: Myth or reality. Curr Oncol Rep. 2000;2:394–401. doi: 10.1007/s11912-000-0058-0. [DOI] [PubMed] [Google Scholar]
- 3.Canil CM, Tannock IF. Is there a role for chemotherapy in prostate cancer? Br J Cancer. 2004;91:1005–11. doi: 10.1038/sj.bjc.6601850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Assikis VJ, Simons JW. Novel therapeutic strategies for androgen-independent prostate cancer: An update. Semin Oncol. 2004;31:26–32. doi: 10.1053/j.seminoncol.2004.02.008. [DOI] [PubMed] [Google Scholar]
- 5.Ryan CJ, Small EJ. Advances in prostate cancer. Curr Opin Oncol. 2004;16:242–46. doi: 10.1097/00001622-200405000-00009. [DOI] [PubMed] [Google Scholar]
- 6.Kasamon KM, Dawson NA. Update on hormone-refractory prostate cancer. Curr Opin Urol. 2004;14:185–93. doi: 10.1097/00042307-200405000-00008. [DOI] [PubMed] [Google Scholar]
- 7.Gulley J, Dahut W. Novel clinical trials in androgen-independent prostate cancer. Clin Prostate Cancer. 2002;1:51–7. doi: 10.3816/cgc.2002.n.007. [DOI] [PubMed] [Google Scholar]
- 8.Gulley J, Dahut WL. Novel approaches to treating the asymptomatic hormone-refractory prostate cancer patient. Urology. 2003;62:147–54. doi: 10.1016/j.urology.2003.08.003. [DOI] [PubMed] [Google Scholar]
- 9.La Porta CA. Cellular targets for anticancer strategies. Curr Drug Targets. 2004;5:347–55. doi: 10.2174/1389450043345434. [DOI] [PubMed] [Google Scholar]
- 10.Engel R, Valkov NI, Gump JL, Hazlehurst L, Dalton WS, Sullivan DM. The cytoplasmic trafficking of DNA topoisomerase II correlates with etoposide resistance in human myeloma cells. Exp Cell Res. 2004;295:421–31. doi: 10.1016/j.yexcr.2004.01.012. [DOI] [PubMed] [Google Scholar]
- 11.Van Dalen EC, Van der Pal HJ, Bakker PJ, Caron HN, Kremer LC. Cumulative incidence and risk factors of mitoxantrone-induced cardiotoxicity in children: A systematic review. Eur J Cancer. 2004;40:643–52. doi: 10.1016/j.ejca.2003.12.006. [DOI] [PubMed] [Google Scholar]
- 12.Goodin S, Rao KV, DiPaola RS. State-of-the-art treatment of metastatic hormone-refractory prostate cancer. Oncologist. 2002;7:360–70. doi: 10.1634/theoncologist.7-4-360. [DOI] [PubMed] [Google Scholar]
- 13.Albrecht W, Van Poppel H, Horenblas S, Mickisch G, Horwich A, Serretta V, Casetta G, Maréchal JM, Jones WG, Kalman S, Sylvester R. Randomized Phase II trial assessing estramustine and vinblastine combination chemotherapy vs estramustine alone in patients with progressive hormone-escaped metastatic prostate cancer. Br J Cancer. 2004;90:100–5. doi: 10.1038/sj.bjc.6601468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McMenemin R, Macdonald G, Moffat L, Bissett D. A phase II study of caelyx (liposomal doxorubicin) in metastatic carcinoma of the prostate: Tolerability and efficacy modification by liposomal encapsulation. Invest New Drugs. 2002;20:331–7. doi: 10.1023/a:1016225024121. [DOI] [PubMed] [Google Scholar]
- 15.Hamilton M, Dahut W, Brawley O, Davis P, Wells-Jones T, Kohler D, Duray P, Liewehr DJ, Lakhani N, Steinberg SM, Figg WD, Reed E. A phase I/II study of high-dose tamoxifen in combination with vinblastine in patients with androgen-independent prostate cancer. Acta Oncol. 2003;42:195–201. doi: 10.1080/02841860310010718. [DOI] [PubMed] [Google Scholar]
- 16.Papandreou CN, Daliani DD, Thall PF, Tu SM, Wang X, Reyes A, Troncoso P, Logothetis CJ. Results of a phase II study with doxorubicin, etoposide, and cisplatin in patients with fully characterized small-cell carcinoma of the prostate. J Clin Oncol. 2002;20:3072–80. doi: 10.1200/JCO.2002.12.065. [DOI] [PubMed] [Google Scholar]
- 17.Vaishampayan U, Fontana J, Du W, Hussain M. An active regimen of weekly paclitaxel and estramustine in metastatic androgen-independent prostate cancer. Urology. 2002;60:1050–54. doi: 10.1016/s0090-4295(02)01990-8. [DOI] [PubMed] [Google Scholar]
- 18.Armstrong AJ, Carducci MA. Chemotherapy for advanced prostate cancer: Results of new clinical trials and future studies. Curr Oncol Rep. 2005;7:220–27. doi: 10.1007/s11912-005-0077-y. [DOI] [PubMed] [Google Scholar]
- 19.Goodin S, Medina P, Capanna T, Shih WJ, Abraham S, Winnie J, Doyle-Lindrud S, Todd M, DiPaola RS. Effect of docetaxel in patients with hormone-dependent prostate-specific antigen progression after local therapy for prostate cancer. J Clin Oncol. 2005;23:3352–57. doi: 10.1200/JCO.2005.11.111. [DOI] [PubMed] [Google Scholar]
- 20.Hussain M, Petrylak D, Fisher E, Tangen C, Crawford D. Docetaxel (Taxotere) and estramustine versus mitoxantrone and prednisone for hormone-refractory prostate cancer: Scientific basis and design of Southwest Oncology Group study 9916. Semin Oncol. 1999;26:55–60. [PubMed] [Google Scholar]
- 21.Petrylak DP, Tangen C, Hussain MH, Lara PN, Jr, Jones JA, Taplin ME, Burch PA, Berry D, Moinpour C, Kohli M, Benson MC, Small EJ, et al. Docetaxel and estramustine compared with mitoxantrone and prednisone for advanced refractory prostate cancer. N Engl J Med. 2004;351:1513–20. doi: 10.1056/NEJMoa041318. [DOI] [PubMed] [Google Scholar]
- 22.Walczak JR, Carducci MA. Phase 3 randomized trial evaluating second-line hormonal therapy versus docetaxel-estramustine combination chemotherapy on progression-free survival in asymptomatic patients with a rising prostate-specific antigen level after hormonal therapy for prostate cancer: An Eastern Cooperative Oncology Group (E1899), intergroup/clinical trials support unit study. Urology. 2003;62:141–6. doi: 10.1016/j.urology.2003.09.006. [DOI] [PubMed] [Google Scholar]
- 23.Small EJ, Baron A, Bok R. Simultaneous antiandrogen withdrawal and treatment with ketoconazole and hydrocortisone in patients with advanced prostate carcinoma. Cancer. 1997;80:1755–9. doi: 10.1002/(sici)1097-0142(19971101)80:9<1755::aid-cncr9>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- 24.Trachtenberg J, Halpern N, Pont A. Ketoconazole: A novel and rapid treatment for advanced prostatic cancer. J Urol. 1983;130:152–3. doi: 10.1016/s0022-5347(17)51007-1. [DOI] [PubMed] [Google Scholar]
- 25.Kelly WK, Curley T, Slovin S, Heller G, McCaffrey J, Bajorin D, Ciolino A, Regan K, Schwartz M, Kantoff P, George D, Oh W, et al. Paclitaxel, estramustine phosphate, and carboplatin in patients with advanced prostate cancer. J Clin Oncol. 2001;19:44–53. doi: 10.1200/JCO.2001.19.1.44. [DOI] [PubMed] [Google Scholar]
- 26.Pawlak JW, Pawlak K, Konopa J. The mode of action of cytotoxic and antitumor 1-nitroacridines. II. In vivo enzyme-mediated covalent binding of a 1-nitroacridine derivative, Ledakrin or Nitracrine, with DNA and other macromolecules of mammalian or bacterial cells. Chem Biol Interact. 1983;43:151–73. doi: 10.1016/0009-2797(83)90093-5. [DOI] [PubMed] [Google Scholar]
- 27.Konopa J, Pawlak JW, Pawlak K. The mode of action of cytotoxic and antitumor 1-nitroacridines. III. In vivo interstrand cross-linking of DNA of mammalian or bacterial cells by 1-nitroacridines. Chem Biol Interact. 1983;43:175–97. doi: 10.1016/0009-2797(83)90094-7. [DOI] [PubMed] [Google Scholar]
- 28.Pawlak K, Pawlak JW, Konopa J. Cytotoxic and antitumor activity of 1-nitroacridines as an aftereffect of their interstrand DNA cross-linking. Cancer Res. 1984;44:4289–96. [PubMed] [Google Scholar]
- 29.Skladanowski A. Modulation of G2arrest enhances cell death induced by the antitumor 1-nitroacridine derivative, Nitracrine. Apoptosis. 2002;7:347–59. doi: 10.1023/a:1016127513947. [DOI] [PubMed] [Google Scholar]
- 30.Gorlewska K, Mazerska Z, Sowinski P, Konopa J. Products of metabolic activation of the antitumor drug ledakrin (nitracrine) in vitro. Chem Res Toxicol. 2001;14:1–10. doi: 10.1021/tx000081c. [DOI] [PubMed] [Google Scholar]
- 31.Gniazdowski M, Szmigiero L. Nitracrine and its congeners—an overview. Gen Pharmacol. 1995;26:473–81. doi: 10.1016/0306-3623(94)00143-b. [DOI] [PubMed] [Google Scholar]
- 32.Mazerska Z, Lukowicz J, Konopa J. Antitumor activity of 1-nitro-9-aminoacridines including nitracrine against some ascitic experimental tumors. Arzneimittelforschung. 1990;40:472–77. [PubMed] [Google Scholar]
- 33.Ashok BT, Tadi K, Garikapaty VPS, Chen Y, Huang Q, Banerjee D, Konopa J, Tiwari RK. Pre-clinical toxicological evaluation of a putative prostate cancer specific 1-nitroacridine derivative in rodents. Anti-Cancer Drugs. 2007;18:87–94. doi: 10.1097/01.cad.0000236316.04199.20. [DOI] [PubMed] [Google Scholar]
- 34.Ashok BT, Tadi K, Banerjee D, Konopa J, Iatropoulos M, Tiwari RK. Pre-clinical toxicology and pathology of 9-(2′-hydroxyethylamino)-4-methyl-1-nitroacridine (C-1748), a novel anti-cancer agent in male Beagle dogs. Life Sci. 2006;79:1334–42. doi: 10.1016/j.lfs.2006.03.043. [DOI] [PubMed] [Google Scholar]
- 35.Narayanan R, Tiwari P, Inoa D, Ashok BT. Comparative analysis of mutagenic potency of 1-nitro-acridine derivatives. Life Sci. 2005;77:2312–23. doi: 10.1016/j.lfs.2005.03.020. [DOI] [PubMed] [Google Scholar]
- 36.Szostek A, Wysocka-Skrzela B, Konopa J, Tiwari RK. Potent anti tumor 4-substituted 1-nitroacridines, DNA cross linking of lowered toxicity. Proceedings of the AACR-NCI-EORTC International Conference; 2003; p. Abstr.65. [Google Scholar]
- 37.van Bokhoven A, Varella-Garcia M, Korch C, Miller GJ. TSU-Pr1 and JCA-1 cells are derivatives of T24 bladder carcinoma cells and are not of prostatic origin. Cancer Res. 2001;61:6340–4. [PubMed] [Google Scholar]
- 38.Ashok BT, Kim E, Mittelman A, Tiwari RK. Proteasome inhibitors differentially affect heat shock protein response in cancer cells. Int J Mol Med. 2001;4:385–90. doi: 10.3892/ijmm.8.4.385. [DOI] [PubMed] [Google Scholar]
- 39.Yedavelli SP, Guo L, Daou ME, Srivastava PK, Mittelman A, Tiwari RK. Preventive and therapeutic effect of tumor derived heat shock protein, gp96, in an experimental prostate cancer model. Int J Mol Med. 1999;3:243–8. doi: 10.3892/ijmm.4.3.243. [DOI] [PubMed] [Google Scholar]
- 40.Huang X, Halicka HD, Traganos F, Tanaka T, Kurose A, Darzynkiewicz Z. Cytometric assessment of DNA damage in relation to cell cycle phase and apoptosis. Cell Prolif. 2005;38:223–43. doi: 10.1111/j.1365-2184.2005.00344.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Huang X, Traganos F, Darzynkiewicz Z. DNA damage induced by DNA topoisomerase I- and topoisomerase II-inhibitors detected by histone H2AX phosphorylation in relation to the cell cycle phase and apoptosis. Cell Cycle. 2003;2:614–19. [PubMed] [Google Scholar]
- 42.Tadi K, Chen YG, Ashok BT, Garikapaty VPS, Banerjee D, Wysocka-Skrzela B, Konopa J, Tiwari RK. Capridine (C-1748): A novel 1-nitroacridine derivative targets the cyclin dependent kinase, cdk4 and the tumor suppressor gene, p16 in prostate cancer. Proc Amer Assoc Cancer Res. 2004;45:Abstract # 3036. [Google Scholar]
- 43.Redaelli A, Laskin BL, Stephens JM, Botteman MF, Pashos CL. A systematic literature review of the clinical and epidemiological burden of acute lymphoblastic leukaemia (ALL) Eur J Cancer Care (Engl) 2005;14:53–62. doi: 10.1111/j.1365-2354.2005.00513.x. [DOI] [PubMed] [Google Scholar]
- 44.Sharkey J, Cantor A, Solc Z, Huff W, Chovnick SD, Behar RJ, Perez J, Otheguy J, Rabinowitz R. (103)Pd brachytherapy versus radical prostatectomy in patients with clinically localized prostate cancer: A 12-year experience from a single group practice. Brachytherapy. 2005;4:34–44. doi: 10.1016/j.brachy.2004.12.001. [DOI] [PubMed] [Google Scholar]
- 45.Hamilton D, Batist G. Anthracyclines. Cancer Chemother Biol Response Modif. 2005;22:19–33. [PubMed] [Google Scholar]
- 46.Liu X, Yao J, Pisha E, Yang Y, Hua Y, van Breemen RB, Bolton JL. Oxidative DNA damage induced by equine estrogen metabolites: Role of estrogen receptor alpha. Chem Res Toxicol. 2002;15:512–19. doi: 10.1021/tx0101649. [DOI] [PubMed] [Google Scholar]
- 47.Crowe DL, Lee MK. New role for nuclear hormone receptors and coactivators in regulation of BRCA1-mediated DNA repair in breast cancer cell lines. Breast Cancer Res. 2006;8:R1. doi: 10.1186/bcr1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bhandari MS, Crook J, Hussain M. Should intermittent androgen deprivation be used in routine clinical practice? J Clin Oncol. 2005;23:8212–18. doi: 10.1200/JCO.2005.03.2557. [DOI] [PubMed] [Google Scholar]
- 49.Koehler KF, Helguero LA, Haldosen LA, Warner M, Gustafsson JA. Reflections on the discovery and significance of estrogen receptor beta. Endocr Rev. 2005;26:465–78. doi: 10.1210/er.2004-0027. [DOI] [PubMed] [Google Scholar]