

Abstract
Transcription is silenced during mitosis and re-activated at mitotic exit. The dynamics and identities of “bookmarking” transcription factors and chromatin marks that mediate reactivation often recapitulate that observed during cell identity establishment in development. Thus, features of post-mitotic gene re-activation can provide insights into mechanisms of developmental cell fate establishment.
Keywords: mitosis, bookmarking, memory, pioneer factors, chromatin
It has long been known that during mitosis, the chromosomes condense, RNA polymerases exit the chromatin, and transcription stops. Yet within about an hour post-mitosis, the transcriptional program specific to a cell is faithfully re-activated, allowing the cell to maintain its identity and continue to function (Egli et al., 2008). We take for granted the remarkable precision with which transcriptional programs are re-established after mitosis, in comparison to the attention paid to the regulation of cell fate specification. During cell fate specification, pioneer transcription factors bind to silent genes in progenitor cells, imparting the potential for gene activation upon inductive signaling (Zaret and Carroll, 2011). Cell fate can be further regulated by the binding of repressive complexes, in place of pioneer factors, to exclude certain cell fates. In addition, cell fates are influenced by transcriptional pausing, whereby control is exerted at the level of transcriptional elongation by RNA polymerases that were engaged at promoters by earlier inductive events (Nechaev and Adelman, 2011).
Yet genome reactivation post-mitosis represents a more dramatic version of what occurs during developmental cell fate specification. Virtually the entire genome is shut down during mitosis and, upon mitotic exit, the cell has to completely regenerate the pattern of regulatory complexes in chromatin, recruit RNA polymerases de novo, and establish the proper extent of transcriptional pausing and elongation, genome-wide. Indeed, recent 5C and Hi-C studies show that most of the higher-order “looping” interactions across the chromosomes in interphase appear to be lost during mitosis (Naumova et al., 2013). How does the newly divided cell re-establish chromatin states that elicit the requisite transcriptional networks? What do details of the process teach us about establishing cell fates in development and reprogramming cell fates at will?
Maintaining cell identity during global loss of transcription
Although histone acetylation is frequently characterized as an epigenetic mark, the bulk of chromatin acetylation is lost in mitosis and histone acetyltransferases are excluded from chromatin (Table S1A). In contrast, repressive methyl histone marks such as histone H3 lysine 9 trimethylation (H3K9me3) and H3K27 trimethylation (H3K27me3) (Follmer et al., 2012), as originally discovered in embryonic development, are retained through mitosis and could therefore play an epigenetic role in keeping target genes silent during mitotic exit (Table S1A).
In terms of enabling post-mitotic gene activation, in vivo footprinting methodologies in the 1990s showed that while most transcription factors are lost from mitotic chromatin, like RNA polymerase itself, a subset of transcription factors are retained at their specific target sites (Martinez-Balbas et al., 1995; Michelotti et al., 1997). Scanning the literature, about 20% of the transcription factors and chromatin binding proteins that have been tested are retained on mitotic chromatin (Egli et al., 2008). Again, various proteins that are presumed to be epigenetic because of their association with chromatin, such as the heterochromatin binding proteins HP1, the chromatin remodeling factor BRG1, and the Polycomb subunit BMI1, are actually excluded from mitotic chromatin (Egli et al., 2008). Phosphorylation by mitotic kinases can abolish DNA binding by transcription factors (Dovat et al., 2002) and ubiquitin and ubiquitin-like modifications in mitosis can further alter protein stability (Merbl et al., 2013). While mitotic chromatin condensation could inhibit factor binding, linker and core histones still exchange in chromatin, though more slowly than in interphase (Chen et al., 2005). Canonical nucleosomes appear to lose their positioning as transcription ceases (Komura and Ono, 2005) and H2A.Z variant-containing nucleosomes shift upstream of their interphase position, so that they now cover the mitotically inactive transcription start site (Kelly et al., 2010). Thus, we now appreciate that mitotic chromatin is not as inaccessible as once assumed and that a degree of histone and nucleosome dynamics persists (Table S1A). In addition, the exclusion of factors from mitotic chromatin can involve their post-translational modifications as well as the compacted state of chromatin.
It is interesting to compare the dynamics in chromatin accessibility that occur in mitosis with the changes that occur during development and cellular reprogramming. Histone exchange appears more dynamic in embryonic stem cells than in differentiated cells (Meshorer et al., 2006), suggesting that the ability of factors to move in and out of chromatin may contribute to the pluripotent state. During somatic cell nuclear reprogramming, the linker histone variant B4 and the nuclear protein Wave1 (involved in actin cytoskeletal organization) promote large scale decompaction and changes in the somatic nuclear architecture that facilitate reprogramming (Jullien et al., 2010; Miyamoto et al., 2013). Although these large-scale changes do not resemble those in mitosis, somatic cell nuclear transfer is more efficient when the transplanted nuclear chromosomes first assume a mitotic-like condensation (Egli et al., 2008). Continued growth allows the cells to undergo a controlled chromosomal decondensation, a process that may enhance reprogramming. Megabase-scale chromatin domains with the repressive H3K9me3 modification in somatic cells can physically occlude transcription factor binding and impair reprogramming (Soufi et al., 2012). As noted above, HP1, which binds H3K9me3 domains, and BMI1, a Polycomb subunit associated with H3K27me3 domains, appear to be excluded from chromatin during mitosis, despite the presence of their associated histone marks. Understanding the basis for their exclusion could provide insight into ways to overcome repressive chromatin during cellular reprogramming.
As for the transcription factors that are retained on mitotic chromatin, knock-down studies and mitotic-specific ablation demonstrate that these factors help promote the timely re-activation of target genes during mitotic exit, and thus are true “mitotic bookmarking” factors (Figure 1, Table S1B). Various features are striking with regard to bookmarking factors in mitotic chromatin. First, while they are retained in mitotic chromatin, global assessments of DNA binding by ChIP-Seq show that the factors only associate with a subset of the target sites that they bind in interphase cells (Table S1B). Significantly, the mitotic targets include genes for transcriptional regulators that are essential for cell fate maintenance. Thus, by remaining bound to such genes in mitosis and promoting their rapid re-activation during mitotic exit, the bookmarking factors can help reset a specific transcriptional program. When the activation of genes by bookmarking transcription factors is considered with the repressive epigenetic marks that are retained in mitosis, it would seem that the fundamental mechanisms of re-establishing cell identity during mitotic exit are in view.
Figure 1.
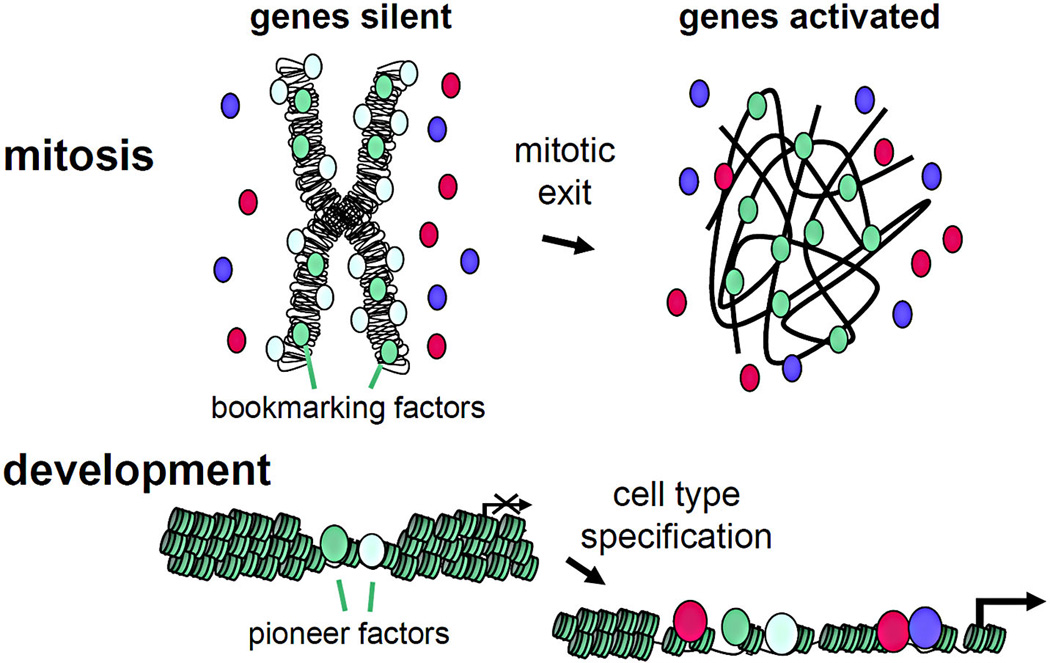
(Top) A transcriptionally silent, compacted mitotic chromosome (left) with bookmarking transcription factors bound (green shades) and other factors not bound (red and purple). During mitotic exit, the bookmarking factors facilitate the rapid activation of the genes to which they are bound in mitosis. (Bottom) A transcriptionally silent, compacted gene with pioneer transcription factors bound in early development. During cell type specification, pioneer factors facilitate the binding of other factors (red, purple) and the activation of genes to which they are bound. The text discusses the recently discovered similarities between bookmarking and pioneer transcription factors.
Mitotic exit appears to recapitulate development
There is an additional distinguishing feature of mitotic bookmarking transcription factors: They also typically function in early development to help specify cell fates. For example, FoxA1 and GATA1, which have been mapped genomically to chromatin in mitosis by ChIP-Seq, were previously shown to function as pioneer factors by being able to bind target sites on nucleosomal DNA as well as to function early in development, prior to gene activity (Table S1B). In a side-by-side comparison of diverse GFP-tagged liver transcription factors in live hepatoma cells, FoxA1 bound quantitatively to mitotic chromosomes, similar to that seen for linker histone, whereas GATA4 was distributed about equally on and off the chromosomes (similar to what was seen with GATA1 in another study (Kadauke et al., 2012)), and NF1 and c-Myc were excluded from the mitotic chromatin (Caravaca et al., 2013). C/EBPα also exhibited greatly diminished binding, though this was due to high turnover of the protein in mitotic hepatoma cells. Remarkably, the strong binding by FoxA1, partial retention of GATA4, and greatly diminished binding by the other factors in mitotic chromatin reflects the developmental hierarchy of the factors as they enter chromatin in liver development (Bossard and Zaret, 1998; Gualdi et al., 1996). Thus, mitotic exit may briefly recapitulate the sequence of transcription factor engagement in development. Indeed, given the rapidity with which signaling can induce cell fate control in rapidly growing embryos, the time course of events during mitotic exit could be similar.
Roles of specific and nonspecific DNA binding by mitotic bookmarking factors
While FoxA1-GFP bound quantitatively to mitotic chromosomes with no total diminution of FoxA1 protein, only 15% of FoxA1 interphase target sites are occupied in mitosis, as noted above (Caravaca et al., 2013). Detailed analysis of the ChIP-Seq tracks revealed a higher background of signals across the genome for FoxA1 (not at motifs) in mitosis than in interphase, suggesting a greater extent of nonspecific chromatin binding. Fluorescence recovery after photobleaching (FRAP) experiments demonstrated that FoxA1 is more mobile in mitotic chromatin than in interphase, whereas linker histone was less mobile in mitotic chromatin. Finally, point mutations in FoxA1 that disrupted sequence-specific DNA binding did not measurably impair global retention on mitotic chromosomes, whereas point mutations in FoxA1 that disrupted nonspecific DNA binding greatly perturbed mitotic binding. Thus, a substantial portion of FoxA1 is bound nonspecifically to mitotic chromosomes, in addition to the factor’s specific binding to a subset of its interphase sites.
Knock-down studies showed that genes that were bound by wild type FoxA1 in mitosis were clearly dependent upon the factor for timely reactivation of transcription during mitotic exit. In addition, genes that were bound by FoxA1 only in interphase, but not in mitosis, were still dependent upon FoxA1 for timely re-activation during mitotic exit. However, these genes showed a wider variation in their dependency for FoxA1 than for genes bound by FoxA1 in mitosis. As expected, genes not bound by FoxA1 in interphase or mitosis were not dependent upon the factor. Thus it appears that the nonspecific chromosome binding by FoxA1 contributes to post-mitotic gene reactivation, but with slower or more variable kinetics than the gene re-activation associated with specific mitotic FoxA1 binding. We speculated that the nonspecific binding of FoxA1 to mitotic chromosomes functions as a storage mechanism (Caravaca et al., 2013). This would give the factor immediate proximity to the genome during mitotic exit, thereby allowing it to scan for targets prior to other factors that first need to re-gain chromatin binding capacity.
Presently, it is unclear to what extent other bookmarking factors bind nonspecifically to the genome in mitosis. Furthermore, the role of nonspecific DNA binding for transcription factors in development or cellular reprogramming is essentially unexplored. We have observed that the pioneer activity of FoxA1 relates to high nucleosome affinity and low mobility in nuclear chromatin, whereas non-pioneer factors have lower nucleosome affinity and higher mobility in nuclear chromatin (Sekiya et al., 2009). Understanding how developmental transcription factors scan the genome and gain access to new targets buried in silent chromatin seems likely to provide insight into improving directed reprogramming of cell fates.
Future directions – additional thoughts
In terms of keeping lineage-inappropriate genes properly repressed during mitotic exit, the locations of H3K9me3 and H3K27me3 have not yet been reported at the genome-wide level in mitotic versus interphase cells. It seems likely that most of the marks will be stable in location in mitosis and thus directly epigenetic for their target genes. If new sites are found to gain H3K9me3 and/or H3K27me3 specifically in mitosis and then lose the mark in G1, the sites (or genes) could be transiently repressed solely during the initial phase of mitotic exit. While purely speculative, such dynamics in studies of mitosis could provide insight into transient states that occur when cell programs are initially specified in development or when they are re-wired during cell reprogramming.
For transcription factors and chromatin modifiers that are excluded from mitotic chromatin via a post-translational modification, it will be useful to learn how a shift, during mitotic exit, from mitotic kinases to phosphatases, among other shifts in protein modifiers, could help orchestrate the ability of excluded chromatin factors to re-engage the genome. Conceivably, similar rapid modification changes, either within or outside of mitosis, will be found to operate during cell fate control.
How general is the model that transcription factor hierarchies in development are played out transiently during mitotic exit? It will be important to compare diverse factors that together specify different cell fates for binding to mitotic chromatin, in order to determine whether other bookmarking factors and their non-mitotic binding partners exhibit the same developmental hierarchy as seen for FoxA1, GATA4, and the liver transcription factors described above. Interestingly, careful analyses of primary transcripts of about 20 genes in studies of hepatoma and erythroid cell lines revealed diverse patterns of re-activation (Caravaca et al., 2013; Kadauke et al., 2012). Some genes reactivated early during mitotic exit, while some reactivated later; and some exhibited a burst in expression that then declined. Genome-wide studies of transcriptome re-activation in mitotic exit are needed, and it will be interesting to compare the resultant network dynamics with those seen in the relevant cells as they are specified in development. Such analyses could definitively assess the extent to which mitotic exit recapitulates development.
As discussed in this Forum, understanding how only a subset of chromatin binding factors function in mitosis, along with the dynamics of gene reactivation during mitotic exit, has provided unexpected insights about fate-determining transcription factors and their functions in chromatin. Given the rapidity with which cell programming occurs within embryos and during reprogramming in vitro, further insights into the unusual genomic periods of mitosis and mitotic exit will continue to shed light on activities and behaviors of regulatory components that we will need to understand to ultimately control cell function precisely.
Supplementary Material
Acknowledgements
The author thanks Katherine Palozola, Justin Becker, Maki Iwafuchi-Doi, and Marie Bao for helpful comments and Eileen Hulme for preparing the manuscript. Work was supported by NIH grant R37GM36477.
References
- Bossard P, Zaret KS. Development. 1998;125:4909–4917. doi: 10.1242/dev.125.24.4909. [DOI] [PubMed] [Google Scholar]
- Caravaca JM, Donahue G, Becker JS, He X, Vinson C, Zaret KS. Genes Dev. 2013;27:251–260. doi: 10.1101/gad.206458.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dovat S, Ronni T, Russell D, Ferrini R, Cobb BS, Smale ST. Genes Dev. 2002;16:2985–2990. doi: 10.1101/gad.1040502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egli D, Birkhoff G, Eggan K. Nat Rev Mol Cell Biol. 2008;9:505–516. doi: 10.1038/nrm2439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gualdi R, Bossard P, Zheng M, Hamada Y, Coleman JR, Zaret KS. Genes Dev. 1996;10:1670–1682. doi: 10.1101/gad.10.13.1670. [DOI] [PubMed] [Google Scholar]
- Jullien J, Astrand C, Halley-Stott RP, Garrett N, Gurdon JB. Proc Natl Acad Sci U S A. 2010;107:5483–5488. doi: 10.1073/pnas.1000599107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Balbas MA, Dey A, Rabindran SK, Ozato K, Wu C. Cell. 1995;83:29–38. doi: 10.1016/0092-8674(95)90231-7. [DOI] [PubMed] [Google Scholar]
- Merbl Y, Refour P, Patel H, Springer M, Kirschner MW. Cell. 2013;152:1160–1172. doi: 10.1016/j.cell.2013.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meshorer E, Yellajoshula D, George E, Scambler PJ, Brown DT, Misteli T. Dev Cell. 2006;10:105–116. doi: 10.1016/j.devcel.2005.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michelotti EF, Sanford S, Levens D. Nature. 1997;388:895–899. doi: 10.1038/42282. [DOI] [PubMed] [Google Scholar]
- Miyamoto K, Teperek M, Yusa K, Allen GE, Bradshaw CR, Gurdon JB. Science. 2013;341:1002–1005. doi: 10.1126/science.1240376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nechaev S, Adelman K. Biochim Biophys Acta. 2011;1809:34–45. doi: 10.1016/j.bbagrm.2010.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekiya T, Muthurajan UM, Luger K, Tulin AV, Zaret KS. Genes Dev. 2009;23:804–809. doi: 10.1101/gad.1775509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soufi A, Donahue G, Zaret KS. Cell. 2012;151:994–1004. doi: 10.1016/j.cell.2012.09.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaret KS, Carroll JS. Genes Dev. 2011;25:2227–2241. doi: 10.1101/gad.176826.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.