

Abstract
A Mo/P catalytic system for an efficient gram-scale oxidation of a variety of nitrogen heterocycles to N-oxides with hydrogen peroxide as terminal oxidant has been investigated. Combined spectroscopic and crystallographic studies point to the tetranuclear Mo4P peroxo complex as one of the active catalytic species present in the solution. Based on this finding an optimized catalytic system has been developed. The utility and chemoselectivity of the catalytic system has been demonstrated by the synthesis of over 20 heterocyclic N-oxides.
Introduction
Heterocyclic N-oxides have significant synthetic value as intermediates en route to diversely functionalized N-heterocycles.1 In particular, recent examples of regioselective C–H-arylation,2 as well syntheses of 2-halo3 2-amino,4 and 2-cyano azines5 constitute an important domain of modern heterocyclic synthesis (Fig. 1). In addition, N-oxides have been employed as ligands and catalysts, as synthetic intermediates in the industrial syntheses of pharmaceuticals (e.g. pranoprofen and omeprazole), as well as new pharmacophores in drug discovery. In the course of studies aimed at development of new reactions of heterocyclic N-oxides6 we had to prepare multigram quantities of various compounds of this class. A survey of the literature showed that current approaches to heterocyclic N-oxides are largely limited to stoichiometric oxidations by peroxyacids, either generated in situ from large excess of hydrogen peroxide in acetic or trifluoroacetic acids at elevated temperatures,7 or as a commercially available reagent (e.g. meta-chloroperbenzoic acid).8 Both methods have significant drawbacks. The in situ method furnishes impure products that have been reported to detonate shortly after isolation.9 On the other hand, stable peroxyacids are more expensive and produce significant amounts of waste. Other less common methods include oxidation with dimethyldioxirane (DMDO)10 and HOF·MeCN.11 There is, therefore, an unmet need for inexpensive and environmentally benign methods of azine and azole N-oxide synthesis based on readily available catalysts.12 Herein, we report a combined mechanistic and synthetic study of the oxidation of a variety of azines and azoles catalyzed by molybdenum-phosphate catalysts with hydrogen peroxide as a terminal oxidant.
Fig. 1.

Examples of applications of heterocyclic N-oxides in organic chemistry and drug discovery.
Catalytic oxidation reactions with hydrogen peroxide as a terminal oxidant have recently gained significant attention due to their low environmental impact, as well as low cost (<0.7 USD kg−1) and high atom efficiency (47%) of hydrogen peroxide.13 In addition, such oxidations are generally carried out under mild conditions, and require less intensive purification, as the only by-product that stems from the oxidant is water. Most of the produced H2O2 (3.8 million metric tons per year) is currently used for bleaching and detoxification,14 but the number of industrial synthetic processes utilizing this environmentally benign oxidant has been growing at a fast pace, e.g. hydrogen peroxide is currently used as an oxidant in the multi-ton scale propylene oxidation3 and caprolactam production.15 Although redox potential of H2O2 is relatively high (E0 = 1.76 V for the half-reaction H2O2 + 2H+ + 2e− → 2H2O),5 most oxidations of typical organic substrates (alcohols, aldehydes, alkenes, sulfides, phenols, etc.) by H2O2 are slow and require some activation of the O–O-bond.13a The O–O-activation can be effected by Brønsted acids via protonation or formation of peracids,16 as well as transition metal catalysts17 (e.g. Fe, Mn, V, Ti, Mo, Re, W) via formation of highly active oxo and peroxymetal species. While significant progress has been achieved in the catalytic hydrogen peroxide-mediated oxidations of alkenes,18 sulfides19 and alcohols,20 including asymmetric modifications,21 other less readily oxidizable substrates,22 such as N-heterocycles, have generally been prepared using stronger oxidants, in part due to substantially higher oxidation potentials (e.g. E0 = 1.41 V for pyridine N-oxide/pyridine,23 versus 0.16 V for (CH3)2SO/(CH3)2S24). Examples of catalytic H2O2-based methods include methyltrioxorhenium (MTO)-catalyzed oxidation of azines developed by Sharpless,25 and Mn(TCDPP)Cl26-catalyzed reaction reported by Mansuy.27 However, methyltrioxorhenium is expensive and undergoes Re–C bond cleavage that leads to a decrease in the catalytic activity and prevents catalyst recycling. On the other hand, Mn(TCDPP)Cl is not commercially available and can only be prepared in low yields.28 Hence, more recent approaches have focused on polyoxometallates as catalysts29 for the oxidation of pyridines by H2O2. Some of the catalysts studied include Na12[(WZn3(H2O)2][(ZnW9O34)2],30 various mixed W/V/Mo-based heteropolyacids,31 M8[BW11O39H] (M = K or R4N),32 Δ-Na8HPW9O34,33 [(C18H37)2(CH3)2N]7[PW11O39],34 and K6[PW9V3O40].35 Notwithstanding this progress, significant problems remain unaddressed. Thus, the synthetic utility and the scope of these catalytic systems have not been assessed, and practical procedures amenable to multi-gram preparation of heterocyclic N-oxides have not been described. These deficiencies may have contributed to the persistence of the less efficient and less environmentally benign N-oxidation methods. Furthermore, while oxidations of pyridines have been studied to some extent, applicability of polyoxometallate catalysis to the synthesis of N-oxides of five-membered N-heterocycles (azoles) has not been investigated. Although, N-methyl-imidazole is known to undergo uncatalyzed oxidation by H2O2, other less electron-rich imidazoles and thiazoles require use of such harsh and non-chemoselective reagents as HOF·MeCN36 and peroxyacids at high temperatures.37 In addition, most of the catalytic systems are based on tungsten that has recently been implicated as carcinogen.38 On the other hand, more environmentally benign molybdenum catalysis of N-oxidation and the mechanistic aspects thereof remain poorly studied. Early pioneering studies by Griffith, Galindo and Longo indicate that H2O2-mediated N-oxidation of pyridine can be catalyzed by molybdate either in stoichiometric or in catalytic fashion,39 but little is known about the scope and applicability of this catalytic system.
Results and discussion
Initial comparison of catalyst performance in acetonitrile at 50 °C with quinoline as a test substrate has revealed that molybdenum-based catalysts have consistently outperformed other catalytic systems. Thus, neither triflic nor trifluoroacetic acids were catalytically active (Table 1). Interestingly, Sc(OTf)3 mediated the oxidation, however the conversion was low. Fe(III), Cu(II), [Ru(p-cumene)Cl]2 and Na2WO4 were also catalytically inactive. On the other hand, Mimoun’s catalyst, oxodiperoxymolybdenum(VI) HMPA complex,40 provided an encouraging result with 22% yield of quinoline N-oxide 2. Subsequent investigation of catalytic activities of other molybdenum compounds has shown that sodium molybdate, molybdenum(VI) oxide, or several other molybdenum compounds did not offer substantial improvements in catalytic efficiency. Phosphomolybdic acid, on the contrary, has lead to clean conversion of 1 to quinoline N-oxide.
Table 1.
Catalytic N-oxidation of quinolinea
| |||
|---|---|---|---|
| Entry | Catalyst (x mol%) | Solvent (temperature/°C) | Yield(%) |
| 1 | — | CH3CN (50) | 0 |
| 2 | — | C2H5OH (50) | 0 |
| 3 | CF3SO3H (10) | CH3CN (50) | 0 |
| 4 | CF3CO2H (10) | CH3CN (50) | 0 |
| 5 | Sc(O3SCF3)3 (10) | CH3CN (50) | 13 |
| 6 | FeCl3 (10) | CH3CN (50) | 0 |
| 7 | CuCl2 (10) | CH3CN (50) | 0 |
| 8 | [Ru(p-cumene)Cl]2 (10) | CH3CN (50) | 0 |
| 9 | Na2WO4 (10) | CH3CN (50) | 0 |
| 10 | Mo(O2)2O·HMPA (10) | CH3CN (50) | 22 |
| 11 | MoO3 (10) | CH3CN (50) | 19 |
| 12 | Na2MoO4 (10) | CH3CN (50) | 33 |
| 13 | MoO2Cl2 (10) | CH3CN (50) | 13 |
| 14 | Mo(CO)6 (10) | CH3CN (50) | 14 |
| 15 | (NH4)2Mo7O24 (10) | CH3CN (50) | 16 |
| 16 | H3PMo12O40 (10) | C2H5OH (50) | 39 |
| 17 | H3PMo12O40 (10) | CH3CN (70) | 95 |
| 18b | H3PMo12O40 (1) | CH3CN (50) | 97 |
| 19 | H3PMo12O40 (0.5) | CH3CN (50) | 99 |
Reaction conditions: csubstrate = 1 mol L−1, except if stated otherwise;
30% aqueous H2O2. csubstrate = 2 mol L−1.
Encouragingly, the amount of the phosphomolybdic acid can be reduced to 0.5 mol% without jeopardizing the yield. Acetonitrile proved to be the solvent of choice, as neither alcohols (ethanol, methanol) nor water led to any improvement. Freshly prepared41 phosphomolybdic acid has exhibited higher catalytic activity than a commercially available solution in ethanol.
With these results we have set out to investigate the scope of the catalytic process (Table 2).
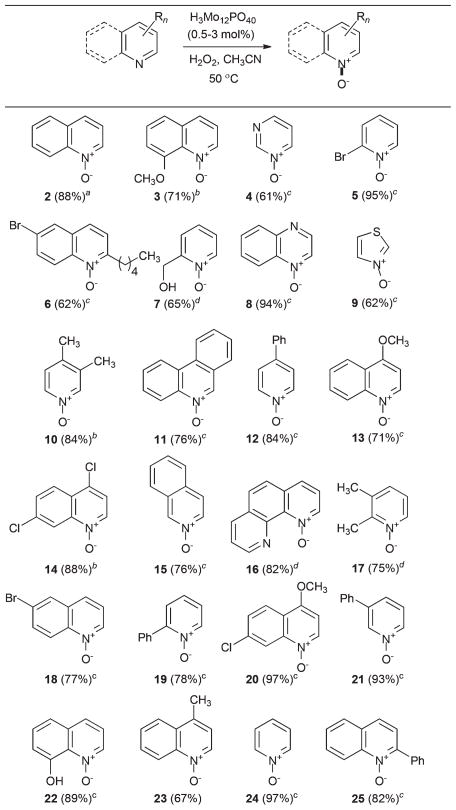
Table 2.
Substrate scope of the catalytic oxidation of N-heterocyclesa
|
0.5 mol% catalyst, 1.5 equiv. H2O2.
1 mol% catalyst, 1.5 equiv. H2O2.
2 mol% catalyst, 3 equiv. H2O2.
3 mol% catalyst, 4.5 equiv. H2O2.
A variety of substituted quinolines, including 8-hydroxyquinoline, that is prone to phenolic oxidation,42 have undergone a smooth and chemoselective transformation to the corresponding N-oxides. Similarly, substituted pyridines are also viable substrates for the reaction. Less easily oxidizable tricyclic systems, such as phenanthridine and 1,10-phenanthroline, readily undergo N-oxidation under these conditions as well.
In addition, pyrimidine and quinoxaline have furnished the corresponding N-oxides in high yields. Thiazole undergoes efficient oxidation, thus eliminating the necessity of using harsh oxidation conditions, such as HOF·MeCN. In none of the cases have we observed benzylic oxidation of α-alkyl substituents. Advantageously, phenols and benzylic alcohols have also proved resistant to this N-oxidation method. The isolation of the products is generally accomplished by treatment of the reaction mixture with aqueous ammonium chloride or manganese dioxide, if aqueous work-up has to be avoided. This oxidation has been routinely performed on gram-scale. Thus, quinoline N-oxide was prepared on a 10 g scale, and 2-phenyl-quinoline was oxidized on a 5 g scale. The oxidation reactions of less reactive substrates proceeded at slower rates than with quinoline, but could be brought to completion by using additional amounts of hydrogen peroxide and the catalyst (Table 2).
In an effort to characterize molybdenum-containing peroxo complexes with heterocyclic N-oxides that can be produced under the reaction conditions, we have studied products formed in a reaction of heterocyclic N-oxides with MoO3, hydrogen peroxide and phosphoric acid. Several new peroxo complexes have been isolated and characterized by means of single crystal X-ray crystallography (Fig. 2). Thus, two complexes 26 and 27 of the type [Mo(η2-O2)2O]L2 with quinoline N-oxide and 4-methylquinoline-N-oxide as monodentate ligands L, as well as chelate 28 with 2,2′-dipyridyl-N-oxide as a bidentate ligand (Fig. 2 and 3) exhibit several noteworthy structural features. The coordination polyhedron of the molybdenum atom in 26–28 (Fig. 3a and c) can be described as a pseudotrigonal bipyramid with two η2-peroxo ligands in the equatorial plane and the oxo ligand occupying one of the axial positions. The Mo–O bonds of the Mo–L units trans to the oxo ligands in 26 and 27 are consistently longer then the Mo–O bonds of the cis ligands due to the trans effect of the oxo ligand and in line with the previous observations for the structurally similar pyridine-N-oxide complex.39 It is interesting that the difference in the lengths of the Mo–Otrans and Mo–Ocis bonds is larger (0.143 Å) for complex 26 (with quinoline-N-oxide) then for the pyridine-N-oxide complex reported by Griffith (0.131 Å), but smaller (0.073 Å) for 27 (with 4-methylquinoline-N-oxide). The crystals of complexes 26 and 27 are not isostructural. For 26 the asymmetric unit consists of the complex and a water molecule (Fig. 3b). The solvent molecule plays a vital role in the packing of this structure by forming strong hydrogen bonds between the peroxo and the N-oxide ligands of interlinked complexes. These strong hydrogen bonds result in an extended hydrogen-bonded chain which propagates along the b-axis. These chains are linked to one another through a weak C–H···O interaction between the quinoline moiety in the Mo complex, and the water molecule extending along the a-axis. This weak interaction results in the formation of a 2D hydrogen-bonded layer (Fig. 3b). In the case of 27 the asymmetric unit comprises the Mo complex (Fig. 4d). There are no hydrogen-bonding interactions present due to the absence of a water molecule in the crystal lattice. The packing is dominated by π–π interactions (3.53 Å) between 4-methylquinoline moieties which result in the formation of centrosymmetric dimers.
Fig. 2.
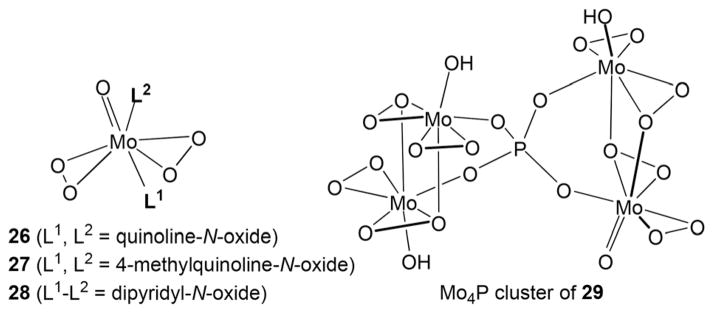
Oxo diperoxo Mo(VI) complexes 26–29.
Fig. 3.

(a) Molecular structure of (quinoline-N-oxide)2[Mo(η2-O2)2O] 26 in the crystal. (b) Two-dimensional hydrogen-bonded layer resulting from the interlinking of hydrogen-bonding chains in the crystal of 26·H2O, viewed down the c-axis. (c) Molecular structure of (4-methylquinoline-N-oxide)2[Mo(η2-O2)2O] 27. (d) Extended chains formed by π–π interactions between molecules of 27, viewed down the b-axis. (e) Molecular structure of (κN,κO-2,2′-dipyridyl-N-oxide)[Mo(η2-O2)2O] 28. (f) Hydrogen-bonded chain of centrosymmetric dimers of 28 viewed down the b-axis. (g) X-ray structure of (4-methoxyquinoline-N-oxide)·H3P[OMo(η2-O2)2O]4 29. Thermal ellipsoids are at 50% probability.
Fig. 4.
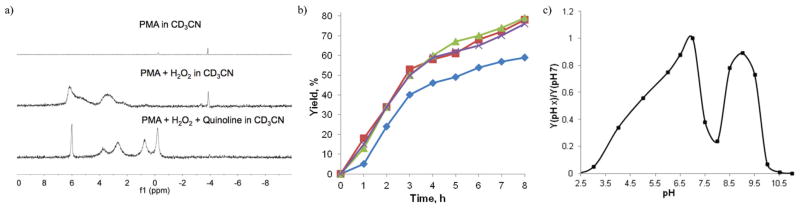
(a) Comparison of the 31P NMR spectra of phosphomolybdic acid in the presence of hydrogen peroxide and during oxidation reaction with quinoline. (b) Comparison of rates of N-oxidation of quinoline with H2O2 (4 equiv.) at 50 °C in acetonitrile in the presence of: 5 mol% of MoO3 (blue); 5 mol% of MoO3–1.25 mol% H3PO4 (Mo/P ratio 4 : 1, red); 5 mol% of MoO3–2.5 mol% H3PO4 (Mo/P ratio 2 : 1, green); and 1.25 mol% [(C12H25)2(CH3)2N]3{P[OMo(η2-O2)2O]4} (purple). (c) Dependence of relative reaction rates of N-oxidation of 1 on the pH of the reaction medium in the presence of 5 mol% of MoO3–1.25 mol% H3PO4 (Mo/P ratio 4 : 1) as a catalyst.
These 4-methylquinoline moieties are involved in other π–π interactions (3.61 Å), which link these dimers and form extended chains along the c-axis (Fig. 3d). In the structure of compound 28 the dipyridyl-N-oxide ligand is coordinated to the Mo atom in a bidentate chelating (N, O) mode (Fig. 3e). The axial position in the pseudotrigonal bipyramid is occupied by the O(1) atom of the N-oxide, and the N(2) atom of the pyridyl moiety is located in the equatorial position. The asymmetric unit in the crystal of 28 is made up of the Mo complex (Fig. 3f). The packing is dominated by centrosymmetric dimers through C–H···O hydrogen bonding interactions. These dimers are linked to one another through another weaker C–H···O interaction that results in the formation of hydrogen-bonded chains. Interestingly, treatment of 4-methoxyquinoline-N-oxide with MoO3, H2O2 and phosphoric acid led to a formation of an adduct of the N-oxide and the tetranuclear complex H3P[OMo(η2-O2)2O]4 29. Crystal structures of compounds containing this PMo4 cluster are exceedingly rare – only one X-ray structure had been reported in the CCDC database to date.43 The Mo complex and the uncoordinated N-oxide ligand occupy the asymmetric unit (Fig. 3g). The phosphorus atom is situated on a fourfold axis. The hydrogen atom that is located on the hydroxide coordinated to molybdenum is disordered, only occupying three of the four symmetry related hydroxide ions at any given time. There were no major long range interactions present in the packing of this complex.
In order to further optimize the reaction and to investigate the mechanism we have carried out several spectroscopic and kinetic experiments. The 31P NMR spectroscopic studies of phosphomolybdic acid (PMA) in the presence of hydrogen peroxide and during the oxidation reaction with quinoline as a substrate (Fig. 4a) suggest that PMA dissociates and forms several phosphorus-containing species. One of them has been identified as P[OMo(η2-O2)2O]43− with δ 6.0 ppm, which was confirmed by comparison with the NMR data of several quaternary ammonium salts with this anion synthesized according to the reported procedure.43 Guided by the indications from our spectroscopic and crystallographic studies that the tetranuclear PMo4 complex can be a key catalytic species in the N-oxidation, we have compared the catalytic performance of MoO3 with that of catalytic systems composed of MoO3–H3PO4 in 4 : 1 and 2 : 1 ratios, as well as preformed [(C12H25)2(CH3)2N]3{P[OMo(η2-O2)2O]4} (Fig. 4b). While the reaction catalyzed by MoO3 proved to be relatively slow and did not go to completion, faster conversion was observed with the other three catalytic systems. More importantly, both 4 : 1 and 2 : 1 MoO3–H3PO4 catalytic systems, and [(C12H25)2(CH3)2N]3-{P[OMo(η2-O2)2O]4} exhibited similar catalytic behavior, faster reaction and achieved >90% conversions within 16 h. It is interesting that no acceleration was observed when phosphoric acid was replaced with boric acid (H3BO3), sulfuric acid, selenium dioxide, or silicic acid (H4SiO4) confirming the important role of Mo/P complexes in the catalytic cycle.
We have also studied the influence of the pH of the reaction media on the reaction rate with 5 mol% of MoO3–1.25 mol% H3PO4 (Mo/P ratio 4 : 1) as a catalyst (Fig. 4c). It was observed that the highest reaction rate is achieved at pH 7. The oxidation is much slower at lower pH and no reaction is observed below pH 2.5, presumably due to protonation of the substrate.
However, an even more rapid decline in reaction rates is observed between pH 7 and 8. It is possible that the catalytic species that were active below pH 7.5 are unstable at higher pH. Subsequent increase in reaction rates between pH 8 and 9.5 may be due to formation of oxo diperoxo Mo complexes similar to 26 and 27 with L = OH−. A rapid deceleration at pH > 9.5 may, on the other hand, be due to the accelerated hydrolysis of acetonitrile that is mediated by hydroperoxide anions.44
The influence of the P=O ligands on the reactivity of the Mo catalyst was studied next (Table 3). The catalytic performance of the phosphoric acid-based system generally exceeded that of phosphine oxides, considering that PO43− ligand can coordinate up to 4 Mo atoms, and thus a 4 : 1 ratio of M : L can be used. Electron rich monophosphine oxides were found to be better ligands (entries 2–6), with tris(p-fluorophenyl)phosphine oxide emerging as the best ligand in the set (entry 4). Steric hindrance on the other hand was detrimental to the catalyst performance, as evidenced by the low activity of the tris(o-tolyl)phosphine oxide system (entry 7). Both triaryl-, trialkyl-, and triaminophosphine oxides were affective ligands (entries 2–6, 9, 10). Interestingly, chelating bisphosphine oxides proved far inferior to monophosphine oxides (entries 11–13), presumably due to increased steric hindrance around the η2-O2 ligands, and in line with the similar negative effect in the monophosphine oxide series.
Table 3.
Influence of the phosphine oxide ligands on the catalyst performance in the N-oxidation of 1a
| Entry | Ligand | TON |
|---|---|---|
| 1b | H3PO4 | 11 |
| 2 | Ph3PO | 12 |
| 3 | (p-FC6H4)3PO | 14.6 |
| 4 | [3,5-(CF3)2C6H3]3PO | 6.9 |
| 5 | Me2PhPO | 7.5 |
| 6 | tBuPh2PO | 8.9 |
| 7 | (o-Tol)3PO | 1.8 |
| 8 | o-C6H4(PPh2O)2 | 1 |
| 9 | Et3PO | 12.9 |
| 10 | (Me2N)3PO | 8.1 |
| 11c | dppe-dioxide | 3.9 |
| 12c | dppm-dioxide | 2.2 |
| 13c | binap-dioxide | 2.1 |
Reaction conditions: 1 (1.4 mmol), MoO3 (5 mol%), ligand (5 mol%) CH3CN (1 mL), H2O2 (30% aq. solution, 4 equiv.), 50 °C, 4 h.
1.25 mol% H3PO4 was used.
dppe = 1,2-bis(diphenylphosphino)-ethane, dppm = bis(diphenylphosphino)methane, binap = 2,2′-bis-(diphenylphosphino)-1,1′-binaphthyl.
Based on the aforementioned observations it is therefore plausible that the Mo4P cluster plays a crucial role as an N-oxidation catalyst in the MoO3–H3PO4 and the PMA systems. Monomeric Mo/P catalytic species may also contribute to the overall N-oxidation as evidenced by the results in Table 3. In this case complexes of type 26 with L1 position occupied by O=PR3 and L2 position occupied by a labile pendant ligand, e.g. solvent or the N-oxide product, may act as active catalysts. Complexes with both L1 and L2 positions occupied by O=PR3 possibly have a very low catalytic activity due to steric hindrance, as evidenced by the results with bidentate bisphosphine dioxides.
Heterocyclic N-oxides are emerging new scaffolds in drug discovery, as exemplified by the structures of the direct Factor Xa inhibitor otamixaban, CCR5 inhibitor ancriviroc, sedative/hypnotic chlordiazepoxide and reverse transcriptase inhibitor BILR 355. Heterocyclic N-oxides are also known to be metabolites of various pharmaceuticals and agrochemicals.
We have employed the developed oxidation procedure for the synthesis of several N-oxides of such biologically active compounds. Thus, N-oxides of selective fungicide quinoxyfen (Quintec) 30, as well antimicrobial drugs chlorquinaldol 31 and cloxyquin 32 have been prepared in high yields using the optimized 5 mol% MoO3–1.25 mol% H3PO4 as a catalyst (Scheme 1). In addition 4-cyanopyridine-N-oxide (33) was prepared (86% yield) using this catalytic system.
Scheme 1.
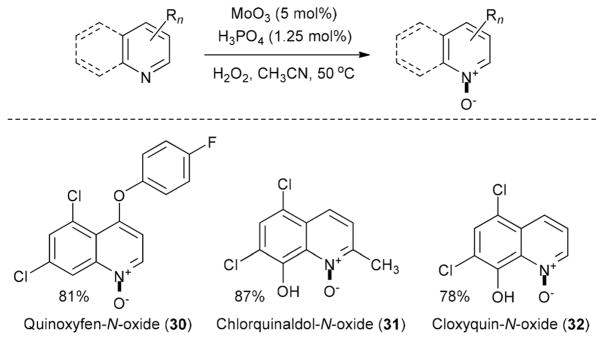
Synthesis of biorelevant heterocyclic N-oxides 30–32.
The Mo/P-catalyzed N-oxidation provided improved yields of the heterocyclic N-oxides when compared with other methods for some substrates. Thus, N-oxide 4 was previously prepared in 34% yield using m-chloroperbenzoic acid,45 while 61% was observed using PMA as a catalyst. Similarly, N-oxide 22 was prepared in 82% using surfactant-encapsulated [(C18H37)2(CH3)2N]8[HBW11O39],32b and 79% with the Fe(acac)3/O2 system,46 whereas Mo/P catalysis afforded the product in an 89% yield.
Conclusions
In conclusion, synthetic scope and the mechanism of the molybdenum(VI)-catalyzed N-oxidation of N-heterocycles have been investigated. The reaction exhibits high chemoselectivity and scope with respect to the heterocyclic backbone and is amenable to gram-scale preparations of various heterocyclic N-oxides. Thus, thiazoles undergo efficient N-oxidation without the concomitant S-oxidation. Benzylic and phenolic oxidations can also be avoided by using this catalytic system. Single crystal X-ray crystallographic studies provided insights into the structure of Mo complexes relevant to the catalytic cycle and the role of the P=O ligand on the catalyst activity. Further experimental evidence points to the influence of the steric and electronic properties of the P=O ligands on the catalytic activity.
Experimental section
General methods
Anhydrous dichloromethane, toluene, tetrahydrofuran and ether were collected under argon from an LC Technologies solvent purification system, having been passed through two columns packed with molecular sieves. Acetonitrile, ethanol, and methanol were dried over 4 Å molecular sieves. All other chemicals were used as commercially available (Sigma-Aldrich, Acros, Alfa Aesar, Combi-Blocks, Strem). Reactions were monitored by TLC until deemed complete using silica gel-coated glass plates (Merck Kieselgel 60 F254). Plates were visualized under ultraviolet light (254 nm). Column chromatography was performed using CombiFlash Rf-200 (Teledyne-Isco) automated flash chromatography system with RediSep columns. 1H, 13C NMR spectra were recorded at 300 and 500 (1H), and 75.5 and 125 MHz on Varian Mercury VX 300 and Agilent Inova 500 instruments in CDCl3 solutions if not otherwise specified. Chemical shifts (δ) are reported in parts per million (ppm) from the residual solvent peak and coupling constants (J) in Hz. Infrared measurements were carried out neat on a Bruker Vector 22 FT-IR spectrometer fitted with a Specac diamond attenuated total reflectance (ATR) module. Compounds 26–29 have been assigned the following CCDC numbers: 26 (CCDC 981527), 27 (CCDC 981529), 28 (CCDC 981530), 29 (CCDC 981531).
General procedure (GP1) for N-oxidation catalyzed by PMA or MoO3–H3PO4
To a solution of azine or azole in acetonitrile (0.5 mL mmol−1 substrate) was added hydrogen peroxide (1.5–3 equiv., 30% in water) and phosphomolybdic acid (0.5–3 mol%) or MoO3 (5 mol%) H3PO4 (4 w/w solution in water, 1.25 mol%) at 50 °C. After 12 h the reaction was diluted with saturated aqueous ammonium chloride (0.1 mL mmol−1 substrate) and extracted with dichloromethane. The combined organic layers were dried over anhydrous sodium sulfate and concentrated under reduced pressure. The crude product was purified by column chromatography [hexanes–EtOAc] to yield the desired N-oxide.
General procedure (GP2) for kinetic experiments
The reactions were carried out as described in GP1. Reaction medium pH was adjusted by adding appropriate amounts of aqueous HBF4 or NaOH solutions. Aliquots of reaction mixtures were drawn at specified times and the conversions were determined by 1H NMR spectroscopy.
Experimental procedures and spectroscopic data
8-Methoxyquinoline 1-oxide3 (3)
According to GP1, S1 (340 mg, 2.12 mmol) was reacted with hydrogen peroxide (313 μL, 3.18 mmol, 1.5 equiv., 30% in H2O) and phosphomolybdic acid (197 μL, 0.21 mmol, 1 mol%, 20% in EtOH) in acetonitrile (1 mL). The product was isolated to yield oxide 3 (260 mg, 71%) as a brown solid. – m.p.: 38–41 °C – 1H NMR (300 MHz): 3.92 (3 H, s), 6.88 (1 H, dd, J = 1.5, 8 Hz), 7.20–7.40 (4 H, m), 7.96 (1 H, dd, J = 1.5, 8 Hz), 8.78 (1 H, dd, J = 1.5, 4 Hz) ppm. – 13C NMR (75 MHz): 55.84, 107.57, 119.42, 121.57, 126.72, 129.19, 136.07, 139.66, 148.89, 155.02 ppm. – IR: 910, 1090, 1232, 1378, 1467, 1504, 2858, 2954, 3037 cm−1.
Pyrimidine 1-oxide47 (4)
According to GP1, pyrimidine (80 mg, 1 mmol) was reacted with hydrogen peroxide (150 μL, 1.5 mmol, 1.5 equiv., 30% in H2O) and phosphomolybdic acid (91 μL, 0.01 mmol, 2 mol%, 20% in EtOH) in acetonitrile (500 μL). The isolated product afforded 4 (59 mg, 61%) as a brown oil. – 1H NMR (300 MHz): 7.33 (1 H, s), 8.27 (1 H, dd, J = 0.5, 2.5 Hz), 8.40 (1 H, d, J = 4 Hz), 9.01 (1 H, s) ppm. – 13C NMR (75 MHz): 121.23, 144.00, 144.35, 149.94 ppm. – IR: 1064, 1179, 1244, 1423, 2958, 2954, 3037 cm−1.
2-Bromopyridine 1-oxide48 (5)
According to GP1, 2-bromopyridine (241 mg, 1.53 mmol) was reacted with hydrogen peroxide (460 μL, 4.59 mmol, 3 equiv., 30% in H2O) and phosphomolybdic acid (279 μL, 0.03 mmol, 2 mol%, 20% in EtOH) in acetonitrile (700 μL). The isolated product afforded 5 (249 mg, 95%) as a brown oil. – 1H NMR (500 MHz): 7.06 (1 H, dd, J = 4.5, 7.5 Hz), 7.26 (1 H, dd, J = 1, 8 Hz), 7.36 (1 H, dt, J = 1, 8 Hz), 8.14 (1 H, dd, J = 1.5, 5 Hz) ppm. – 13C NMR (125 MHz): 122.64, 128.12, 138.53, 141.96, 150.04 ppm. – IR: 1042, 1270, 1414, 1560, 2998, 3049 cm−1.
6-Bromo-2-pentylquinoline 1-oxide (6)
According to GP1, 6-bromo-2-pentylquinoline13 (38 mg, 0.137 mmol) was reacted with hydrogen peroxide (41 μL, 0.411 mmol, 3 equiv., 30% in H2O) and phosphomolybdic acid (25 μL, 0.002 mmol, 2 mol%, 20% in EtOH) in acetonitrile (100 μL). The isolated product afforded 6 (25 mg, 62%) as a brown oil. – 1H NMR (500 MHz): 0.93 (3 H, t, J = 7 Hz), 1.37–1.48 (4 H, m), 1.82 (2 H, quin., J = 7 Hz), 3.10 (2 H, t, J = 8 Hz), 7.32 (1 H, d, J = 8.5 Hz), 7.56 (1 H, d, J = 8.5 Hz), 7.80 (1 H, dd, J = 2, 9 Hz), 7.99 (1 H, d, J = 2 Hz), 8.66 (1 H, d, J = 9.5 Hz) ppm. – 13C NMR (125 MHz): 13.99, 22.47, 25.65, 31.45, 31.77, 121.80, 122.03, 123.13, 123.71, 129.87, 130.19, 133.48, 130.52, 139.68 ppm. – IR: 1064, 1179, 1336, 1449, 1504, 2858, 2954, 3064 cm−1. – MS (ESI): 293.0, HRMS: calcd: 293.0415; found 294.0542 [M + H+].
2-(Hydroxymethyl)pyridine 1-oxide49 (7)
According to GP1, 2-(hydroxymethyl)pyridine (163 mg, 1.53 mmol) was reacted with hydrogen peroxide (690 μL, 6.88 mmol, 4.5 equiv., 30% in H2O) and phosphomolybdic acid (418 μL, 0.045 mmol, 3 mol%, 20% in EtOH) in acetonitrile (750 μL). The isolated product afforded 7 (124 mg, 65%) as a yellow oil. – 1H NMR (500 MHz): 4.78 (2 H, s), 7.24–7.27 (1 H, m), 7.37 (1 H, t, J = 7.5 Hz), 7.45 (1 H, d, J = 7.5 Hz), 8.23 (1 H, d, J = 6 Hz) ppm. – 13C NMR (125 MHz): 60.55, 124.68, 128.13, 139.36, 150.61, 161.10 ppm. – IR: 1064, 1209, 1437, 1491, 1559, 1653, 3049, 3216 cm−1.
Quinoxaline 1-oxide50 (8)
According to GP1, quinoxaline (200 mg, 1.53 mmol) was reacted with hydrogen peroxide (230 μL, 2.29 mmol, 1.5 equiv., 30% in H2O) and phosphomolybdic acid (140 μL, 0.015 mmol, 2 mol%, 20% in EtOH) in acetonitrile (700 μL). The isolated product afforded 8 (210 mg, 94%) as a red solid. – m.p.: 152–155 °C16 – 1H NMR (300 MHz): 7.33 (2 H, d, J = 6.5 Hz), 8.18 (2 H, d, J = 5.5 Hz), 8.45 (2 H, s) ppm. – 13C NMR (75 MHz): 124.70, 125.86, 126.90, 129.37, 135.10, 137.58, 140.36, 149.90 ppm. – IR: 1000, 1169, 1305, 1457, 1565, 2990, 3269 cm−1.
Thiazole 3-oxide51 (9)
According to GP1, thiazole (130 mg, 1.53 mmol) was reacted with hydrogen peroxide (460 μL, 4.59 mmol, 3 equiv., 30% in H2O) and phosphomolybdic acid (280 μL, 0.03 mmol, 2 mol%, 20% in EtOH) in acetonitrile (750 μL). The isolated product afforded 9 (96 mg, 62%) as a colorless oil. – 1H NMR (500 MHz): 7.40 (1 H, s), 7.94 (1 H, s), 8.86 (1 H, s) ppm. – 13C NMR (125 MHz): 118.87, 143.28, 153.09 ppm. – IR: 909, 1106, 123, 1414, 1560, 3028, 3049 cm−1.
3,4-Dimethylpyridine 1-oxide52 (10)
According to GP1, 3,4-lutidine (500 mg, 4.67 mmol) was reacted with hydrogen peroxide (702 μL, 7 mmol, 1.5 equiv., 30% in H2O) and phosphomolybdic acid (430 μL, 0.046 mmol, 1 mol%, 20% in EtOH) in acetonitrile (650 μL). The isolated product afforded 10 (480 mg, 84%) as a colorless solid. – m.p.: 126–128 °C21 –1H NMR (300 MHz): 2.19 (3 H, s), 2.23 (3 H, s), 7.01 (1 H, d, J = 6.5 Hz), 7.97 (1 H, d, J = 6.5 Hz), 8.02 (1 H, s) ppm. – 13C NMR (75 MHz): 16.81, 18.40, 126.57, 135.49, 136.24, 137.41, 138.73 ppm. – IR: 910, 1090, 1209, 1394, 1449, 1554, 2857, 2931, 3135 cm−1.
Phenanthridine 5-oxide53 (11)
According to GP1, phenanthridine (50 mg, 0.279 mmol) was reacted with hydrogen peroxide (83 μL, 0.837 mmol, 3 equiv., 30% in H2O) and phosphomolybdic acid (25 μL, 0.005 mmol, 2 mol%, 20% in EtOH) in acetonitrile (200 μL). The isolated product afforded 11 (41 mg, 76%) as a brown oil. – 1H NMR (500 MHz): 7.25–8.09 (5 H, m), 8.53–8.63 (2 H, m), 8.97 (1 H, d, J = 2 Hz), 9.19 (1 H, d, J = 2 Hz) ppm. – 13C NMR (125 MHz): 120.67, 122.13, 122.77, 127.06, 128.18, 128.99, 129.53, 129.71, 129.91, 130.15, 130.46, 133.83, 169.27 ppm. – IR: 1071, 1191, 1473, 1559, 1647, 3071 cm−1.
4-Phenylpyridine 1-oxide48 (12)
According to GP1, 4-phenylpyridine (237 mg, 1.53 mmol) was reacted with hydrogen peroxide (460 μL, 4.59 mmol, 3 equiv., 30% in H2O) and phosphomolybdic acid (280 μL, 0.03 mmol, 2 mol%, 20% in EtOH) in acetonitrile (750 μL). The isolated product afforded 12 (220 mg, 84%) as a red oil. – 1H NMR (500 MHz): 7.43–7.67 (5 H, m), 8.29 (4 H, dd, J = 6 Hz) ppm. – 13C NMR (125 MHz): 115.58, 123.81, 126.50, 129.39, 129.46, 139.37, 157.21 ppm. –IR: 1043, 1236, 1430, 1592, 2981, 3053 cm−1.
4-Methoxyquinoline 1-oxide3 (13)
According to GP1, 4-methoxyquinoline (2.76 g, 17.36 mmol) was reacted with hydrogen peroxide (5.3 mL, 52.07 mmol, 3 equiv., 30% in H2O) and phosphomolybdic acid (3.16 mL, 0.34 mmol, 2 mol%, 20% in EtOH) in acetonitrile (9 mL). The isolated product afforded 13 (2.14 g, 71%) as a red oil. – 1H NMR (500 MHz): 4.00 (3 H, s), 6.74 (1 H, d, J = 6.5 Hz), 7.48 (1 H, dt, J = 1, 7 Hz), 7.68 (1 H, dt, J = 1, 7 Hz), 8.08 (1 H, d, J = 8.5 Hz), 8.14 (1 H, dd, J = 1, 8.5 Hz), 8.73 (1 H, d, J = 4.5 Hz) ppm. – 13C NMR (125 MHz): 56.09, 121.96, 122.63, 126.15, 127.31, 128.27, 130.56, 131.13, 150.11, 163.42 ppm. – IR: 1156, 1394, 1507, 2999, 3125 cm−1.
4,7-Dichloroquinoline 1-oxide54 (14)
According to GP1, 4,7-dichloroquinoline (2 g, 10.2 mmol) was reacted with hydrogen peroxide (1.55 mL, 15.30 mmol, 1.5 equiv., 30% in H2O) and phosphomolybdic acid (930 μL, 0.102 mmol, 1 mol%, 20% in EtOH) in acetonitrile (5 mL). The isolated product afforded 14 (1.9 g, 88%) as a tan solid. – m.p.: 164–165 °C24 – 1H NMR (500 MHz): 7.36 (1 H, d, J = 6.5 Hz), 7.68 (1 H, d, J = 9 Hz), 8.13 (1 H, d, J = 9 Hz), 8.43 (1 H, d, J = 6.5 Hz), 8.77 (1 H, s) ppm. – 13C NMR (125 MHz): 119.93, 121.22, 121.42, 125.60, 126.52, 126.75, 128.68, 129.85, 130.80, 135.96, 138.22, 142.33, 150.88 ppm. – IR: 829, 1091, 1291, 1367, 1412, 1555, 1609, 3025, 3094 cm−1.
1,10-Phenanthroline 1-oxide55 (16)
According to GP1, 1,10-phenanthroline (275 mg, 1.53 mmol) was reacted with hydrogen peroxide (690 μL, 6.88 mmol, 4.5 equiv., 30% in H2O) and phosphomolybdic acid (418 μL, 0.045 mmol, 3 mol%, 20% in EtOH) in acetonitrile (750 μL). The isolated product afforded 16 (246 mg, 82%) as a yellow oil. – 1H NMR (300 MHz): 7.45 (1 H, t, J = 6.5 Hz), 7.63–7.81 (4, m), 8.22 (1 H, dd, J = 1.5, 8 Hz), 8.74 (1 H, dd, J = 1, 6.5 Hz), 9.30 (1 H, dd, J = 1.5, 4 Hz) ppm. – 13C NMR (75 MHz): 122.85, 123.04, 123.19, 124.64, 126.50, 128.91, 129.04, 133.25, 135.93, 140.75, 149.92, 150.30 ppm. – IR: 1068, 1249, 1436, 1559, 1653, 2981, 3042 cm−1.
2,3-Dimethylpyridine 1-oxide56 (17)
According to GP1, 2,3-lutidine (163 mg, 1.53 mmol) was reacted with hydrogen peroxide (690 μL, 6.88 mmol, 4.5 equiv., 30% in H2O) and phosphomolybdic acid (418 μL, 0.045 mmol, 3 mol%, 20% in EtOH) in acetonitrile (750 μL). The isolated product afforded 17 (141 mg, 75%) as a yellow oil. – 1H NMR (500 MHz): 1.91 (3 H, s), 2.05 (3 H, s), 6.65–6.72 (2 H, m), 7.71 (1 H, d, J = 6.5 Hz) ppm. – 13C NMR (75 MHz): 13.21, 18.95, 122.00, 131.86, 137.84, 144.81, 155.96 ppm. – IR: 1018, 1260, 1457, 1540, 2961, 3026 cm−1.
6-Bromoquinoline 1-oxide3 (18)
According to GP1, 6-bromoquinoline (50 mg, 0.24 mmol) was reacted with hydrogen peroxide (36 μL, 0.36 mmol, 1.5 equiv., 30% in H2O) and phosphomolybdic acid (20 μL, 0.002 mmol, 2 mol%, 20% in EtOH) in acetonitrile (100 μL). The isolated product afforded 18 (41 mg, 77%) as a red solid. – m.p.: 110–112 °C – 1H NMR (300 MHz): 7.31 (1 H, dd, J = 1, 2.5 Hz), 7.63 (1 H, d, J = 8.5 Hz), 7.79 (1 H, dd, J = 2, 9.5 Hz), 8.02 (1 H, d, J = 2 Hz), 8.52 (1 H, d, J = 6 Hz), 6.60 (1 H, d, J = 9.5 Hz) ppm. – 13C NMR (75 MHz): 121.72, 122.19, 123.29, 124.98, 130.08, 131.57, 133.83, 135.90, 140.25 ppm. – IR: 1143, 1266, 1421, 1569, 2999, 3026 cm−1.
2-Phenylpyridine 1-oxide48 (19)
According to GP1, 2-phenylpyridine (200 mg, 1.29 mmol) was reacted with hydrogen peroxide (418 μL, 4.17 mmol, 3 equiv., 30% in H2O) and phosphomolybdic acid (235 μL, 0.25 mmol, 2 mol%, 20% in EtOH) in acetonitrile (650 μL). The isolated product afforded 19 (210 mg, 78%) as a colorless solid. – m.p.: 150–152 °C12 – 1H NMR (300 MHz): 7.21–7.31 (2 H, m), 7.40–7.49 (4 H, m), 7.80 (2 H, dd, J = 2.5, 5.5 Hz), 8.32 (1 H, d, J = 5.5 Hz) ppm. –13C NMR (75 MHz): 124.51, 125.76, 127.38, 128.27, 129.25, 129.58, 132.59, 140.39, 149.25 ppm. – IR: 995, 1073, 1158, 1259, 1416, 1476, 2925, 3032 cm−1.
7-Chloro-4-methoxyquinoline 1-oxide (20)
According to GP1, S3 (1.6 g, 5.54 mmol) was reacted with hydrogen peroxide (1.7 mL, 16.62 mmol, 3 equiv., 30% in H2O) and phosphomolybdic acid (1 mL, 0.118 mmol, 2 mol%, 20% in EtOH) in acetonitrile (2.5 mL). The isolated product afforded 20 (1.12 g, 97%) as a colorless solid. – m.p.: 145–147 °C – 1H NMR (500 MHz): 4.06 (3 H, s), 6.64 (1 H, d, J = 7 Hz), 7.59 (1 H, d, J = 2 Hz), 8.15 (1 H, d, J = 9 Hz), 8.46 (1 H, d, J = 7 Hz), 8.77 (1 H, d, J = 2 Hz) ppm. – 13C NMR (125 MHz): 56.34, 99.84, 119.57, 120.99, 123.39, 124.28, 129.06, 136.99, 137.88, 154.26 ppm. – IR: cm−1. – MS (ESI): found 212.9. MS (HRMS): calc. 210.0316, found 213.9937 [M + H+].
3-Phenylpyridine 1-oxide57 (21)
According to GP1, 2-phenylpyridine (200 mg, 1.29 mmol) was reacted with hydrogen peroxide (418 μL, 4.17 mmol, 3 equiv., 30% in H2O) and phosphomolybdic acid (235 μL, 0.25 mmol, 2 mol%, 20% in EtOH) in acetonitrile (650 μL). The isolated product afforded 21 (250 mg, 93%) as a colorless solid. – m.p.: 116–118 °C –1H NMR (300 MHz): 7.71–7.84 (5 H, m), 8.11 (1 H, dd, J = 1, 8.5 Hz), 8.34 (1 H, d, J = 3.5 Hz), 8.55 (1 H, dd, J = 1, 8.5 Hz), 8.66 (1 H, d, J = 3.5 Hz) ppm. – 13C NMR (75 MHz): 118.88, 127.95, 129.24, 129.94, 130.07, 130.24, 131.75, 137.44, 145.92 ppm. – IR: 1015, 1131, 1314, 1497, 1575, 1639, 2995, 3079 cm−1.
8-Hydroxyquinoline 1-oxide58 (22)
According to GP1, 8-hydroxyquinoline (221 mg, 1.53 mmol) was reacted with hydrogen peroxide (460 μL, 4.59 mmol, 3 equiv., 30% in H2O) and phosphomolybdic acid (280 μL, 0.03 mmol, 2 mol%, 20% in EtOH) in acetonitrile (750 μL). The isolated product afforded 22 (220 mg, 89%) as a red oil. – 1H NMR (300 MHz): 7.05 (1 H, d, J = 5.5 Hz), 7.21–7.27 (2 H, m), 7.48 (1 H, t, J = 8.5 MHz), 7.80 (1 H, d, J = 8.5 Hz), 8.30 (1 H, dd, J = 0.5, 5.5 Hz) ppm. – 13C NMR (75 MHz): 115.06, 116.88, 120.41, 128.11, 130.07, 130.60, 134.76, 153.79, 168.71 ppm. – IR: 1047, 1153, 1201, 1312, 1436, 1598, 1698, 2950, 3066 cm−1.
4-Methylquinoline 1-oxide3 (23)
According to GP1, leptidine (218 mg, 1.53 mmol) was reacted with hydrogen peroxide (690 μL, 6.88 mmol, 4.5 equiv., 30% in H2O) and phosphomolybdic acid (418 μL, 0.045 mmol, 3 mol%, 20% in EtOH) in acetonitrile (750 μL). The isolated product afforded 23 (169 mg, 67%) as a brown oil. – 1H NMR (500 MHz): 2.65 (3 H, s), 7.11 (1 H, d, J = 6 Hz), 7.66 (1 H, t, J = 7 Hz), 7.76 (1 H, t, J = 7 Hz), 7.96 (1 H, d, J = 7), 8.44 (1 H, d, J = 7 Hz), 8.80 (1 H, t, J = 9 Hz) ppm. – 13C NMR (125 MHz): 18.38, 120.32, 121.41, 124.73, 128.51, 129.83, 130.18, 135.02, 135.11, 140.86 ppm. – IR: 1058, 1277, 1394, 1473, 1559, 1653, 2949, 3024, 3081 cm−1.
5,7-Dichloro-4-(4-fluorophenoxy)quinoline 1-oxide6 (30)
To a solution of quinoxyfen (50 mg, 0.162 mmol) in acetonitrile (80 μL) was reacted with hydrogen peroxide (36 μL, 0.324 mmol, 2 equiv., 30% in H2O), phosphoric acid (0.2 mg, 0.002 mmol, 1.25 mol%) and molybdenum(VI) oxide (1 mg, 0.008 mmol, 5 mol%). The crude reaction mixture was diluted with saturated aqueous ammonium chloride (5 mL) and extracted with chloroform (3 × 5 mL), dried over anhydrous sodium sulfate, and concentrated under reduced pressure to yield 30 (42 mg, 81%) as a yellow oil. – 1H NMR (500 MHz): 6.57 (1 H, d, J = 6.5 Hz), 7.06–7.12 (4 H, m), 7.63 (1 H, d, J = 1.5 Hz), 8.34 (1 H, 6.5 Hz), 8.72 (1 H, s) ppm. – 13C NMR (125 MHz): 107.70, 117.19 (d, J = 23.5 Hz), 119.26 (d, J = 51 Hz), 121.75 (d, J = 8.5 Hz), 128.24, 131.77 (d, J = 74 Hz) 137.03, 143.61, 150.05 (d, J = 2 Hz), 153.01, 158.99, 160.94 ppm. – 19F NMR (252 MHz): −116.42 ppm. – IR: 1012, 1091, 1174, 1254, 1303, 1415, 1563, 1641, 2988, 3071, 3102 cm−1.
5,7-Dichloro-8-hydroxy-2-methylquinoline 1-oxide55 (31)
To a solution of 5,7-dichloro-8-hydroxy-2-methylquinoline (217 mg, 0.961 mmol) in acetonitrile (500 μL) was reacted with hydrogen peroxide (150 μL, 1.92 mmol, 2 equiv.), phosphoric acid (1.17 mg, 0.012 mmol, 1.25 mol%) and molybdenum(VI) oxide (4.5 mg, 0.048 mmol, 5 mol%). The crude reaction mixture was diluted with saturated aqueous ammonium chloride (5 mL) and extracted with chloroform (3 × 5 mL), dried over anhydrous sodium sulfate, and concentrated under reduced pressure to yield 31 (203 mg, 87%) as a yellow oil. – 1H NMR (500 MHz): 2.64 (3H, s), 7.32 (1H, d, J = 8.9 Hz), 7.51 (1H, s), 7.92 (1H, d, J = 8.9 Hz). 13C NMR (125 MHz): 18.0, 118.4, 118.5, 123.6, 125.7, 126.9, 130.0, 130.1, 146.2, 149.3. – IR: 3080, 1535, 1454, 1413 cm−1.
5,7-Dichloro-8-hydroxyquinoline 1-oxide6 (32)
To a solution of 5,7-dichloro-8-quinolinol (214 mg, 1.0 mmol) in acetonitrile (500 μL) was reacted with hydrogen peroxide (225 μL, 2.0 mmol, 2 equiv., 30% in H2O), phosphoric acid (1.22 mg, 0.012 mmol, 1.25 mol%) and molybdenum(VI) oxide (7 mg, 0.05 mmol, 5 mol%). The crude reaction mixture was diluted with saturated aqueous ammonium chloride (5 mL) and extracted with chloroform (3 × 5 mL), dried over anhydrous sodium sulfate, and concentrated under reduced pressure to yield 32 (178 mg, 78%) as a yellow oil. – 1H NMR (300 MHz): 7.40 (1 H, dd, J = 6, 9 Hz), 7.70 (1 H, s), 8.17 (1 H, d, J = 9 Hz), 8.33 (1 H, d, J = 6 Hz) ppm. – 13C NMR (75 MHz): 118.89, 118.98, 121.37, 126.96, 128.42, 128.90, 131.39, 135.54, 149.68 ppm. – IR: 906, 1068, 1156, 1208, 1320, 1423, 1513, 2995, 3067, 3078 cm−1.
4-Cyanopyridine 1-oxide59 (33)
To a solution of 4-cyano-pyridine (100 mg, 0.961 mmol) in acetonitrile (500 μL) was reacted with hydrogen peroxide (150 μL, 1.92 mmol, 2 equiv.), phosphoric acid (1.17 mg, 0.012 mmol, 1.25 mol%) and molybdenum(VI) oxide (4.5 mg, 0.048 mmol, 5 mol%). The crude reaction mixture was diluted with saturated aqueous ammonium chloride (5 mL) and extracted with chloroform (3 × 5 mL), dried over anhydrous sodium sulfate, and concentrated under reduced pressure to yield 33 (99 mg, 86%) as a yellow oil. – 1H NMR (500 MHz): δ 8.23 (1 H, d, J = 5 Hz), 8.83 (2 H, d, J = 8.5 Hz). 13C NMR (125 MHz): 109.3, 117.8, 130.2, 140.8 ppm.
Supplementary Material
Acknowledgments
Financial support by the Welch Foundation (AX-1788), the Max and Minnie Tomerlin Voelcker Fund, the San Antonio Life Sciences Institute (SALSI) and the University of Texas at San Antonio is gratefully acknowledged. Mass spectroscopic analysis was supported by a grant from the National Institute on Minority Health and Health Disparities (G12MD007591).
Footnotes
Electronic supplementary information (ESI) available: Experimental procedures and spectroscopic data. CCDC 981527, 981529, 981530 and 981531. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4ob00115j
Notes and references
- 1.(a) Albini A, Pietra S. Heterocyclic N-Oxides. CRC Press; Boca Raton, FL: 1991. [Google Scholar]; (b) Youssif S. ARKIVOC. 2001;2:U2. [Google Scholar]; (c) Begtrup M. Adv Heterocycl Chem. 2012;106:1. [Google Scholar]; (d) Ryzhakov AV, Rodina LL. Heterocycles. 2008;75:2367. [Google Scholar]
- 2.(a) Campeau LC, Rousseaux S, Fagnou K. J Am Chem Soc. 2005;127:18020. doi: 10.1021/ja056800x. [DOI] [PubMed] [Google Scholar]; (b) Leclerc JP, Fagnou K. Angew Chem, Int Ed. 2006;45:7781. doi: 10.1002/anie.200602773. [DOI] [PubMed] [Google Scholar]; (c) Cho SH, Hwang SJ, Chang S. J Am Chem Soc. 2008;130:9254. doi: 10.1021/ja8026295. [DOI] [PubMed] [Google Scholar]; (d) Campeau LC, Stuart DR, Leclerc JP, Bertrand-Laperle M, Villemure E, Sun HY, Lasserre S, Guimond N, Lecavallier M, Fagnou K. J Am Chem Soc. 2009;131:3291. doi: 10.1021/ja808332k. [DOI] [PubMed] [Google Scholar]; (e) Wu ZY, Song HY, Cui XL, Pi C, Du WW, Wu YJ. Org Lett. 2013;15:1270. doi: 10.1021/ol400178k. [DOI] [PubMed] [Google Scholar]
- 3.(a) Wengryniuk SE, Weickgenannt A, Reiher C, Strotman NA, Chen K, Eastgate MD, Baran PS. Org Lett. 2013;15:792. doi: 10.1021/ol3034675. [DOI] [PubMed] [Google Scholar]; (b) Paudler WW, Jovanovic MV. J Org Chem. 1983;48:1064. [Google Scholar]
- 4.(a) Londregan AT, Jennings S, Wei L. Org Lett. 2011;13:1840. doi: 10.1021/ol200352g. [DOI] [PubMed] [Google Scholar]; (b) Keith JM. J Org Chem. 2010;75:2722. doi: 10.1021/jo1001017. [DOI] [PubMed] [Google Scholar]; (c) Storz T, Bartberger MD, Sukits S, Wilde C, Soukup T. Synthesis. 2008:201. [Google Scholar]; (d) Yin J, Xiang B, Huffman MA, Raab CE, Davies IW. J Org Chem. 2007;72:4554. doi: 10.1021/jo070189y. [DOI] [PubMed] [Google Scholar]; (e) Yin J, Xiang B, Huffman MA, Raab CE, Davies IW. J Org Chem. 2007;72:4554. doi: 10.1021/jo070189y. [DOI] [PubMed] [Google Scholar]
- 5.(a) Veerareddy A, Surendrareddy G, Dubey PK. J Heterocycl Chem. 2011;48:961. [Google Scholar]; (b) Fife WK. J Org Chem. 1983;48:1375. [Google Scholar]
- 6.Larionov OV, Stephens D, Mfuh A, Chavez G. Org Lett. 2014;16:864–867. doi: 10.1021/ol403631k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) Caron S, Do NM, Sieser JE. Tetrahedron Lett. 2000;41:2299. [Google Scholar]; (b) Balicki R, Golinski J. Synth Commun. 2000;30:1529. [Google Scholar]; (c) Rosenau T, Potthast A, Kosma P. Synlett. 1999, 1972 [Google Scholar]; (d) Kaczmarek L, Balicki R, Nanka-Namirski P. Chem Ber. 1992;125:1965. [Google Scholar]; (e) Sharma KS, Kumari S, Singh RP. Synthesis. 1981:316. [Google Scholar]
- 8.Broughman P, Cooper PMS, Cummerson DA, Heaney H, Thompson N. Synthesis. 1987:1015.
- 9.(a) Dholakia S, Gillard RD, Lancashire RG. Chem Ind. 1977:963. [Google Scholar]; (b) Wommack JB. Chem Eng News. 1977;55:5. [Google Scholar]
- 10.(a) Zhu Z, Espenson JH. J Org Chem. 1995;60:7728. [Google Scholar]; (b) Murray RW, Singh M. J Org Chem. 1990;55:2954. [Google Scholar]
- 11.Dayan S, Kol M, Rozen S. Synthesis. 1999:1427. [Google Scholar]
- 12.Sivasubramanian G, Parameswaran VR. J Heterocycl Chem. 2007;44:1223. doi: 10.1002/jhet.5570440601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.(a) Jones CW. Applications of Hydrogen Peroxide and Derivatives. Royal Society of Chemistry; Cambridge, U.K: 1999. [Google Scholar]; (b) Strukul G, editor. Catalytic Oxidations with Hydrogen Peroxide as Oxidant. Kluwer Academic; Dordrecht, The Netherlands: 1992. [Google Scholar]; (c) Talsi EP, Bryliakov KP. Coord Chem Rev. 2012;256:1418. [Google Scholar]; (d) Piera J, Backvall JE. Angew Chem, Int Ed. 2008;47:3506. doi: 10.1002/anie.200700604. [DOI] [PubMed] [Google Scholar]; (e) Noyori R, Aoki M, Sato K. Chem Commun. 2003:1977. doi: 10.1039/b303160h. [DOI] [PubMed] [Google Scholar]
- 14.Goor G, Glenneberg J, Jacobi S. Ullmann’s Encyclopedia of Industrial Chemistry. Wiley-VCH; Weinheim, Germany: 2007. Hydrogen Peroxide. [Google Scholar]
- 15.Sheldon RA. Chem Commun. 2008:3352. doi: 10.1039/b803584a. [DOI] [PubMed] [Google Scholar]
- 16.Zhao X, Cheng K, Hao J, Liua D. J Mol Catal A: Chem. 2008;284:58. [Google Scholar]
- 17.(a) Correa A, Mancheno OG, Bolm C. Chem Soc Rev. 2008;37:1108. doi: 10.1039/b801794h. [DOI] [PubMed] [Google Scholar]; (b) Que L, Jr, Tolman WB. Nature. 2008;455:333. doi: 10.1038/nature07371. [DOI] [PubMed] [Google Scholar]; (c) Venkataramanan NS, Kuppuraj G, Rajagopal S. Coord Chem Rev. 2005;249:1249. [Google Scholar]
- 18.Lane BS, Burgess K. Chem Rev. 2003;103:2457. doi: 10.1021/cr020471z. [DOI] [PubMed] [Google Scholar]
- 19.(a) Egami H, Katsuki T. J Am Chem Soc. 2007;129:8940. doi: 10.1021/ja071916+. [DOI] [PubMed] [Google Scholar]; (b) Kaczorowska K, Kolarska Z, Mitka K, Kowalski P. Tetrahedron. 2005;61:8315. [Google Scholar]
- 20.Muzart J. Tetrahedron. 2003;59:5789. [Google Scholar]
- 21.(a) De Faveri G, Ilyashenko G, Watkinson M. Chem Soc Rev. 2011;40:1722. doi: 10.1039/c0cs00077a. [DOI] [PubMed] [Google Scholar]; (b) Wojaczynska E, Wojaczynski J. Chem Rev. 2010;110:4303. doi: 10.1021/cr900147h. [DOI] [PubMed] [Google Scholar]; (c) Egami H, Oguma T, Katsuki T. J Am Chem Soc. 2010;132:5886. doi: 10.1021/ja100795k. [DOI] [PubMed] [Google Scholar]; (d) Egami H, Katsuki T. Angew Chem, Int Ed. 2008;47:5171. doi: 10.1002/anie.200800797. [DOI] [PubMed] [Google Scholar]; (e) Tse MK, Dobler C, Bhor S, Klawonn M, Magerlein W, Hugl H, Beller M. Angew Chem, Int Ed. 2004;43:5255. doi: 10.1002/anie.200460528. [DOI] [PubMed] [Google Scholar]
- 22.Thiel WR. Coord Chem Rev. 2003;245:95. [Google Scholar]
- 23.(a) Kubota T, Miyazaki H. Bull Chem Soc Jpn. 1962;35:1549. [Google Scholar]; (b) Emerson TK, Rees CW. J Chem Soc. 1962:1923. [Google Scholar]
- 24.Vogt C, Rabenstein A, Rethmeier J, Fischer U. Microbiology. 1997;143:767. doi: 10.1099/00221287-143-3-767. [DOI] [PubMed] [Google Scholar]
- 25.Coperet C, Adolfsson H, Khuong TAW, Yudin AK, Sharpless KB. J Org Chem. 1998;63:1740. [Google Scholar]
- 26.Manganese tetrakis(2,6-dichlorophenyl)porphyrin chloride.
- 27.Thellend A, Battioni P, Sanderson W, Mansuy D. Synthesis. 1997:1387. [Google Scholar]
- 28.(a) Van der Made AW, Hoppenbrouwer EJH, Nolte RJM, Drenth W. Recl Trav Chim Pays-Bas. 1988;107:15–16. [Google Scholar]; (b) Naik R, Joshi P, Kaiwar SP, Deshpande RK. Tetrahedron. 2003;59:2207–2213. [Google Scholar]; (c) Jain N, Kumar A, Chauhan SMS. Synth Commun. 2005;35:1223–1230. [Google Scholar]
- 29.(a) Kozhevnikov IV. Chem Rev. 1998;98:171–198. doi: 10.1021/cr960400y. [DOI] [PubMed] [Google Scholar]; (b) Katsoulis DE. Chem Rev. 1998;98:359–388. doi: 10.1021/cr960398a. [DOI] [PubMed] [Google Scholar]
- 30.Sloboda-Rozner D, Witte P, Alsters PL, Neumann R. Adv Synth Catal. 2004;346:339–345. [Google Scholar]
- 31.(a) Bamoharram FF, Heravi MM, Roshani M, Abrishami F. J Mol Catal A: Chem. 2007;267:241–244. [Google Scholar]; (b) Bamoharram FF, Heravi MM, Roshani M, Tavakoli N. J Mol Catal A: Chem. 2006;252:219–225. [Google Scholar]
- 32.(a) Zhao W, Wang X, Yang C. Synth Commun. 2014;44:150–160. [Google Scholar]; (b) Zhao W, Yang C, Ding Y, Ma BC. New J Chem. 2013;37:2614–2618. [Google Scholar]
- 33.Zhao W, Yanga C. New J Chem. 2013;37:1867–1870. [Google Scholar]
- 34.Ding Y, Zhao W. J Mol Catal A: Chem. 2011;337:45–51. [Google Scholar]
- 35.Ding Y, Zhao W, Song W, Zhang Z, Ma B. Green Chem. 2011;13:1486–1489. [Google Scholar]
- 36.Amir E, Rozen S. Chem Commun. 2006:2262–2264. doi: 10.1039/b602594c. [DOI] [PubMed] [Google Scholar]
- 37.Blaszczak-Swiatkiewicz K, Mirowski M, Kaplinska K, Kruszynski R, Trzesowska-Kruszynska A, Mikiciuk-Olasik E. Acta Biochim Pol. 2012;59:279–288. [PubMed] [Google Scholar]
- 38.Wittena ML, Sheppard PR. Chem Biol Interact. 2012;96:87–88. doi: 10.1016/j.cbi.2011.12.002. [DOI] [PubMed] [Google Scholar]
- 39.(a) Griffith WP, Slawin AMZ, Thompson KM, Williams DJ. J Chem Soc, Chem Commun. 1994:569–570. [Google Scholar]; (b) Sensato FR, Cass QB, Longo E, Zukerman-Schpector J, Custodio R, Andrés J, Zaldini Hernandes M, Longo RL. Inorg Chem. 2001;40:6022–6025. doi: 10.1021/ic010020h. [DOI] [PubMed] [Google Scholar]; (c) Herbert M, Montilla F, Galindo A, Moyano R, Pastor A, Alvarez E. Dalton Trans. 2011;40:5210–5219. doi: 10.1039/c1dt10065c. [DOI] [PubMed] [Google Scholar]
- 40.(a) Mimoun H, de Roch IS, Sajus L. Bull Soc Chim Fr. 1964:1481. [Google Scholar]; (B) Mimoun H, de Roch IS, Sajus L. Tetrahedron. 1970;26:37. [Google Scholar]
- 41.Hastings TJ, Frediani HA. Anal Chem. 1948;20:382–383. [Google Scholar]
- 42.Chauhan SMS, Kalra B, Mohapatra PP. J Mol Catal A: Chem. 1999;137:85. [Google Scholar]
- 43.Salles L, Aubry C, Robert F, Chottard G, Thouvenot R, Ledon H, Bregeault JM. New J Chem. 1993;17:367. [Google Scholar]
- 44.Katritzky AR, Pilarski B, Urogdi L. Synthesis. 1989:949–950. [Google Scholar]
- 45.Aitken RA, Fodi B, Palmer MH, Slawin AMZ, Yang J. Tetrahedron. 2012;68:5845–5851. [Google Scholar]
- 46.Dongre R, Sheikh J, Ghugal D, Ali P, Meshram J, Ramteke DS. Indian J Chem Sect B: Org Chem Incl Med Chem. 2012;51:374–379. [Google Scholar]
- 47.Keith JM. J Org Chem. 2007;73:327–330. doi: 10.1021/jo702038g. [DOI] [PubMed] [Google Scholar]
- 48.Kokatla HP, Thomson PF, Bae S, Doddi VR, Lakshman MK. J Org Chem. 2011;76:7842–7848. doi: 10.1021/jo201192c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Landa A, Minkkilä A, Blay G, Jørgensen KA. Chem – Eur J. 2006;12:3472–3483. doi: 10.1002/chem.200501475. [DOI] [PubMed] [Google Scholar]
- 50.Boykin DW, Balakrishnan P, Baumstark AL. J Heterocycl Chem. 1985;22:981–984. [Google Scholar]
- 51.Petit A, Flygare J, Miller AT, Winkel G, Ess DH. Org Lett. 2012;14:3680–3683. doi: 10.1021/ol301521n. [DOI] [PubMed] [Google Scholar]
- 52.Dega-Szafran Z, Szafran M, Sitkowski J, Stefaniak L. J Phys Org Chem. 1996;9:746–750. [Google Scholar]
- 53.Couturier M, Caron L, Tumidajski S, Jones K, White TD. Org Lett. 2006;8:1929–1932. doi: 10.1021/ol060473w. [DOI] [PubMed] [Google Scholar]
- 54.Rong D, Phillips VA, Rubio RS, Angeles Castro M, Wheelhouse RT. Tetrahedron Lett. 2008;49:6933–6935. [Google Scholar]
- 55.Antkowiak R, Antkowiak W. Heterocycles. 1998;47:893–909. [Google Scholar]
- 56.Nakashima Y, Fukunaga M, Suzuki K, Takahashi K. Bull Chem Soc Jpn. 1993;66:2143–2148. [Google Scholar]
- 57.Gong X, Song G, Zhang H, Li X. Org Lett. 2011;13:1766–1769. doi: 10.1021/ol200306y. [DOI] [PubMed] [Google Scholar]
- 58.Yang K, Qiu Y, Li Z, Wang Z, Jiang S. J Org Chem. 2011;76:3151–3159. doi: 10.1021/jo1026035. [DOI] [PubMed] [Google Scholar]
- 59.Eliseeva SV, Ryazanov M, Gumy F, Troyanov SI, Lepnev LS, Bünzli JCG, Kuzmina NP. Eur J Inorg Chem. 2006;23:4809–4820. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.