

Abstract
We have developed a photocatalytic reduction of nitroarenes as an efficient, chemoselective route to biologically important N-phenyl hydroxamic acid scaffolds. Optimal conditions call for 2.5 mol% of a ruthenium photocatalyst, visible light irradiation, and a dihydropyridine terminal reductant. Because of the mild nature of the visible light activation, functional groups that might be sensitive to other non-photochemical reduction methods are easily tolerated.
Keywords: electron transfer, heterocycles, hydroxamic acids, photochemistry, reduction
Hydroxamic acids are high-affinity chelating ligands for a wide range of metal cations.1 Many hydroxamic acid containing secondary metabolites are produced naturally, and they have important biological roles in a variety of contexts including microbial iron metabolism and endogenous chemical defense in plants.2 In medicinal chemistry, cyclic hydroxamic acids have been reported to possess antimicrobial and antifungal activity and have also been investigated as potential treatments for conditions ranging from cancer to schizophrenia.3
The most common strategies for the synthesis of cyclic hydroxamic acids involve reduction of nitroarenes to the corresponding hydroxylamines followed by intramolecular cyclization with a tethered acyl moiety (Scheme 1). A variety of methods to achieve this transformation have been reported, including those using stoichiometric zinc or tin4 as well as palladium5 or platinum6 catalyzed partial reduction. Many of these methods can be somewhat problematic. First, the stoichiometric processes can generate metal-containing byproducts that complicate the isolation and purification of these strong chelators. Second, the strongly reducing conditions used in many of these reactions can be incompatible with sensitive, easily reduced functional groups such as aryl halides. Finally, a significant challenge in this approach to the synthesis of hydroxamic acids is to achieve selective four-electron reduction of the nitroarene to the desired hydroxamic acid without competitive six-electron reduction to the fully reduced quinolinone.
Scheme 1.

Preparation of hydroxamic acids by reduction and cyclization of nitroarenes.
Over the last several years, our laboratory, along with several others, has been investigating the design of synthetically useful new reactions that exploit the photochemical properties of Ru(bpy)32+ and related transition metal chromophores in the visible light regime.7 Our efforts have led to a wide range of cycloaddition reactions that are initiated by photocatalytic oxidation or reduction of alkenes;8 related efforts in other groups investigating photocatalytic redox reactions of amines, arenes, and alkyl halides have resulted in the development of a remarkable diversity of synthetically useful transformations.9 As part of our ongoing efforts to broaden the scope of reactions amenable to visible light photocatalysis, we became interested in designing a selective photocatalytic four-electron reduction of nitroarenes to afford hydroxamic acids.
The use of Ru(bpy)32+ as a photocatalyst for the exhaustive six-electron reduction of nitrobenzene to aniline has been previously reported using hydrazine as the terminal reductant.10 Similarly, the photocatalytic four-electron reduction of nitroalkenes to oximes has been accomplished using EDTA as the terminal reductant.11 To the best of our knowledge, the photocatalytic four-electron reduction of nitrobenzene to a hydroxylamine or hydroxamic acid has not been previously been described.
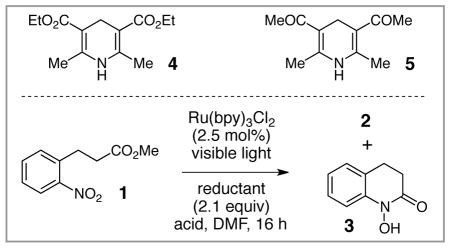
Table 1 summarizes optimization and control experiments for the photocatalytic reductive cyclization of nitroarene 1 to hydroxamic acid 3. We began by applying conditions reported by Stephenson for reductive dehalogenation reactions9b to this reduction. However, when 1 was irradiated in the presence of formic acid, i-Pr2NEt, and 2.5 mol% Ru(bpy)32+, we observed none of the expected hydroxamic acid 3 and only a trace of the intermediate hydroxylamine 2 (entry 1). In a screen of alternate terminal reductants, we observed that while Hantzsch ester 4 provided only a trace of reduction products (entry 2), the related diketone 5 resulted in good conversion of 1 to a mixture of hydroxylamine and hydroxamic acid (entry 3). We speculated that the Brønsted acid could be responsible for the cyclization of 2 to 3; indeed, in the absence of an exogenous Brønsted acid additive, we observed exclusive formation of 2 without any obvious change in the rate of the photoreduction process (entry 4). The use of stronger acids, on the other hand, increased the yield of 3 (Entries 5–7). Optimal results were obtained using camphorsulfonic acid (CSA), and we found that the stoichiometry of this acid could be lowered to 0.1 equiv without affecting the yield of the reaction (entry 8). Finally, control experiments verified the photocatalytic nature of this reaction; in the absence of either Ru(bpy)32+ or light, we observed no significant formation of 3 (entries 9–10).
Table 1.
Optimization studies for photocatalytic hydroxamic acid synthesis
| ||||
|---|---|---|---|---|
| entry | reductant | acid (equiv) | yield 2 (%)a | yield 3 (%)a |
| 1 | i-Pr2NEt | HCO2H (1) | <5 | 0 |
| 2 | 4 | HCO2H (1) | 0 | <5 |
| 3 | 5 | HCO2H (1) | 50 | 20 |
| 4 | 5 | none | 71 | 0 |
| 5 | 5 | AcOH (1) | 52 | 25 |
| 6 | 5 | TFA (1) | 0 | 84 |
| 7 | 5 | CSA (1) | 0 | 89 |
| 8 | 5 | CSA (0.1) | 0 | 88 |
| 9b | 5 | CSA (0.1) | 0 | <5 |
| 10c | 5 | CSA (0.1) | 0 | 0 |
Yield determined by 1H NMR analysis.
Reaction conducted in the absence of Ru(bpy)3Cl2.
Reaction conducted in the dark.
On larger scales, isolation of pure hydroxamic acid 3 could easily be accomplished in good yields by recrystallization. Chromatographic isolation of this material, however, proved to be more challenging; the mass recovery was low, and the eluted product was deeply colored, which we attributed to the ability of this strongly chelating compound to leach metallic impurities from the silica gel. However, treatment of the unpurified reaction mixture with Boc2O and Et3N resulted in the formation of a protected hydroxamic acid that could be easily be purified by standard chromatographic methods.4c
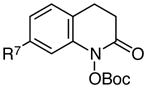
Using these optimized conditions for production and protection of hydroxamic acids, we conducted an exploration of the scope of this process (Table 2). The reaction proved to be relatively insensitive to electronic perturbation at C7; both electron-donating and electron-withdrawing substituents at this position provide similarly good yields of hydroxamic acids (entries 1–6). Importantly, we observed no reduction of potentially reducible functional groups such as aryl bromides or nitriles (entries 5 and 6). The identity of the C6 substituent had a more dramatic effect. While electron-withdrawing groups at this position had little impact (entry 7), the methoxy-substituted substrate cleanly underwent over-reduction to the quinolinone. A similar effect of electron-donating substituents was reported by McAllister,4c who proposed that the accessibility of an iminoquinone intermediate could be responsible for the ease of subsequent overreduction (Scheme 2). Changes to the tethering moiety were also tolerated (entries 9–11), although either introducing a tosyl-protected nitrogen (entry 10) or reducing the length of the tether by one carbon (entry 11) resulted in slower cyclizations that necessitated stoichiometric acid. Finally, these conditions tolerated an α-acetamido substituent (entry 12), which provided access to a privileged scaffold reported to possess a range of biological properties.12
Table 2.
Scope studies for hydroxamic acid synthesis.
| entrya | product | yield (%)b | |
|---|---|---|---|
| 1 |
|
R7 = H | 83 |
| 2 | R7 = OMe | 77 | |
| 3 | R7 = Me | 85 | |
| 4 | R7 = CF3 | 81 | |
| 5 | R7 = CN | 72 | |
| 6 | R7 = Br | 76 | |
| 7 |
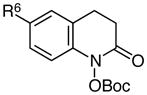
|
R6 = F | 79 |
| 8 | R6 = OMe | 0c | |
| 9 |
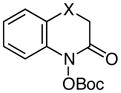
|
X = O | 78 |
| 10d | X = NTs | 53 | |
| 11d |
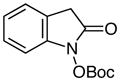
|
64 | |
| 12d |
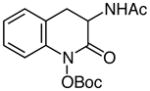
|
58 |
Reactions conducted using 2.5 mol% Ru(bpy)3Cl2, 2.1 equiv 5, and 0.1 equiv CSA unless otherwise noted.
Values represent the averaged isolated yields from two reproducible experiments.
Quinolinone 10 was isolated in 54% yield (Scheme 2).
Reaction conducted using 3 equiv of 5 and 1 equiv of CSA.
Scheme 2.

Origin of over-reduction of 6.
N-Hydroxyindoles have also received considerable attention as potential pharmacophores, and the methods for their synthesis have been similar to those used for the preparation of hydroxamic acids.13 Thus, we examined the photocatalytic reduction of 11 under conditions identical to those optimized for reduction of 1. Indeed, hydroxyindole 12 could be isolated in 88% yield without O-protection (eq 1).
Equation 1.
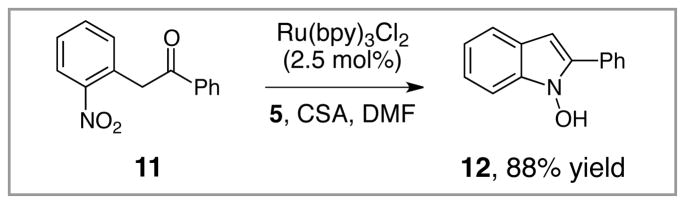
Preparation of N-hydroxyindoles.
Finally, the Boc protecting group can be cleaved in good yield using previously reported conditions (Scheme 3).14 Treatment of 13 with TFA in CH2Cl2 reveals the unprotected hydroxamic acid 3 in 83% yield. Alternatively, the N–O bond of 13 can be cleaved with Fe powder to afford quinolinone 14 in 86% yield. Thus, the easily handled O-Boc hydroxamic acid can be converted to these useful scaffolds with good efficiency.
Scheme 3.

Manipulation of N-Boc hydroxamic acids.
In conclusion, we have developed a mild photocatalytic method for the reduction and cyclization of nitroarenes to hydroxamic acids. This method provides access to a class of biologically relevant scaffolds that should possess utility in drug discovery efforts. In the context of our ongoing studies of visible light-induced organic reactions, this study is significant because it shows that synthetically useful transformations can be initiated by photoreduction of nitroarenes. These results raise intriguing questions concerning the precise mechanism of this process, including the effect of the terminal reductant both of the effectiveness of the reduction and the selectivity between four-electron and six-electron reduction. Studies to further interrogate this reaction and design new transformations initiated by reduction of nitro organics are subjects of continuing interest in our laboratory.
General Information
N,N-Dimethylformamide (DMF), triethylamine, and diisopropylethylamine were purified by distillation from CaH2 prior to use. Dihydropyridines 4 and 5 were prepared using known methods.15 The syntheses of the nitroarene substrates are described in the Supporting Information. All other reagents were purchased from commercial sources and used without further purification. Chromatography was performed with Purasil 60Å silica gel (230–400 mesh). 1H and 13C NMR data for all previously uncharacterized compounds were obtained using Varian Inova-500 spectrometers and are referenced to TMS (0.00 ppm) and CDCl3 (77 ppm), respectively. IR spectral data were obtained using a Bruker Vector 22 spectrometer (thin film, NaCl on NaCl). Melting points were obtained using a Mel-Temp II (Laboratory Devices, Inc., USA) melting point apparatus. Mass spectrometry was performed with a Micromass LCT (electrospray ionization, time-of-flight analyzer).
General Procedure for Photochemical Reactions
A solution of the appropriate nitroarene (1 equiv), Ru(bpy)3Cl2•6H2O (0.025 equiv), CSA (0.10 or 1.0 equiv), and dihydropyridine 5 (2.1, 3.0, or 4.0 equiv) in DMF (0.1 M) was placed in a sealed 25 mL Schlenk flask. The solution was degassed using three freeze-pump-thaw cycles and then irradiated using a household 20 W compact fluorescent light bulb. After 16 h, the reaction was diluted with ethyl acetate, then washed twice with 1 M HCl. The aqueous phases were extracted with ethyl acetate, and the organic phases were combined and washed once with brine, dried over MgSO4, and concentrated in vacuo. A solution of Boc2O (1.1 or 2.2 equiv), Et3N (5.0 equiv), and THF (0.05 M) was added. After 2–24 h, the reaction mixture was concentrated in vacuo and purified by column chromatography.
tert-Butyl (2-oxo-3,4-dihydroquinolin-1(2H)-yl) carbonate (13, Table 2, Entry 1)
Experiment 1: 105 mg (0.500 mmol) of methyl 3-(2-nitrophenyl)propanoate, 9.3 mg (0.012 mmol) Ru(bpy)3Cl2•6H2O, 204 mg (1.05 mmol) 5, 11.3 mg (0.0486 mmol) CSA, 5 mL (0.1 M) DMF, 123 mg (0.562 mmol) Boc2O, 0.35 mL (2.5 mmol) triethylamine, and 10 mL (0.05 M) THF. Purification by column chromatography (8:1 hexanes:ethyl acetate) yielded 111 mg ( 0.42 mmol, 84%) of a white solid. Experiment 2: 105 mg (0.500 mmol) of methyl 3-(2-nitrophenyl)propanoate, 9.5 mg (0.013 mmol) Ru(bpy)3Cl2•6H2O, 203 mg (1.05 mmol) 5, 11.8 mg (0.0508 mmol) CSA, 5 mL (0.1 M) DMF, 122 mg (0.559 mmol) Boc2O, 0.35 mL (2.5 mmol) triethylamine, and 10 mL (0.05 M) THF yielded 106 mg (0.40 mmol, 81%).
mp = 112.6–116.4 °C.
IR (thin film, NaCl): 2983, 1792, 1701, 1247 cm−1
1H NMR (500 MHz, CDCl3) δ 7.28 – 7.22 (m, 1H), 7.18 (dd, J = 7.5, 1.1 Hz, 1H), 7.05 (td, J = 7.5, 1.2 Hz, 1H), 6.99 (dd, J = 8.0, 1.1 Hz, 1H), 3.02 – 2.98 (m, 2H), 2.80 (t, J = 7.4 Hz, 2H), 1.57 (s, 9H).
13C NMR (126 MHz, CDCl3) δ 164.6, 150.7, 138.3, 127.7, 127.7, 123.9, 123.9, 111.7, 86.4, 31.4, 27.5, 24.8.
HRMS (ESI) calc’d for [C14H17NO4+Na]+ requires m/z 288.1050, found m/z 288.1050.
tert-Butyl (7-methoxy-2-oxo-3,4-dihydroquinolin-1(2H)-yl) carbonate (Table 2, Entry 2)
Experiment 1: 117 mg (0.490 mmol) of methyl 3-(4-methoxy-2-nitrophenyl)propanoate, 9.1 mg (0.012 mmol) Ru(bpy)3Cl2•6H2O, 203 mg (1.05 mmol) 5, 11.8 mg (0.0508 mmol) CSA, 5 mL (0.1 M) DMF, 124 mg (0.569 mmol) Boc2O, 0.35 mL (2.5 mmol) triethylamine, and 10 mL (0.05 M) THF. Purification by column chromatography (5:1 hexanes:ethyl acetate) yielded 109 mg (0.37 mmol, 74%) of a white solid. Experiment 2: 120 mg (0.500 mmol) of methyl 3-(4-methoxy-2-nitrophenyl)propanoate, 9.3 mg (0.012 mmol) Ru(bpy)3Cl2•6H2O, 202 mg (1.05 mmol) 5, 11.7 mg (0.0504 mmol) CSA, 5 mL (0.1 M) DMF, 123 mg (0.564 mmol) Boc2O, 0.35 mL (2.5 mmol) triethylamine, and 10 mL (0.05 M) THF yielded 117 mg (0.40 mmol, 80%).
mp = 73.4–74.5 °C.
IR (thin film, NaCl): 2983, 1793, 1713, 1248 cm−1.
1H NMR (500 MHz, CDCl3) δ 7.08 (d, J = 8.0 Hz, 1H), 6.61 – 6.50 (m, 2H), 3.78 (s, 3H), 2.97 – 2.85 (m, 2H), 2.76 (t, J = 7.3 Hz, 2H), 1.57 (s, 9H).
13C NMR (126 MHz, CDCl3) δ 164.7, 159.3, 150.5, 139.1, 128.5, 115.9, 108.2, 98.8, 86.4, 55.4, 31.6, 27.5, 23.9.
HRMS (ESI) calc’d for [C15H19NO5+Na]+ requires m/z 316.1156, found m/z 316.1151.
tert-Butyl (7-methyl-2-oxo-3,4-dihydroquinolin-1(2H)-yl) carbonate (Table 2, Entry 3)
Experiment 1: 108 mg (0.485 mmol) of methyl 3-(4-methyl-2-nitrophenyl)propanoate, 9.1 mg (0.012 mmol) Ru(bpy)3Cl2•6H2O, 203 mg (1.05 mmol) 5, 11.8 mg (0.0508 mmol) CSA, 5 mL (0.1 M) DMF, 124 mg (0.569 mmol) Boc2O, 0.35 mL (2.5 mmol) triethylamine, and 10 mL (0.05 M) THF. Purification by column chromatography (5:1 hexanes:ethyl acetate) yielded 118 mg (0.42 mmol, 87%) of a white solid. Experiment 2: 111 mg (0.499 mmol) of methyl 3-(4-methyl-2-nitrophenyl)propanoate, 9.7 mg (0.013 mmol) Ru(bpy)3Cl2•6H2O, 203 mg (1.05 mmol) 5, 11.6 mg (0.0499 mmol) CSA, 5 mL (0.1 M) DMF, 123 mg (0.564 mmol) Boc2O, 0.35 mL (2.5 mmol) triethylamine, and 10 mL (0.05 M) THF yielded 115 mg (0.42 mmol, 83%).
mp = 96.4–97.0 °C.
IR (thin film, NaCl): 3092, 2959, 1733, 1204 cm−1.
1H NMR (500 MHz, CDCl3) δ 7.05 (d, J = 7.5 Hz, 1H), 6.84 (d, J = 7.5 Hz, 1H), 6.79 (s, 1H), 3.01 – 2.86 (m, 2H), 2.75 (t, J = 7.3 Hz, 2H), 2.33 (s, 3H), 1.57 (s, 9H).
13C NMR (126 MHz, CDCl3) δ 164.6, 150.6, 138.0, 137.5, 127.5, 124.4, 120.8, 112.3, 86.3, 31.5, 27.4, 24.3, 21.3.
HRMS (ESI) calc’d for [C15H19NO4+NH4]+ requires m/z 295.1653, found m/z 295.1664.
tert-Butyl (2-oxo-7-(trifluoromethyl)-3,4-dihydroquinolin-1(2H)-yl) carbonate (Table 2, Entry 4)
Experiment 1: 141 mg (0.507 mmol) of methyl 3-(2-nitro-4-(trifluoromethyl)phenyl)propanoate, 9.7 mg (0.013 mmol) Ru(bpy)3Cl2•6H2O, 204 mg (1.06 mmol) 5, 11.8 mg (0.0508 mmol) CSA, 5 mL (0.1 M) DMF, 120 mg (0.550 mmol) Boc2O, 0.35 mL (2.5 mmol) triethylamine, and 10 mL (0.05 M) THF. Purification by column chromatography (5:1 hexanes:ethyl acetate) yielded 139 mg (0.42 mmol, 82%) of a white solid. Experiment 2: 139 mg (0.501 mmol) of methyl 3-(2-nitro-4-(trifluoromethyl)phenyl)propanoate, 9.4 mg (0.013 mmol) Ru(bpy)3Cl2•6H2O, 204.2 mg (1.06 mmol) 5, 11.6 mg (0.0499 mmol) CSA, 5 mL (0.1 M) DMF, 122 mg (0.559 mmol) Boc2O, 0.35 mL (2.5 mmol) triethylamine, and 10 mL (0.05 M) THF yielded 132 mg (0.40 mmol, 79%).
mp = 70.0–73.8 °C.
IR (thin film, NaCl): 2986, 1794, 1716, 1335, 1248 cm−1.
1H NMR (500 MHz, CDCl3) δ 7.35 – 7.29 (m, 2H), 7.23 – 7.18 (m, 1H), 3.12 – 3.03 (m, 2H), 2.83 (t, J = 7.4 Hz, 2H), 1.58 (s, 9H).
13C NMR (126 MHz, CDCl3) δ 164.3, 150.3, 138.8, 130.3 (q, J = 32.9 Hz), 128.2, 127.6, 123.8 (q, J = 272.2 Hz), 120.6 (q, J = 3.8 Hz), 108.7 (q, J = 3.9 Hz), 87.1, 30.8, 27.4, 24.7.
HRMS (ESI) calc’d for [C15H16F3NO4+Na]+ requires m/z 354.0924, found m/z 354.0932.
tert-Butyl (7-cyano-2-oxo-3,4-dihydroquinolin-1(2H)-yl) carbonate (Table 2, Entry 5)
Experiment 1: 118 mg (0.503 mmol) of methyl 3-(4-cyano-2-nitrophenyl)propanoate, 9.6 mg (0.013 mmol) Ru(bpy)3Cl2•6H2O, 204 mg (1.06 mmol) 5, 11.6 mg (0.0499 mmol) CSA, 5 mL (0.1 M) DMF, 121 mg (0.554 mmol) Boc2O, 0.35 mL (2.5 mmol) triethylamine, and 10 mL (0.05 M) THF. Purification by column chromatography (2:1 hexanes:ethyl acetate) yielded 102 mg (0.35 mmol, 70%) of a white solid. Experiment 2: 117 mg (0.501 mmol) of methyl 3-(4-cyano-2-nitrophenyl)propanoate, 9.7 mg (0.013 mmol) Ru(bpy)3Cl2•6H2O, 204 mg (1.06 mmol) 5, 11.9 mg (0.0512 mmol) CSA, 5 mL (0.1 M) DMF, 123 mg (0.565 mmol) Boc2O, 0.35 mL (2.5 mmol) triethylamine, and 10 mL (0.05 M) THF yielded 105 mg (0.36 mmol, 73%).
mp = 199.4–200.2 °C.
IR (thin film, NaCl): 2984, 2231, 1794, 1717, 1249 cm−1.
1H NMR (500 MHz, CDCl3) δ 7.36 (dd, J = 7.7, 1.4 Hz, 1H), 7.31 (dd, J = 7.7, 1.0 Hz, 1H), 7.24 (d, J = 1.3 Hz, 1H), 3.14 – 3.02 (m, 2H), 2.83 (t, J = 7.4 Hz, 2H), 1.59 (s, 9H).
13C NMR (126 MHz, CDCl3) δ 163.9, 150.3, 139.2, 129.1, 128.7, 127.6, 118.2, 114.7, 111.8, 87.4, 30.5, 27.5, 25.0.
HRMS (ESI) calc’d for [C15H16N2O4+NH4]+ requires m/z 306.1449, found m/z 306.1447.
tert-Butyl (7-bromo-2-oxo-3,4-dihydroquinolin-1(2H)-yl) carbonate (Table 2, Entry 6)
Experiment 1: 145 mg (0.503 mmol) of methyl 3-(4-bromo-2-nitrophenyl)propanoate, 9.7 mg (0.013 mmol) Ru(bpy)3Cl2•6H2O, 205 mg (1.06 mmol) 5, 12.2 mg (0.0525 mmol) CSA, 5 mL (0.1 M) DMF, 121 mg (0.554 mmol) Boc2O, 0.35 mL (2.5 mmol) triethylamine, and 10 mL (0.05 M) THF. Purification by column chromatography (6:1 hexanes:ethyl acetate) yielded 125 mg (0.36 mmol, 72%) of a white solid. Experiment 2: 145 mg (0.504 mmol) of methyl 3-(4-bromo-2-nitrophenyl)propanoate, 9.5 mg (0.013 mmol) Ru(bpy)3Cl2•6H2O, 204 mg (1.06 mmol) 5, 12.5 mg (0.0538 mmol) CSA, 5 mL (0.1 M) DMF, 122 mg (0.560 mmol) Boc2O, 0.35 mL (2.5 mmol) triethylamine, and 10 mL (0.05 M) THF yielded 137 mg (0.40 mmol, 79%).
mp = 108.2–109.7 °C.
IR (thin film, NaCl): 2982, 1793, 1716, 1247 cm−1.
1H NMR (500 MHz, CDCl3) δ 7.18 (dd, J = 7.9, 1.9 Hz, 1H), 7.14 (d, J = 1.9 Hz, 1H), 7.05 (d, J = 7.9 Hz, 1H), 3.03 – 2.89 (m, 2H), 2.78 (t, J = 6.8 Hz, 2H), 1.58 (s, 9H).
13C NMR (126 MHz, CDCl3) δ 164.4, 150.4, 139.4, 129.1, 126.7, 122.8, 121.1, 115.0, 86.9, 31.1, 27.5, 24.4.
HRMS (ESI) calc’d for [C14H16BrNO4+NH4]+ requires m/z 359.0601, found m/z 359.0597.
tert-Butyl (6-fluoro-2-oxo-3,4-dihydroquinolin-1(2H)-yl) carbonate (Table 2, Entry 7)
Experiment 1: 114 mg (0.500 mmol) of methyl 3-(5-fluoro-2-nitrophenyl)propanoate, 9.5 mg (0.013 mmol) Ru(bpy)3Cl2•6H2O, 204 mg (1.06 mmol) 5, 11.7 mg (0.0504 mmol) CSA, 5 mL (0.1 M) DMF, 124 mg (0.566 mmol) Boc2O, 0.35 mL (2.5 mmol) triethylamine, and 10 mL (0.05 M) THF. Purification by column chromatography (6:1 hexanes:ethyl acetate) yielded 109 mg (0.39 mmol, 78%) of a white solid. Experiment 2: 114 mg (0.502 mmol) of methyl 3-(5-fluoro-2-nitrophenyl)propanoate, 9.4 mg (0.013 mmol) Ru(bpy)3Cl2•6H2O, 205 mg (1.06 mmol) 5, 12.2 mg (0.0525 mmol) CSA, 5 mL (0.1 M) DMF, 121 mg (0.554 mmol) Boc2O, 0.35 mL (2.5 mmol) triethylamine, and 10 mL (0.05 M) THF yielded 112 mg (0.40 mmol, 79%).
mp = 94.6–95.4 °C.
IR (thin film, NaCl): 2984, 1793, 1707, 1248 cm−1.
1H NMR (500 MHz, CDCl3) δ 6.98 – 6.89 (m, 3H), 3.08 – 2.90 (m, 2H), 2.79 (t, J = 7.4 Hz, 2H), 1.57 (s, 9H).
13C NMR (126 MHz, CDCl3) δ 164.2, 159.1 (d, J = 243.6 Hz), 150.6, 134.6 (d, J = 2.6 Hz), 126.1 (d, J = 7.8 Hz), 114.9 (d, J = 23.6 Hz), 114.1 (d, J = 23.0 Hz), 113.2 (d, J = 8.3 Hz), 86.6, 31.2, 27.5, 24.9 (d, J = 1.2 Hz).
HRMS (ESI) calc’d for [C14H16FNO4+NH4]+ requires m/z 299.1402, found m/z 299.1415.
6-methoxy-2-oxo-3,4-dihydroquinolin-1(2H)-yl (10, Table 2, Entry 8)
Following general procedure but without protection after aqueous workup. 121 mg (0.506 mmol) of methyl 3-(5-methoxy-2-nitrophenyl)propanoate, 9.7 mg (0.013 mmol) Ru(bpy)3Cl2•6H2O, 388 mg (1.96 mmol) 5, 113 mg (0.486 mmol) CSA, 5 mL (0.1 M) DMF. Purification by column chromatography (1:1 hexanes:ethyl acetate, 0.5% triethylamine) yielded 60.2 mg (0.27 mmol, 54%) of a white solid. All spectra data were consistent with reported values.16
tert-Butyl (3-oxo-2H-benzo[b][1,4]oxazin-4(3H)-yl) carbonate (Table 2, Entry 9)
Experiment 1: 106 mg (0.502 mmol) of methyl 2-(2-nitrophenoxy)acetate, 9.5 mg (0.013 mmol) Ru(bpy)3Cl2•6H2O, 203 mg (1.05 mmol) 5, 11.3 mg (0.0486 mmol) CSA, 5 mL (0.1 M) DMF, 122 mg (0.559 mmol) Boc2O, 0.35 mL (2.5 mmol) triethylamine, and 10 mL (0.05 M) THF. Purification by column chromatography (5:1 hexanes:ethyl acetate) yielded 98.0 mg (0.37 mmol, 74%) of a white solid. Experiment 2: 106 mg (0.501 mmol) of methyl 2-(2-nitrophenoxy)acetate, 9.3 mg (0.012 mmol) Ru(bpy)3Cl2•6H2O, 203 mg (1.05 mmol) 5, 11.9 mg (0.0512 mmol) CSA, 5 mL (0.1 M) DMF, 122 mg (0.559 mmol) Boc2O, 0.35 mL (2.5 mmol) triethylamine, and 10 mL (0.05 M) THF yielded 107 mg (0.41 mmol, 81%).
mp = 89.1–90.5 °C.
IR (thin film, NaCl): 2979, 1700, 1685, 1244 cm−1.
1H NMR (500 MHz, CDCl3) δ 7.14 – 6.85 (m, 4H), 4.76 (s, 2H), 1.58 (s, 9H).
13C NMR (126 MHz, CDCl3) δ 159.4, 149.9, 143.5, 127.8, 124.8, 122.9, 116.9, 112.1, 87.2, 68.2, 27.4.
HRMS (ESI) calc’d for [C13H15NO5+Na]+ requires m/z 288.0843, found m/z 288.0842.
tert-Butyl (2-oxo-4-tosyl-3,4-dihydroquinoxalin-1(2H)-yl) carbonate (Table 2, Entry 10)
Experiment 1: Following general procedure without aqueous workup before protection. After protection is complete, the reaction mixture is diluted with ethyl acetate and washed twice with water and once with brine. 182 mg (0.499 mmol) of methyl 2-(4-methyl-N-(2-nitrophenyl)phenylsulfonamido)acetate, 9.4 mg (0.013 mmol) Ru(bpy)3Cl2•6H2O, 290 mg (1.50 mmol) 5, 116 mg (0.498 mmol) CSA, 5 mL (0.1 M) DMF, 242 mg (1.11 mmol) Boc2O, 0.35 mL (2.5 mmol) triethylamine, and 10 mL (0.05 M) THF. Purification by column chromatography (9:1 hexanes:ethyl acetate) yielded 109 mg (0.26 mmol, 52%) of a white solid. Experiment 2: 182 mg (0.500 mmol) of methyl 2-(4-methyl-N-(2-nitrophenyl)phenylsulfonamido)acetate, 9.4 mg (0.013 mmol) Ru(bpy)3Cl2•6H2O, 289 mg (1.50 mmol) 5, 116 mg (0.500 mmol) CSA, 5 mL (0.1 M) DMF, 237 mg (1.09 mmol) Boc2O, 0.35 mL (2.5 mmol) triethylamine, and 10 mL (0.05 M) THF yielded 112 mg (0.27 mmol, 53%).
mp = 117.2–117.8 °C.
IR (thin film, NaCl): 2983, 1798, 1721, 1361, 1248 cm−1.
1H NMR (500 MHz, CDCl3) δ 7.78 (dd, J = 8.0, 1.4 Hz, 1H), 7.35 (d, J = 8.3 Hz, 2H), 7.29 (td, J = 7.8, 1.4 Hz, 1H), 7.20 (td, J = 7.8, 1.4 Hz, 1H), 7.16 (d, J = 8.4 Hz, 2H), 6.76 (dd, J = 8.1, 1.4 Hz, 1H), 4.56 (apparent s, 1H), 2.36 (s, 3H), 1.51 (s, 9H).
13C NMR (126 MHz, CDCl3) δ 160.1, 149.6, 144.7, 133.8, 132.8, 129.8, 128.1, 127.6, 126.9, 124.4, 124.0, 112.0, 87.1, 49.6, 27.4, 21.6.
HRMS (ESI) calc’d for [C20H22N2O6S+NH4]+ requires m/z 436.1537, found m/z 437.1555.
tert-Butyl (2-oxoindolin-1-yl) carbonate (Table 2, Entry 11)
Experiment 1: Following general procedure without aqueous workup before protection. After protection is complete, the reaction mixture is diluted with ethyl acetate and washed twice with water and once with brine. 97.3 mg (0.499 mmol) of methyl 2-(2-nitrophenyl)acetate, 9.3 mg (0.012 mmol) Ru(bpy)3Cl2•6H2O, 291 mg (1.50 mmol) 5, 117 mg (0.503 mmol) CSA, 5 mL (0.1 M) DMF, 241 mg (1.11 mmol) Boc2O, 0.35 mL (2.5 mmol) triethylamine, and 10 mL (0.05 M) THF. Purification by column chromatography (4:1 hexanes:ethyl acetate) yielded 76.9 mg (0.31 mmol, 62%) of a white solid. Experiment 2: 97.0 mg (0.497 mmol) of methyl 2-(2-nitrophenyl)acetate, 9.7 mg (0.013 mmol) Ru(bpy)3Cl2•6H2O, 290 mg (1.50 mmol) 5, 116 mg (0.500 mmol) CSA, 5 mL (0.1 M) DMF, 240 mg (1.10 mmol) Boc2O, 0.35 mL (2.5 mmol) triethylamine, and 10 mL (0.05 M) THF yielded 81.9 mg (0.33 mmol, 66%).
mp = 88.1–89.4 °C.
IR (thin film, NaCl): 2984, 1796, 1743, 1247 cm−1.
1H NMR (500 MHz, CDCl3) δ 7.29 (td, J = 7.7, 1.0 Hz, 1H), 7.25 (d, J = 7.6 Hz, 1H), 7.08 (td, J = 7.6, 1.0 Hz, 1H), 6.85 (d, J = 7.8 Hz, 1H), 3.60 (s, 2H), 1.57 (s, 9H).
13C NMR (126 MHz, CDCl3) δ 169.1, 150.2, 141.2, 128.1, 124.9, 123.3, 120.1, 107.2, 87.1, 33.6, 27.4.
HRMS (ESI) calc’d for [C13H19NO4+Na]+ requires m/z 229.0819, found m/z 229.0809.
tert-Butyl 3-acetamido-2-oxo-3,4-dihydroquinolin-1(2H)-yl carbonate (Table 2, Entry 12)
Experiment 1: Following general procedure without aqueous workup before protection. After protection is complete, the reaction mixture is diluted with ethyl acetate and washed twice with water and once with brine. 140 mg (0.500 mmol) of ethyl 2-acetamido-3-(2-nitrophenyl)propanoate, 9.7 mg (0.013 mmol) Ru(bpy)3Cl2•6H2O, 290 mg (1.50 mmol) 5, 116 mg (0.501 mmol) CSA, 5 mL (0.1 M) DMF, 240 mg (1.10 mmol) Boc2O, 0.35 mL (2.5 mmol) triethylamine, and 10 mL (0.05 M) THF. Purification by column chromatography (3:1 to 0:1 hexanes:ethyl acetate) yielded 92.2 mg (0.29 mmol, 58%) of a white solid. Experiment 2: 141 mg (0.503 mmol) of ethyl 2-acetamido-3-(2-nitrophenyl)propanoate, 9.9 mg (0.013 mmol) Ru(bpy)3Cl2•6H2O, 290 mg (1.50 mmol) 5, 117 mg (0.502 mmol) CSA, 5 mL (0.1 M) DMF, 246 mg (1.12 mmol) Boc2O, 0.35 mL (2.5 mmol) triethylamine, and 10 mL (0.05 M) THF yielded 94.8 mg (0.29 mmol, 58%).
mp = 112.6–113.9 °C.
IR (thin film, NaCl): 3308, 2984, 1795, 1715, 1246 cm−1.
1H NMR (500 MHz, CDCl3) δ 7.29 (t, J = 7.8 Hz, 1H), 7.24 (d, J = 7.5 Hz, 1H), 7.11 (td, J = 7.5, 1.1 Hz, 1H), 7.10–6.82 (m, 1H), 6.61 (d, J = 5.5 Hz, 1H), 4.78 (dt, J = 14.1, 5.8 Hz, 1H), 3.51 (dd, J = 15.1, 6.0 Hz, 1H), 3.01 – 2.85 (m, 1H), 2.08 (s, 3H), 1.56 (s, 9H).
13C NMR (126 MHz, CDCl3) δ 170.3, 163.9, 150.3, 128.5, 128.1, 124.8, 112.7, 86.9, 65.8, 49.5, 31.7, 27.4, 23.1, 15.2.
HRMS (ESI) calc’d for [C16H20N2O5+NH4]+ requires m/z 338.1711, found m/z 338.1718.
2-Phenyl-1H-indol-1-ol (13)
Experiment 1: Following general procedure but without protection after aqueous workup. 121 mg (0.500 mmol) of 2-(2-nitrophenyl)-1-phenylethanone 11, 10.0 mg (0.013 mmol) Ru(bpy)3Cl2•6H2O, 205 mg (1.06 mmol) 5, 11.6 mg (0.0499 mmol) CSA, 5 mL (0.1 M) DMF. Purification by column chromatography (20:1 hexanes:ethyl acetate) yielded 94.3 mg (0.45 mmol, 90%) of a white solid. Experiment 2: 122 mg (0.505 mmol) of 2-(2-nitrophenyl)-1-phenylethanone 11, 9.2 mg (0.012 mmol) Ru(bpy)3Cl2•6H2O, 205 mg (1.06 mmol) 5, 11.7 mg (0.0504 mmol) CSA, 5 mL (0.1 M) DMF yielded 90.2 mg (0.43 mmol, 85%).
mp = 149.1–150.3 °C.
IR (thin film, NaCl): 3277, 3053, 2923, 2520, 1532 cm−1.
1H NMR (500 MHz, DMSO-d6) δ 11.17 (s, 1H), 7.88 (d, J = 7.3 Hz, 2H), 7.54 (d, J = 7.9 Hz, 1H), 7.49 (t, J = 7.7 Hz, 2H), 7.44 (d, J = 8.1 Hz, 1H), 7.38 (t, J = 7.4 Hz, 1H), 7.18 (t, J = 7.5 Hz, 1H), 7.05 (t, J = 7.4 Hz, 1H), 6.63 (s, 1H).
13C NMR (126 MHz, DMSO-d6) δ 136.9, 135.5, 130.9, 128.6, 127.7, 123.0, 121.8, 120.2, 119.7, 108.8, 96.2.
HRMS (ESI) calc’d for [C14H11NO+H]+ requires m/z 210.0914, found m/z 210.0919.
1-Hydroxy-3,4-dihydroquinolin-2(1H)-one (3)
A round-bottom flask was charged with 100 mg (0.380 mmol) 13, 7.5 mL (0.05 M) CH2Cl2, and 7.5 mL (0.05M) trifluoroacetic acid. The reaction was stirred for 1 hour before diluting with 50 mL CH2Cl2 and pouring onto 50 mL water. The reaction was washed twice with 50 mL CH2Cl2. The organic layers were combined, dried over MgSO4, and concentrated in vacuo. Purification by recrystallization in ether yielded 51.7 mg (0.317 mmol, 83%) of a tan solid. All spectra data were consistent with reported values.17
3,4-Dihydroquinolin-2(1H)-one (14)
A 2 dram vial was charged with 100 mg (0.380 mmol) 13, 41.9 mg (0.776 mmol) iron metal, 1.0 mL (0.4 M) ethanol, and 1.0 mL (0.4 M) acetic acid. The reaction was heated to 80 °C for 1.5 h before cooling to room temperature. A saturated solution of Na2CO3 was added, and the reaction was extracted with three 50 mL portions of ether. The organic layers were combined, dried over MgSO4, and concentrated in vacuo. Purification by recrystallization in ether yielded 48.0 mg (0.326 mmol, 86%) of a white solid. All spectra data were consistent with reported values.18
Supplementary Material
Acknowledgments
M.A.C. is grateful for a graduate fellowship from the NSF. Funding for this project was provided by the NIH (GM095666) and the Sloan Foundation.
Footnotes
Supporting Information for this article is available online at http://www.thieme-connect.com/ejournals/toc/synthesis.
References
- 1.Kurzak B, Kozlowski H, Farkas E. Coord Chem Rev. 1992;114:169. [Google Scholar]
- 2.(a) Weisburger JH, Weisburger EK. Pharmacol Rev. 1973;25:1. [PubMed] [Google Scholar]; (b) Hider RC, Kong X. Nat Prod Rep. 2010;27:637. doi: 10.1039/b906679a. [DOI] [PubMed] [Google Scholar]
- 3.(a) Miller MJ. Chem Rev. 1989;89:1563. [Google Scholar]; (b) Muri EMF, Nieto MJ, Sindelar RD, Williamson JS. Curr Med Chem. 2002;9:1631. doi: 10.2174/0929867023369402. [DOI] [PubMed] [Google Scholar]
- 4.(a) Wright WB, Collins KH. J Am Chem Soc. 1956;78:221. [Google Scholar]; (b) Sicker D, Prätorius B, Mann G, Meyer L. Synthesis. 1989:211. [Google Scholar]; (c) McAllister LA, Bechle BM, Dounay AB, Evrard E, Gan X, Ghosh S, Kim JY, Parikh VD, Tuttle JB, Verhoest PR. J Org Chem. 2011;76:3484. doi: 10.1021/jo200530j. [DOI] [PubMed] [Google Scholar]
- 5.Coutts RT, Wibberley DG. J Chem Soc. 1963:4610. [Google Scholar]
- 6.Davis AL, Choun OHP, Cook DE, McCord TJ. J Med Chem. 1964;7:632. doi: 10.1021/jm00335a014. [DOI] [PubMed] [Google Scholar]
- 7.For reviews, see: Zeitler K. Angew Chem Int Ed. 2009;48:9785. doi: 10.1002/anie.200904056.Yoon TP, Ischay MA, Du J. Nat Chem. 2010;2:527. doi: 10.1038/nchem.687.Narayanam JMR, Stephenson CRJ. Chem Soc Rev. 2011;40:102. doi: 10.1039/b913880n.Telpý F. Collect Czech Chem Commun. 2011;76:859.Tucker JW, Stephenson CRJ. J Org Chem. 2012;77:1617. doi: 10.1021/jo202538x.Xuan J, Xiao WJ. Angew Chem Int Ed. 2012;51:6828. doi: 10.1002/anie.201200223.
- 8.(a) Ischay MA, Anzovino ME, Du J, Yoon TP. J Am Chem Soc. 2008;130:12886. doi: 10.1021/ja805387f. [DOI] [PubMed] [Google Scholar]; (b) Du J, Yoon TP. J Am Chem Soc. 2009;131:14604. doi: 10.1021/ja903732v. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Ischay MA, Lu Z, Yoon TP. J Am Chem Soc. 2010;132:8572. doi: 10.1021/ja103934y. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Du J, Ruiz Espelt L, Guzei IA, Yoon TP. Chem Sci. 2011;2:2115. doi: 10.1039/C1SC00357G. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Ischay MA, Ament MS, Yoon TP. Chem Sci. 2012;3:2807. doi: 10.1039/C2SC20658G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.For leading examples from other groups, see: Nicewicz DA, MacMillan DWC. Science. 2008;322:77. doi: 10.1126/science.1161976.Narayanam JMR, Tucker JW, Stephenson CRJ. J Am Chem Soc. 2009;131:8756. doi: 10.1021/ja9033582.McNally A, Prier CK, MacMillan DWC. Science. 2011;334:1114. doi: 10.1126/science.1213920.Kalyani D, McMurtrey KB, Neufeldt SR, Sanford MS. J Am Chem Soc. 2011;133:18566. doi: 10.1021/ja208068w.Freeman DB, Furst L, Condie AG, Stephenson CRJ. Org Lett. 2012;14:94. doi: 10.1021/ol202883v.Maity S, Zhu M, Shinabery RS, Zheng N. Angew Chem Int Ed. 2012;51:222. doi: 10.1002/anie.201106162.Leung JCT, Chatalova-Sazepin C, West JG, Rueda-Becerril M, Paquin JF, Sammis GM. Angew Chem Int Ed. 2012;51:10804. doi: 10.1002/anie.201206352.
- 10.Hirao T, Shiori J, Okahata N. Bull Chem Soc Jpn. 2004;77:1763. [Google Scholar]
- 11.Tomioka H, Ueda K, Ohi H, Izawa Y. Chem Lett. 1986;15:1359. [Google Scholar]
- 12.(a) Davis AL, Choun OHP, Cook DE, McCord TJ. J Med Chem. 1964;7:632. doi: 10.1021/jm00335a014. [DOI] [PubMed] [Google Scholar]; (b) Davis AL, Chambers WH, Kelley DH, Fell DA, Haynes JR, Hulme KL, Gage LD, McCord TJ. J Med Chem. 1975;18:752. doi: 10.1021/jm00241a022. [DOI] [PubMed] [Google Scholar]; (c) Erhardt S, Olsson SK, Engberg G. CNS Drugs. 2009;23:91. doi: 10.2165/00023210-200923020-00001. [DOI] [PubMed] [Google Scholar]
- 13.(a) Dong W, Jimenez LS. J Org Chem. 1999;64:2520. [Google Scholar]; (b) Wong A, Kuethe JT, Davies IW. J Org Chem. 2003;68:9865. doi: 10.1021/jo035351l. [DOI] [PubMed] [Google Scholar]; (c) Wong A, Kuethe JT, Davies IW, Hughes DL. J Org Chem. 2004;69:7761. doi: 10.1021/jo048843m. [DOI] [PubMed] [Google Scholar]; (d) Nicolaou KC, Lee SH, Estrada AA, Zak M. Angew Chem Int Ed. 2005;44:3736. doi: 10.1002/anie.200500724. [DOI] [PubMed] [Google Scholar]
- 14.Claffey MM, Dounay AB, Gan X, Hayward MM, Rong S, Tuttle JB, Verhoest PR. WO2010/146488. PCT In Appl. 2010 Dec 23;
- 15.Wang GW, Xia JJ, Miao CB, Wu XL. Bull Chem Soc Jpn. 2006;79:454. [Google Scholar]
- 16.Zhou W, Zhang L, Jiao N. Tetrahedron. 2009;65:1982. [Google Scholar]
- 17.Murahashi SI, Oda T, Sugahara T, Masui Y. J Org Chem. 1990;55:1744. [Google Scholar]
- 18.Liu B, Hu L. Bioorg Med Chem. 2003;11:3889. doi: 10.1016/s0968-0896(03)00426-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.