

Abstract
The scope and mechanistic implications of the direct transformation of heterocyclic N-oxides to 2-trifluoromethyl-, and related perfluoroalkyl- and perfluoroaryl-substituted N-heterocycles has been studied. The reaction is effected by perfluoroalkyl- and perfluorophenyltrimethylsilane in the presence of strong base. In situ displacement of the para-fluoro substituent in the pentafluorophenyl ring and the methoxy group in 8-methoxyquinolines with additional nucleophiles allows for further site-selective refunctionalization of the N-heterocyclic products.
Introduction
Aromatic compounds bearing fluorine-containing alkyl and aryl substituents are important structural motifs of numerous bioactive compounds1 and advanced functional materials.2 Due to the unique electronic properties of fluorine, introduction of fluorinated alkyl groups can significantly improve metabolic stability, lipophilicity and binding properties of small organic molecules. As a consequence of the central position of N-heterocycles in medicinal and agricultural chemistry, introduction of trifluoromethyl and related groups into the N-heteroarene core has been successfully employed for the development of a number of drugs and agrochemicals (Figure 1). Hence, regioselective trifluoromethylation reactions of N-heterocycles have recently attracted significant attention.3 In particular, introduction of trifluoromethyl and difluoromethyl substituents in the C2-position of pyridines, quinolines and related six-membered nitrogenous aromatic heterocycles is a potentially useful synthetic strategy that continues to pose a significant methodological challenge.4
Fig. 1.

Representative bioactive N-heterocycles bearing trifluoro- and difluoromethyl groups.
Heterocyclic N-oxides are important and readily available5 synthetic intermediates that have recently been used as substrates for the regioselective transition metal-catalyzed preparation of various N-heterocycles bearing aryl-, alkyl- and heteroatom-substituents in the C2 position.6 In addition, a number of non-catalytic transformations, such as halogenations7 and aminations,8 have also been developed.
Trimethyl(trifluoromethyl)silane (1) and related perfluoroalkyl/arylsilanes are among the most cost-effective sources of the corresponding fluorinated alkyl and aryl groups for organic synthesis.9 However their application is sometimes hindered by the unproductive α-elimination of the fluoride and other side processes, and a careful selection of reaction conditions is required to achieve high yields.10
We herein report a study of the reaction of heterocyclic N-oxides with fluorinated alkylsilanes and trimethyl(pentafluorophenyl)-silane that directly affords 2-trifluoromethyl- and related substituted N-heterocycles regioselectively and in good to excellent yields. A survey of the patent literature shows that, although a reaction of 1 with two substituted quinoline N-oxides was used to access the corresponding 2-trifluoromethylquinolines,11 the scope and utility of this transformation were not systematically explored, and potential use of other perfluoroalkylsilanes was not investigated, which prevented its wider application in the mainstream synthetic organic, medicinal and materials chemistry. Recently, Kuninobu, Kanai and co-workers conducted an elegant study on the reaction of 1 with heterocyclic N-oxides that is mediated by a combination of cesium fluoride and (trifluoromethyl)difluoroboron as a Lewis acidic N-oxide activator.12 However, this method is limited to trifluoromethylation, and, in addition, the (trifluoromethyl)difluoroboron has to be generated from expensive potassium (trifluoromethyl)trifluoroborate. Another interesting approach to C2-trifluoromethylation of azines was described by Makosza and co-workers.13 In this case azines were first transformed into the corresponding N-(p-methoxybenzyl)azinium salts that were reacted with silane 1 in the presence of potassium fluoride. Treatment of the addition products with cerium ammonium nitrate led to removal of the PMB group and oxidative aromatization to give 2-trifluoromethyalted azines.
We also describe several unexpected concomitant dual substitution processes that can be used to increase structural diversity of the N-heterocyclic motifs. In particular, a facile substitution of 8-methoxy group in the quinoline core by tert-butoxide represents an unusual example of aromatic nucleophilic substitution.
Results and Discussion
We began our study by examining the reaction between quinoline N-oxide (2) and silane 1 (Table 1) in the presence of several fluoride salts and bases. While formation of product 3 (11%) was observed upon reacting 1 and 2 in the presence of 0.2 equiv of TBAF in THF, the reaction was accompanied by formation of quinoline in comparable amounts. The yield of 3 was improved in toluene/THF (1:1), albeit with concomitant increase in formation of quinoline (entry 2).
Table 1.
Optimization of the reaction conditions.a
| |||||
|---|---|---|---|---|---|
| Entry | Base/fluoride (equiv) | Solvent | T (°C) | 2 : 3 : quinolineRatiob | Yieldb of 3, % |
| 1 | TBAF (0.2) | THF | 23 | 7 : 1 : 1 | 11 |
| 2 | TBAF (0.2) | PhCH3/THF (1:1) | 23 | 1.7 : 1.5 : 1 | 34 |
| 3 | TBAF (0.2) | PhCH3/THF (1:1) | 65 | 10 : 1 : 1.5 | 8 |
| 4 | TASF (1.1) | PhCH3/THF (1:1) | 23 | 2 : 2.5 : 1 | 44 |
| 5 | CsF (1.1) | PhCH3/THF (1:1) | 23 | 1.6 : 1.7 : 1 | 34 |
| 6c | TASF (1.1) | PhCH3/THF (1:1) | 23 | 1.3 : 2.5 : 1 | 50 |
| 7d | TASF (1.1) | THF | 23 | 1.5 : 2.1 : 1 | 44 |
| 8 | KOt-Bu (1.1) | THF | 23 | 1 : 6.8 : 17.3 | 26 |
| 9 | KOt-Bu (3) | THF | 23 | 1 : 3.2 : 5.8 | 32 |
| 10e | KOt-Bu (3) | THF | −20 | 1 : 29 : 3.2 | 87 |
Reaction conditions: 2 (0.35 or 1 mmol), 1 (2.5 equiv), in the solvent (c=0.7 M) under Ar for 2 h.
Determined by 1H NMR.
(CuOTf)2·PhH (5 mol %) was added.
MgCl2 (2 equiv) was added.
c=0.2 M.
No improvement was observed at elevated temperatures (e.g. entry 3). Further increase in yields of 3 was achieved with TASF [tris(dimethylamino)sulfonium difluorotrimethylsilicate] and cesium fluoride as fluoride sources. Both salts also gave higher 3/quinoline. Attempts to catalyze the reaction by transition metal salts (e.g. copper(I) triflate) or increase the electrophilicity of the N-oxide via coordination to a Lewis acid (e.g. MgCl2) proved unsuccessful (entries 6 and 7). On the other hand, use of potassium tert-butoxide (1.1 equiv) led to faster conversion, however with concomitant acceleration of the formation of quinoline. Further optimization was achieved by increasing the amount of the base and lowering the temperature to −20 °C. This way the formation of quinoline was effectively minimized, and a clean conversion to product 3 took place.
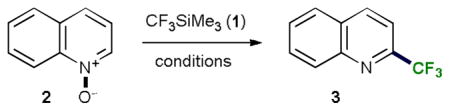
After completion of the optimization study we set out to explore the scope of the novel trifluoromethylation method with other heterocyclic N-oxides (Table 2).
Table 2.
Scope of the trifluoromethylation reaction of heterocyclic N-oxides.a
|
Reaction conditions: heterocyclic N-oxide (0.2 or 0.5 mmol), 1 (1.5 equiv), KOt-Bu (3 equiv), 3Å MS, THF (c=0.2 M), at −20 °C under Ar for 50 min.
The reaction generally exhibits a broad scope of applicable substrates. Thus, silane 1 reacted readily with substituted quinoline, pyridine, phenanthridine and isoquinoline N-oxides giving rise to the corresponding products in a highly regioselective fashion. In none of the cases were any other regioisomers observed or isolated. Halogenated substrates are well tolerated. N-Oxides that have a labile 4-thio and 4-oxy substituents that are known to be prone to facile nucleophilic displacement of the C4-group,14 afford the desired 2-trifluoromethylquinolines in good yields. In general, bi- and tricyclic N-heterocyclic N-oxides reacted more readily and gave the products in higher yields, while 4-phenylpyridine N-oxide was less reactive and afforded the trifluoromethylated product 10 in a 28% yield. The reaction is best carried out at −20 °C for all substrates.
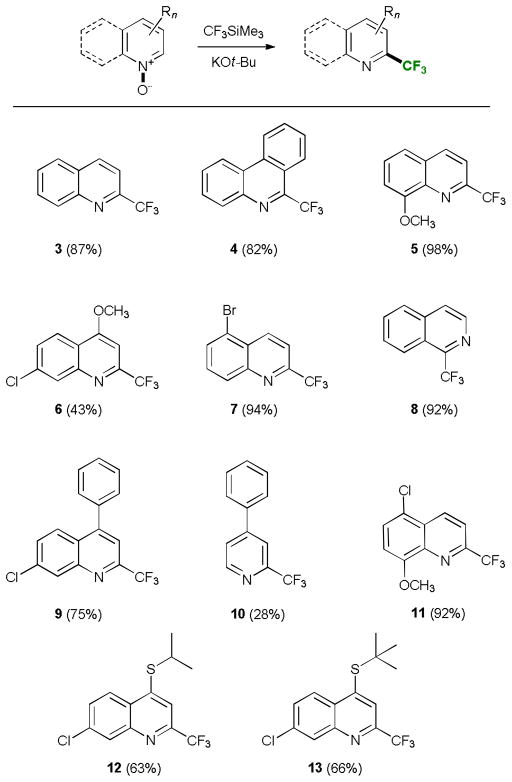
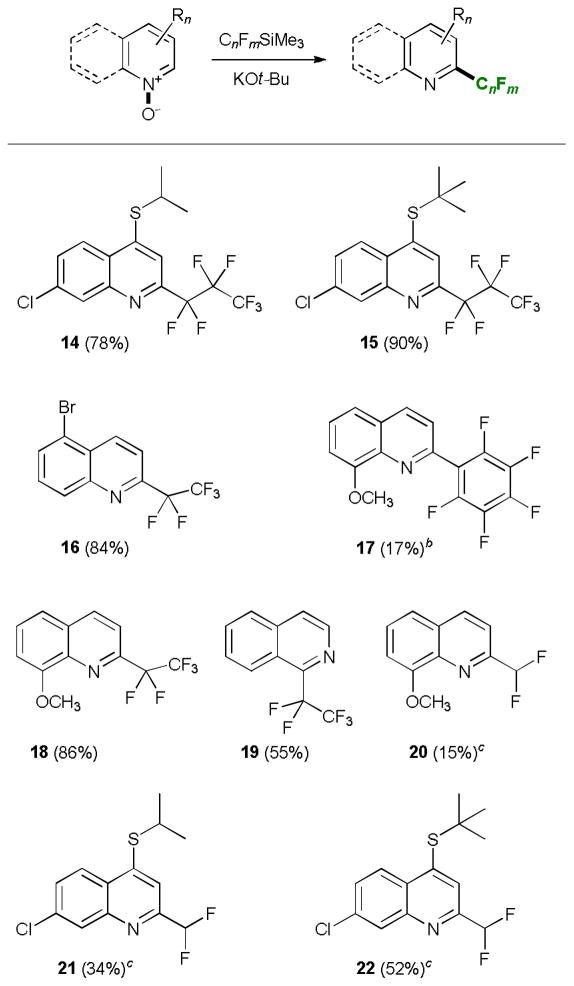
We have also studied the performance of other fluorinated alkylsilanes in the base-mediated reaction with N-oxides (Table 3). In general, both (pentafluoroethyl)- and (heptafluoropropyl)-trimethylsilane proved to be very efficient perfluoroalkylating reagents. The products were obtained in good to excellent yields under the optimized conditions at −20 °C. (Difluoromethyl)trimethylsilane, on the other hand, was less efficient, with somewhat lower yields of the products and higher loadings of the silane and potassium tert-butoxide. Trimethyl(pentafluorophenyl)-silane afforded the desired product 17 with one equivalent of potassium tert-butoxide in 17% yield. When the previously optimized amount of the base (3 equiv) was used an unexpected displacement of the fluoride in the para-position with respect to the N-heterocyclic residue by tert-butoxide took place (Table 4). The reaction also worked well with sodium thiophenolate and sodium ethanethiolate as nucleophiles. This reactivity is in line with previously reported observations of facile and regioselective nucleophilic para-substitution of the fluoride in other pentafluoroarenes.15
Table 3.
Reaction of perfluoroalkyl/phenylsilanes with heterocyclic N-oxides.a
|
Reaction conditions: heterocyclic N-oxide (0.2–0.5 mmol), CnFmSiMe3 (1.5 equiv), KOt-Bu (3 equiv), 3Å MS, THF (c=0.2 M), at −20 °C under Ar for 50 min.
1 equiv KOt-Bu was used.
CHF2SiMe3 (5 equiv), KOt-Bu (4 equiv), THF (c=0.1 M) was used.
Table 4.
Dual substitution reaction of heterocyclic N-oxides with (pentafluorophenyl)trimethylsilane and additional nucleophiles.a
|
Reaction conditions: heterocyclic N-oxide (0.2 mmol), C6F5SiMe3 (1.5 equiv), KOt-Bu (3 equiv), 3Å MS, THF (c=0.1 M), at −20 °C under Ar for 50 min.
NaSPh (6 equiv) was added.
NaSC2H5 (6 equiv) was added.
Another unusual substitution reaction was observed with 5,7-dichloro-8-methoxyquinoline N-oxide (27). In this case, a facile substitution of the 8-methoxy group by the tert-butoxide took place with concomitant perfluoroalkylation (Scheme 1).
Scheme 1.
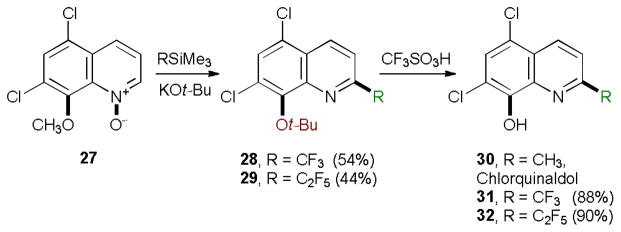
Dual substitution reaction of 8-methoxyquinoline N-oxides with perfluoroalkylsilanes.
Substitution of the 8-methoxy group in the 8-alkoxyquinoline series has previously been achieved only at elevated temperatures,16 and in the presence of a transition-metal catalyst.17
It is possible that introduction of the electron-withdrawing 2-perfluoroalkyl group in the quinoline facilitates the nucleophilic aromatic substitution. The reaction takes place after installation of the perfluoroalkyl group, since no 8-methoxy group displacement as observed when N-oxide 27 was treated with potassium tert-butoxide in the absence of 1 under the standard reaction conditions.
The reaction was exploited for the synthesis of new analogues of antimicrobial drug chlorquinaldol (30). The tert-butyl group removal with triflic acid afforded chlorquinaldol derivatives 31 and 32 in 88 and 90% yields.
Based on the previous studies18,10b it is plausible that tert-butoxide activates silane 1 by forming a pentacoordinated silicon nucleophile that subsequently binds to the oxygen atom of the N-oxide. The rate-limiting transfer of the trifluoromethyl group to C2 is then followed by fast deprotonation and elimination of the trimethylsiloxide. This rationale is supported by a relatively small kinetic isotope effect (kH/kD = 1.2) in the reaction of isoquinoline N-oxide (33) with 1 (Scheme 2).
Scheme 2.
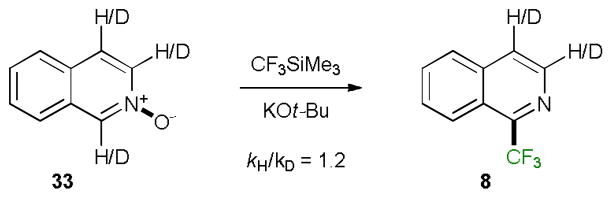
Kinetic isotope effect in the trifluoromethylation reaction of isoquinoline N-oxide.
Conclusions
In conclusion, we have investigated the scope and synthetic utility of a reaction of heterocyclic N-oxides and perfluoroalkylsilanes that affords 2-perfluoroalkyl/aryl-substituted nitrogenous heterocycles in good to excellent yields. Potassium tert-butoxide has been identified as the base of choice for the nucleophilic activation of the silane. The reported transformation of heterocyclic N-oxides to 2-trifluoromethyl N-heterocycles and related compounds can be used to access fluorinated heterocyclic scaffolds of practical synthetic and medicinal value, as demonstrated by the synthesis of two new fluorine-containing analogues of the antimicrobial drug chlorquinaldol. In addition, small kinetic isotope effect points toward the addition of the trifluoromethyl anion as the rate-determining step that is followed by fast elimination.
Experimental section
General methods
Tetrahydrofuran was distilled from sodium benzophenone ketyl. Isoquinoline-N-oxide was purchased from Alfa Aesar, 4-phenylpyridine N-oxide, (pentafluorophenyl)trimethylsilane, and (pentafluoroethyl)trimethylsilane were purchased from TCI. (Trifluoromethyl)trimethylsilane was purchased from Matrix Scientific, and (difluoromethyl)trimethylsilane was purchased from Oakwood Chemicals. All other chemicals were used as commercially available (Sigma-Aldrich, Acros, Alfa Aesar, Combi-Blocks, Strem). All reactions were conducted with continuous magnetic stirring under an atmosphere of argon in oven-dried glassware. Low-temperature experiments were conducted using a Neslab Cryotrol CB-80 cryostat. Reactions were monitored by TLC until deemed complete using silica gel-coated glass plates (Merck Kieselgel 60 F254). Plates were visualized under ultraviolet light (254 nm). Column chromatography was performed using CombiFlash Rf-200 (Teledyne-Isco) automated flash chromatography system with RediSep columns. 1H, 13C and 19F NMR spectra were recorded at 300 and 500 (1H), and 75.5, 125 MHz, and 282 MHz (19F) on Varian Mercury VX 300 and Agilent Inova 500 instruments in CDCl3 solutions if not otherwise specified. Chemical shifts (δ) are reported in parts per million (ppm) from the residual solvent peak and coupling constants (J) in Hz. Infrared measurements were carried out neat on a Bruker Vector 22 FT-IR spectrometer fitted with a Specac diamond attenuated total reflectance (ATR) module.
General procedure (GP1) for the reaction of heterocyclic N-oxides with perfluoroalkyl- and pentafluorophenylsilanes
To a stirred solution of heterocyclic N-oxide (0.2 mmol), 3Å molecular sieves (25 mg), and trimethyl(trifluoromethyl)silane (45 μL, 0.3 mmol, 1.5 equiv.) in tetrahydrofuran (1 mL) at −20 °C was added potassium tert-butoxide (66 mg, 0.6 mmol, 3 equiv.) in 1 equiv. portions every 10 min. The reaction was allowed to stir an additional 10 min before being diluted with a saturated aqueous solution of ammonium chloride (2 mL) and the aqueous layer extracted with dichloromethane (3 × 5 mL). The combined organic layers were dried over anhydrous sodium sulfate, concentrated under reduced pressure, and purified by column chromatography [hexanes/EtOAc/silica gel] to yield the desired product.
Experimental procedures and spectroscopic data
2-(Trifluoromethyl)quinoline13 (3)
According to GP1, 2 (50 mg, 0.34 mmol), 3Å molecular sieves (50 mg), and trimethyl(trifluoromethyl)silane (76 μL, 0.51 mmol, 1.5 equiv.) in tetrahydrofuran (1.8 mL) at −20 °C was added potassium tert-butoxide (116 mg, 1.03 mmol, 3 equiv.) in 3 poritions over 30 min. After 10 min the reaction was worked up and the crude product purified by column chromatography to yield 2-(trifluoromethyl)quinoline (3) (59 mg, 87%). – 1H NMR (500 MHz): 7.69 (1 H, td, J = 1, 7 Hz), 7.75 (1 H, d, J = 8.5 Hz), 7.84 (1 H, td, J = 1.5, 7 Hz), 7.92 (1 H, dd, J = 1.5, 8.5 Hz), 8.25 (1 H, dd, J = 1, 8.5 Hz), 8.37 (1 H, d, J = 8.5 Hz) ppm. – 13C NMR (125 MHz): 116.7, 116.8, 127.7, 128.6, 130.1, 130.8, 138.1, 147.2 ppm. – 19F NMR (282 MHz): −67.4 ppm. – IR: 910, 1087, 1128, 1214, 1274, 1303, 1431, 2930, 3063 cm−1.
6-(Trifluoromethyl)phenanthridine14 (4)
According to GP1, phenanthridine N-oxide (39 mg, 0.20 mmol), 3Å molecular sieves (25 mg), and trimethyl(trifluoromethyl)silane (40 μL, 0.22 mmol, 1.1 equiv.) at −20 °C was reacted with potassium tert-butoxide (66 mg, 0.60 mmol, 3 equiv.) in 3 portions over 30 min. The reaction was allowed to stir an additional 10 min before being worked up and purified by column chromatography to yield phenanthridine 4 (41 mg, 82 %). – 1H NMR (500 MHz): 7.79 – 7.85 (3 H, m), 7.96 (1 H, dt, J = 1.5 Hz, 8.5 Hz), 8.32 (1 H, dd, J = 1.5, 7 Hz), 8.41 (1 H, dd, J = 0.5, 8.5 Hz), 8.65 (1 H, dd, J = 1, 7 Hz), 8.74 (1 H, d, J = 8.5 Hz) ppm. – 13C NMR (75 MHz): 120.1, 121.8, 122.1, 122.6, 125.1, 125.9, 125.9, 126.0, 128.1, 128.3, 129.2, 129.4, 131.1, 131.4, 134.0, 141.7 ppm. – 19F NMR (282 MHz): −63.5 ppm. – IR: 1179, 1395, 1418, 1506, 2988, 3026 cm−1.
8-Methoxy-2-(trifluoromethyl)quinoline15 (5)
According to GP1, to a stirred solution of 8-methoxyquinoline N-oxide (35 mg, 0.20 mmol) and trimethyl(trifluoromethyl)silane (45 μL, 0.30 mmol, 1.50 equiv.) in tetrahydrofuran (1 mL) was added potassium tert-butoxide (66 mg, 0.60 mmol, 3 equiv.) in 3 portions over 30 min. After 10 min the reaction was worked up and the crude product purified by column chromatography to yield quinoline 5 (45 mg, 98%). – 1H NMR (500 MHz): 4.12 (3 H, s), 7.15 (1 H, d, J = 7.5 Hz), 7.48 (1 H, d, J = 8 Hz), 7.61 (1 H, t, J = 8 Hz), 7.77 (1 H, d, J = 8.5 Hz), 8.34 (1 H, d, J = 8.5 Hz) ppm. – 13C NMR (125 MHz): 56.4, 109.2, 118.5, 119.2, 129.1, 130.0, 137.7, 139.5, 145.8, 136.2 (d), 156.0 ppm. – 19F NMR (282 MHz): −67.0 ppm. – IR: 1067, 1110, 1213, 1259, 1329, 1432, 1445, 1561, 2937, 3046 cm−1.
7-Chloro-4-methoxy-2-(trifluoromethyl)quinoline (6)
According to GP1, to a stirred solution of 7-chloro-4-methoxyquinoline N-oxide (42 mg, 0.20 mmol) and trimethyl(trifluoromethyl)silane (45 μL, 0.30 mmol, 1.5 equiv.) in tetrahydrofuran (1 mL) was added potassium tert-butoxide (66 mg, 0.60 mmol, 3 equiv.) in 3 portions over 30 min. After 10 min the reaction was worked up and the crude product purified by column chromatography to yield quinoline 6 (23 mg, 43 %). – 1H NMR (500 MHz): 4.14 (3 H, s), 7.05 (1 H, s), 7.57 (1 H, dd, J = 1, 8 Hz), 8.14–8.20 (2 H, m) ppm. – 13C NMR (125 MHz): 56.3, 96.4, 120.1, 120.2, 122.4, 123.4, 128.5, 137.1, 148.6, 150.1, 150.4, 163.9 ppm. – 19F NMR (282 MHz): −68.0 ppm. – IR: 1110, 1215, 1325, 1448, 2988, 2999, 3026 cm−1. – MS (ESI): 261.0 [M], HRMS: 262.0449, calcd: 262.0241 [M+H+].
5-Bromo-2-(trifluoromethyl)quinoline16 (7)
According to GP1, 5-bromoquinoline N-oxide (111 mg, 0.50 mmol), 3Å molecular sieves (100 mg), and trimethyl(trifluoromethyl)silane (115 μL, 0.75 mmol, 1.5 equiv.) at −20 °C were reacted with potassium tert-butoxide (56 mg, 1.50 mmol, 3 equiv.) in 3 portions over 30 min. After 10 min the reaction was worked up and the crude product purified by column chromatography to yield quinoline 7 (123 mg, 94 %). – 1H NMR (300 MHz): 7.68 (1 H, dt, J = 2, 8 Hz), 7.83 (1 H, dd, J = 2, 9.5 Hz), 7.95 (1 H, d, J = 8 Hz), 8.20 (1 H, d, J = 8 Hz), 8.75 (1 H, dd, J = 2, 9.5 Hz) ppm. – 13C NMR (125 MHz): 117.9, 120.2, 121.8, 122.4, 124.6, 130.0, 132.2, 137.9, 147.9, 148.5 (quart., J = 2.5 Hz) ppm. – 19F NMR (282 MHz): −67.6 ppm. – IR: 1102, 1207, 1321, 1357, 1434, 1514, 2931, 2970, 3025 cm−1.
1-(Trifluoromethyl)isoquinoline17 (8)
According to GP1, isoquinoline-N-oxide (207 mg, 1.42 mmol), 3Å molecular sieves (100 mg), and trimethyl(trifluoromethyl)silane (315 μL, 2.13 mmol, 1.5 equiv.) at −20 °C were reacted with potassium tert-butoxide (477 mg, 4.26 mmol, 3 equiv.) in 3 portions over 30 min. After 10 min the reaction worked up and the crude product purified by column chromatography to yield isoquinoline 8 (259 mg, 92 %). – 1H NMR (500 MHz): 7.71 (1 H, dt, J = 2, 8.5 Hz), 7.78 (1 H, t, J = 8 Hz), 7.84 (1 H, d, J = 5.5 Hz), 7.92 (1 H, d, J = 8 Hz), 8.30 (1 H, d, J = 8.5 Hz), 8.59 (1 H, d, J = 5.5 Hz) ppm. – 13C NMR (125 MHz): 124.6 (m), 127.5, 128.8, 130.9, 137.1, 140.7, 146.3 (quart., J = 33 Hz) ppm. – 19F NMR (282 MHz): −63.0 ppm. – IR: 1024, 1141, 1192, 1233, 1303, 1363, 1400, 1507, 2879, 2974, 3021 cm−1.
7-Chloro-4-phenyl-2-(trifluoromethyl)quinoline18 (9)
According to GP1, to a stirred solution of 7-chloro-4-phenylquinoline N-oxide (51 mg, 0.20 mmol) and trimethyl(trifluoromethyl)silane (45 μL, 0.30 mmol, 1.5 equiv.) in tetrahydrofuran (1 mL) was added potassium tert-butoxide (66 mg, 0.60 mmol, 3 equiv.) in 3 portions over 30 min. After 10 min the reaction was worked up and the crude product purified by column chromatography to yield quinoline 9 (46 mg, 75 %). – 1H NMR (300 MHz): 7.24 (1 H, d, J = 4 Hz), 7.45–7.56 (5 H, m), 7.88 (1 H, d, J = 5.5 Hz), 8.55 (1 H, d, J = 4 Hz), 8.85 (1 H, d, J = 1.5 Hz) ppm. – 13C NMR (125 MHz): 117.2, 120.3, 122.5, 125.9, 127.3, 128.3, 128.9, 1293, 129.3, 129.4, 129.6, 136.7 (d, J = 1 Hz), 148.2, 148.7 (m), 151.1 ppm. – 19F NMR (282 MHz): −67.7 ppm. – IR: 1099, 1167, 1266, 1384, 1448, 1502, 2878, 2938, 3041 cm−1.
4-Phenyl-2-(trifluoromethyl)pyridine19 (10)
According to GP1, 4-phenylpyridine N-oxide (35 mg, 0.20 mmol), 3Å molecular sieves (25 mg), and trimethyl(trifluoromethyl)silane (45 μL, 0.30 mmol, 1.5 equiv.) at −20 °C were reacted with potassium tert butoxide (66 mg, 0.60 mmol, 3 equiv.) in 3 portions over 10 min. After 10 min the reaction was worked up and the crude product purified by column chromatography to yield pyridine 10 (12 mg, 28 %). – 1H NMR (300 MHz): 7.45 – 7.57 (3 H, m), 7.63–7.71 (3 H, m), 7.90 (1 H, s), 8.78 (1 H, d, J = 5 Hz) ppm. – 13C NMR (75 MHz): 118.1 (m), 120.6 (m), 121.7, 123.8, 126.7, 123.8, 126.7, 129.0, 129.5, 136.5, 150.1 ppm. – 19F NMR (282 MHz): −68.0 ppm. – IR: 1023, 1149, 1184, 1261, 1336, 1460, 2854, 2926, 3026 cm−1.
5-Chloro-8-methoxy-2-(trifluoromethyl)quinoline (11)
According to GP1, to a stirred solution of 5-chloro-8-methoxyquinoline N-oxide (42 mg, 0.20 mmol) and trimethyl(trifluoromethyl)silane (45 μL, 0.30 mmol, 1.5 equiv.) in tetrahydrofuran (1 mL) was added potassium tert-butoxide (66 mg, 0.60 mmol, 3 equiv.) in 3 portions over 30 min. After 10 min the reaction was worked up and the crude product purified by column chromatography to yield quinoline 11 (48 mg, 92 %). – 1H NMR (500 MHz): 4.11 (3 H, s), 7.08 (1 H, dd, J = 2.5, 8.5 Hz), 7.68 (1 H, dd, J = 2, 8.5 Hz), 7.89 (1 H, dd, J = 2.5, 8.5 Hz), 8.75 (1 H, dd, J = 2, 8.5 Hz) ppm. – 13C NMR (125 MHz): 56.5, 108.8, 110.0, 118.3, 120.3, 122.5, 127.8, 128.6, 135.4, 139.7, 144.3 (d, J = 2 Hz), 155.1 ppm. – 19F NMR (282 MHz): −67.1 ppm. – IR: 1106, 1186, 1213, 1310, 1372, 1463, 1509, 2842, 2936, 3003 cm−1. – MS (ESI): 262.0, HRMS: 262.0300, calcd: 262.0241 [M+H+].
7-Chloro-4-(isopropylthio)-2-(trifluoromethyl)quinoline (12)
According to GP1, to a stirred solution of 7-chloro-4-(isopropylthio)quinoline N-oxide (51 mg, 0.20 mmol) and trimethyl(trifluoromethyl)silane (45 μL, 0.30 mmol, 1.5 equiv.) in tetrahydrofuran (1 mL) was added potassium tert-butoxide (66 mg, 0.6 mmol, 3 equiv.) in 3 portions over 30 min. After 10 min the reaction was worked up and the crude product purified by column chromatography to yield quinoline 12 (61 mg, 63 %). – 1H NMR (500 MHz): 1.52 (6 H, d, J = 7 Hz), 3.79 (1 H, sept., J = 7 Hz), 7.51 (1 H, s), 7.60 (1 H, dd, J = 2, 9 Hz), 8.14 (1 H, d, J = 9 Hz), 8.18 (1 H, d, J = 2 Hz) ppm. – 13C NMR (125 MHz): 22.6, 36.1, 112.4, 120.3, 122.4, 125.0, 125.7, 129.0, 129.5, 137.0, 147.0, 138.0 (d), 150.8 ppm. – 19F NMR (282 MHz): −67.9 ppm. – IR: 1027, 1105, 1197, 1238, 1340, 1560, 2956, 3010, 3345 cm−1. – MS (ESI): 305.8, HRMS: 306.2045, calcd: 306.0326 [M+H+].
4-(tert-Butylthio)-7-chloro-2-(trifluoromethyl)quinoline (13)
According to GP1, to a stirred solution of 4-(tert-butylthio)-7-chloroquinoline N-oxide (64 mg, 0.20 mmol) and trimethyl(trifluoromethyl)silane (45 μL, 0.30 mmol, 1.5 equiv.) in tetrahydrofuran (1 mL) was added potassium tert-butoxide (66 mg, 0.60 mmol, 3 equiv.) in 3 portions over 30 min. After 10 min the reaction was worked up and the crude product purified by column chromatography to yield quinoline 13 (42 mg, 66 %). – 1H NMR (500 MHz): 1.70 (9 H, s), 7.64 (1 H, dd, J = 2, 9 Hz), 7.91 (1 H, s), 8.23 (1 H, dd, J = 2 Hz), 8.56 (1 H, d, J = 9 Hz) ppm. – 13C NMR (75 MHz): 31.3, 49.5, 119.4, 123.2, 127.6, 129.3, 129.8, 129.9, 137.1, 146.4, 147.7 (m) ppm. – IR: 1144, 1213, 1345, 2899, 2987, 3033 cm−1. – MS (ESI): 319.8, HRMS: 319.3671, calcd: 320.0482 [M+H+].
7-Chloro-4-(isopropylthio)-2-(perfluoropropyl)quinoline (14)
According to GP1, to a stirred solution of 7-chloro-4-(isopropylthio)quinoline N-oxide (51 mg, 0.20 mmol), 3Å molecular sieves (50 mg), and trimethyl(heptafluoropropyl)silane (61 μL, 0.30 mmol, 1.5 equiv.) in tetrahydrofuran (1 mL) was added potassium tert-butoxide (67 mg, 0.60 mmol, 3 equiv.) in 3 portions over 30 min at −20 °C. After 10 min the reaction was worked up and the crude product purified by column chromatography to yield quinoline 14 (63 mg, 78 %). – 1H NMR (500 MHz): 7.49 (1 H, s), 7.61 (1 H, dd, J = 2, 9 Hz), 8.14 (1 H, d, J = 9 Hz), 8.20 (1 H, d, J = 2 Hz) ppm. – 13C NMR (125 MHz): 22.4, 36.1, 113.9, 125.0, 125.6, 128.3, 129.2, 129.7, 137.0, 147.1, 147.5, 150.4 ppm. – 19F NMR (282 MHz): −80.1, −114.9, −126.1 ppm. – IR: 1076, 1153, 1231, 1304, 1371, 1431, 1567, 2928, 2970, 3035 cm−1.
4-(tert-Butylthio)-7-chloro-2-(perfluoropropyl)quinoline (15)
According to GP1, to a solution of 4-(tert-butylthio)-7-chloroquinoline N-oxide (54 mg, 0.20 mmol), 3Å molecular sieves (50 mg), and trimethyl(heptafluoropropyl)silane (61 μL, 0.30 mmol, 1.5 equiv.) in tetrahydrofuran (1 mL) was added potassium tert-butoxide (67 mg, 0.60 mmol, 3 equiv.) in 3 portions over 30 min at −20 °C. After 10 min the reaction was worked up and the crude product purified by column chromatography to yield quinoline 15 (75 mg, 90 %). – 1H NMR (500 MHz): 1.42 (9 H, s), 7.65 (1 H, dd, J = 2, 9 Hz), 7.91 (1 H, s), 8.26 (1 H, d, J = 2 Hz), 8.58 (1 H, d, J = 9 Hz) ppm. – 13C NMR (125 MHz): 31.3, 49.5, 109.2 (m), 112.6 (m), 116.0 (m), 119.7 (m), 124.8, 127.6, 128.3, 129.5, 129.8, 129.9, 137.1, 146.0, 147.4 (m), 147.9 ppm. – 19F NMR (282 MHz): −80.1, −114.9, −126.2 ppm. – IR: 1063, 1122, 1191, 1231, 1305, 1408, 1558, 2901, 2970, 3060 cm−1. – MS (ESI): 457.9, HRMS: 457.9973, calcd: 457.9977 [M+K+].
5-Bromo-2-(perfluoroethyl)quinoline (16)
According to GP1, to a stirred solution of 5-bromoquinoline N-oxide (45 mg, 0.20 mmol), 3Å molecular sieves (50 mg), and trimethyl(pentafluoroethyl)silane (53 μL, 0.30 mmol, 1.5 equiv.) in tetrahydrofuran (1 mL) was added potassium tert-butoxide (67 mg, 0.60 mmol, 3 equiv.) in 3 portions over 30 min at −20 °C. After 10 min the reaction was worked up and the crude product purified by column chromatography to yield quinoline 16 (54 mg, 84 %). – 1H NMR (500 MHz): 7.69 (1 H, t, J = 7.5 Hz), 7.86 (1 H, d, J = 8.5 Hz), 7.97 (1 H, d, J = 7.5 Hz), 8.23 (1 H, d, J = 8.5 Hz), 8.76 (1 H, d, J = 8.5 Hz) ppm. – 13C NMR (125 MHz): 111.2 (d), 113.2 (d), 117.8 (t), 119.0, 120.1 (t), 121.8, 128.2, 130.2, 130.9, 132.3, 137.6, 148.0, 148.3 (t) ppm. – 19F NMR (282 MHz): −80.7, −114.8 ppm. – IR: 1069, 1122, 1199, 1254, 1319, 1417, 2956, 3006, 3042 cm−1. – MS (ESI): 326.0, HRMS: 325.9560, calcd: 325.9598 [M+H+].
8-Methoxy-2-(perfluorophenyl)quinoline (17)
According to GP1, to a stirred solution of 8-methoxyquinoline N-oxide (35 mg, 0.20 mmol), 3Å molecular sieves (50 mg), and trimethyl(pentafluorophenyl)silane (57 μL, 0.30 mmol, 1.5 equiv.) in tetrahydrofuran (1 mL) was added potassium tert-butoxide (23 mg, 0.20 mmol, 1 equiv.) in 3 portions over 30 min at −20 °C. After 10 min the reaction was worked up and the crude product purified by column chromatography to yield quinoline 17 (11 mg, 17 %). – 1H NMR (500 MHz): 4.10 (3 H, s), 7.13 (1 H, dd, J = 1, 8 Hz), 7.43 – 7.57 (3 H, m), 8.27 (1 H, d, J = 8.5 Hz) ppm. – 13C NMR (125 MHz): 56.3, 108.6, 119.3, 123.3, 124.9, 127.4, 128.0, 128.4, 128.7, 135.7, 136.8, 140.3, 145.7, 155.6 ppm. – 19F NMR (282): −161.2 (m), −153.0 (t), −140.1 (m) ppm. – IR: 1110, 1176, 1213, 1315, 1380, 1446, 1505, 2842, 2937, 3046 cm−1. – MS (ESI): 325.9, HRMS: 326.0649, calcd: 326.0599 [M+H+].
8-Methoxy-2-(perfluoroethyl)quinoline (18)
According to GP1, to a stirred solution of 8-methoxyquinoline N-oxide (35 mg, 0.20 mmol), 3Å molecular sieves (50 mg), and trimethyl(pentafluoroethyl)silane (53 μL, 0.30 mmol, 1.5 equiv.) in tetrahydrofuran (1 mL) was added potassium tert-butoxide (67 mg, 0.60 mmol, 3 equiv.) in 3 portions over 30 min at −20 °C. After 10 min the reaction was worked up and the crude product was purified by column chromatography to yield quinoline 18 (49 mg, 86 %). – 1H NMR (500 MHz): 4.10 (3 H, s), 7.15 (1 H, d, J = 8 Hz), 7.46 (1 H, d, J = 8.5 Hz), 7.61 (1 H, t, J = 8 Hz), 7.77 (1 H, d, J = 8.5 Hz), 8.32 (1 H, d, J = 8.5 Hz) ppm. – 13C NMR (125 MHz): 56.4, 109.2, 111.2 (d), 113.5 (d), 117.9 (t), 118.5, 119.2, 120.18 (t), 128.3, 129.1, 130.0, 137.7, 139.5, 146.0 (t), 156.0 ppm. – 19F NMR (282 MHz): –116.5 (t), −82.8 (quart.) ppm. – IR: 1213, 1314, 1445, 2899, 2985, 3025 cm−1. – MS (ESI): 277.9, HRMS: 278.054, calcd: 278.0599 [M+H+].
1-(Perfluoroethyl)isoquinoline20 (19)
According to GP1, to a stirred solution of isoquinoline-N-oxide (30 mg, 0.20 mmol), 3Å molecular sieves (50 mg), and trimethyl(pentafluoroethyl)silane (53 μL, 0.30 mmol, 1.5 equiv.) in tetrahydrofuran (1 mL) was added potassium tert-butoxide (67 mg, 0.60 mmol, 3 equiv.) in 3 portions over 30 min at −20 °C. After 10 min the reaction was worked up and the crude product purified by column chromatography to yield isoquinoline 19 (27 mg, 55 %). – 1H NMR (500 MHz): 7.73 (1 H, dt, J = 1, 6.5 Hz), 7.79 (1 H, dt, J = 1, 6.5 Hz), 7.87 (1 H, d, J = 5.5 Hz), 7.96 (1 H, d, J = 8 Hz), 8.40 (1 H, d, J = 8.5 Hz), 8.63 (1 H, d, J = 5.5 Hz) ppm. – 13C NMR (125 MHz): 111.5 (d), 113.5 (d), 115.5 (m), 118.1 (t), 120.4 (t), 124.4, 124.8, 125.9, 127.7, 128.8, 130.7, 137.2, 140.8, 146.3 (m) ppm. – 19F NMR (282 MHz): −81.2, −107.7 ppm. – IR: 1071, 1122, 1174, 1260, 1319, 1474, 2920, 2963, 3028 cm−1.
2-(Difluoromethyl)-8-methoxyquinoline (20)
According to GP1, to a stirred solution of 7-chloro-4-(isopropylthio)quinoline N-oxide (53 mg, 0.30 mmol), 3Å molecular sieves (50 mg), and trimethyl(difluoromethyl)silane (150 μL, 0.45 mmol, 5 equiv.) in tetrahydrofuran (3 mL) was added potassium tert-butoxide (135 mg, 0.90 mmol, 4 equiv.) in 3 portions over 30 min at −20 °C. After 30 min the reaction was worked up and the crude product purified by column chromatography to yield quinoline 20 (51 mg, 81 %). – 1H NMR (500 MHz): 3.89 (3 H, s), 4.77 (1 H, br s), 5.43 (1 H, dd, J = 2.5, 10 Hz), 5.69 – 5.95 (1 H, m), 6.63 – 6.76 (4 H, m) ppm. – 13C NMR (125 MHz): 55.6, 110, 111.2, 111.8, 111.9, 113.8, 113.9, 115.9, 117.5, 117.7, 119.9, 130.4, 131.9, 145.1 ppm. – 19F NMR (282 MHz): −132.8 (d) ppm. – IR: 1110, 1213, 1345, 1415, 1556, 2898, 3026 cm−1. – MS (ESI): 210.0, HRMS: 210.0721, calcd: 210.0725 [M+H+].
7-Chloro-2-(difluoromethyl)-4-(isopropylthio)quinoline (21)
According to GP1, to a stirred solution of 7-chloro-4-(isopropylthio)quinoline N-oxide (76 mg, 0.30 mmol), 3Å molecular sieves (50 mg), and trimethyl(difluoromethyl)silane (150 μL, 1.50 mmol, 5 equiv.) in tetrahydrofuran (3 mL) was added potassium tert-butoxide (134 mg, 1.20 mmol, 4 equiv.) in 3 portions over 30 min at −20 °C. After 30 min the reaction was worked up and the crude product purified by column chromatography to yield quinoline 21 (30 mg, 34 %). – 1H NMR (500 MHz): 1.36 (6 H, d, J = 6.5 Hz), 3.31 (1 H, sept., J = 6.5 Hz), 4.37 (1 H, br s), 5.45 (1 H, d, J = 2 Hz), 5.83 (1 H, m), 6.60 (1 H, d, J = 2 Hz), 6.71 (1 H, dd, J = 2, 8 Hz), 7.43 (1 H, d, J = 8 Hz) ppm. – 13C NMR (125 MHz): 22.6, 36.4, 110.8, 111.6, 113.1, 113.6, 115.6, 116.7, 118.9, 126.9, 136.0, 137.5, 141.8 ppm. – 19F NMR (282 MHz): −132.2 (dd) ppm. – IR: 1093, 1263, 1368, 1414, 2905, 2964, 3013 cm−1. – MS (ESI): 287.0, HRMS: 287.0167, calcd: 287.0347 [M+H+].
4-(tert-Butylthio)-7-chloro-2-(difluoromethyl)quinoline (22)
According to GP1, to a stirred solution of 4-(tert-butylthio)-7-chloroquinoline N-oxide (80 mg, 0.3 mmol), 3Å molecular sieves (50 mg), and trimethyl(difluoromethyl)silane (150 μL, 1.50 mmol, 5 equiv.) in tetrahydrofuran (3 mL) was added potassium tert-butoxide (134 mg, 1.20 mmol, 4 equiv.) in 3 portions over 30 min at −20 °C. After 30 min the reaction was worked up and the crude product purified by column chromatography to yield quinoline 22 (47 mg, 52 %). – 1H NMR (500 MHz): 1.35 (9 H, s), 4.34 (1 H, br s), 5.72–5.97 (2 H, m), 6.58 (1 H, d, J = 2 Hz), 6.72 (1 H, dd, J = 2, 8.5 Hz), 7.70 (1 H, d, J = 8 Hz) ppm. – 13C NMR (75 MHz): 31.3, 112.9, 113.4, 116.7, 118.6, 119.9, 122.2, 129.2, 131.0, 135.9, 141.8 ppm. – 19F NMR (282 MHz): −132.1 (d) ppm. – IR: 1093, 1213, 1368, 1475, 2995, 3025 cm−1. – MS (ESI): 302.1, HRMS: 302.0599, calcd: 302.0576 [M+H+].
8-Methoxy-2-(2,3,5,6-tetrafluoro-4-(phenylthio)phenyl)-quinoline (23)
According to GP1, to a stirred solution of 8-methoxyquinoline N-oxide (35 mg, 0.20 mmol), 3Å molecular sieves (50 mg), sodium thiophenolate (158 mg, 1.20 mmol, 6 equiv.), and trimethyl(pentafluorophenyl)silane (57 μL, 0.30 mmol, 1.5 equiv.) in tetrahydrofuran (2 mL) was added potassium tert-butoxide (67 mg, 0.60 mmol, 3 equiv.) in 3 portions over 30 min at −20 °C. After 10 min the reaction was worked up and the crude product purified by column chromatography to yield quinoline 23 (72 mg, 86 %). – 1H NMR (500 MHz): 4.10 (3 H, s), 7.14 (1 H, d, J = 7.5 Hz), 7.31 – 7.37 (5 H, m), 7.42 (1 H, d, J = 7.5 Hz), 7.45 (1 H, d, J = 8 Hz), 7.55 – 7.61 (1 H, m), 8.28 (1 H, d, J = 8.5 Hz) ppm. – 13C NMR (125 MHz): 56.3, 108.3 (m), 119.4, 123.3, 124.7, 127.2, 127.5, 127.8, 129.2, 129.4, 129.6, 130.4, 135.7, 135.7, 139.9, 146.3, 155.6 (m) ppm. – 19F NMR (282 MHz): −133.0 (dd), −142.3 (dd) ppm. – IR: 1111, 1260, 1323, 1379, 1479, 1561, 2874, 2942, 3024 cm−1. – MS (ESI): 415.8, HRMS: 416.0784, calcd: 416.0727 [M+H+].
2-(4-(tert-Butoxy)-2,3,5,6-tetrafluorophenyl)-8-methoxy-quinoline (24)
According to GP1, to a stirred solution of 8-methoxyquinoline N-oxide (35 mg, 0.20 mmol), 3Å molecular sieves (50 mg), and trimethyl(pentafluorophenyl)silane (57 μL, 0.3 mmol, 1.50 equiv.) in tetrahydrofuran (2 mL) was added potassium tert-butoxide (67 mg, 0.60 mmol, 3 equiv.) in 3 portions over 30 min at −20 °C. After 10 min the reaction was worked up and the crude product purified by column chromatography to yield quinoline 24 (63 mg, 83 %). – 1H NMR (500 MHz): 1.46 (9 H, s), 4.08 (3 H, s), 7.11 ( 1H, d, J = 7.5 Hz), 7.45 (1 H, d, J = 8.5 Hz), 7.54 (1 H, t, J = 7.5 Hz), 7.58 (1 H, d, J = 7.5 Hz), 8.25 (1 H, d, J = 8.5 Hz) ppm. – 13C NMR (125 MHz): 56.3, 108.6, 119.4, 123.4, 128.0, 128.3, 128.7, 136.8, 140.3, 145.7, 155.6 ppm. – 19F NMR (282 MHz): −140.1 (m), −161.3 (m) ppm. – IR: 1110, 1211, 1345, 1445, 2899, 3026 cm−1. – MS (ESI): 401.9 [M+Na+], HRMS: 380.0725, calcd: 380.1268 [M+H+].
2-(4-(tert-Butoxy)-2,3,5,6-tetrafluorophenyl)-5-chloro-8-methoxyquinoline (25)
According to GP1, to a stirred solution of 5-chloro-8-methoxyquinoline N-oxide (42 mg, 0.20 mmol), 3Å molecular sieves (50 mg), and trimethyl(pentafluorophenyl)silane (57 μL, 0.30 mmol, 1.5 equiv.) in tetrahydrofuran (2 mL) was added potassium tert-butoxide (67 mg, 0.60 mmol, 3 equiv.) in 3 portions over 30 min at −20 °C. After 10 min the reaction was worked up and the crude product purified by column chromatography to yield quinoline 25 (69 mg, 83 %). – 1H NMR (300 MHz): 1.45 (9 H, s), 4.08 (3 H, s), 7.03 (1 H, d, J = 9.5 Hz), 7.61 (1 H, d, J = 9.5 Hz), 7.71 (1 H, dt, J = 1.5, 9.5 Hz), 8.66 (1 H, d, J = 9.5 Hz) ppm. – 13C NMR (125 MHz): ppm. – 19F NMR (282 MHz): −140.5 (m), −151.5 (m) ppm. – IR: 1092, 1104, 1159, 1246, 1310, 1372, 1423, 1488, 2963, 2995, 3042 cm−1. – MS (ESI): 414.1, HRMS: 414.0907, calcd: 414.0878 [M+H+].
5-Bromo-2-(4-(ethylthio)-2,3,5,6-tetrafluorophenyl)quinoline (26)
According to GP1, to a stirred solution of 5-bromoquinoline N-oxide (45 mg, 0.20 mmol), 3Å molecular sieves (50 mg), sodium ethanethiolate (101 mg, 1.20 mmol, 6 equiv.), and trimethyl(difluoromethyl)silane (60 μL, 0.3 mmol, 1.50 equiv.) in tetrahydrofuran (2 mL) was added potassium tert-butoxide (90 mg, 0.8 mmol, 4 equiv.) in 3 portions over 30 min at −20 °C. After 10 min the reaction was worked up and the crude product purified by column chromatography to yield quinoline 26 (37 mg, 45 %). – 1H NMR (300 MHz): 1.33 (3 H, t, J = 4.5 Hz), 3.05 (1 H, quart., J = 4.5 Hz), 7.61 – 7.71 (2 H, m), 7.91 (1 H, dd, J = 1, 8.5 Hz), 8.15 (1 H, dd, J = 1, 9.5 Hz), 8.68 (1 H, d, J = 9.5 Hz) ppm. – 13C NMR (75 MHz): 15.1, 28.9, 121.9, 123.9, 127.0 (d), 129.7, 130.5, 131.4, 138.2 (d), 148.8 (m) ppm. – 19F NMR (282 MHz): −133.7 (dd), −143.2 (dd) ppm. – IR: 1110, 1213, 1333, 1415, 1554, 2989, 3014 cm−1. – MS (ESI): 416.0, HRMS: 415.9701, calcd: 415.9726 [M+H+].
5,7-Dichloro-8-methoxyquinoline 1-oxide (27)
To a stirred solution of 5,7-dichloro-8-methoxyquinoline (500 mg, 2.19 mmol) in dichloromethane (5 mL) was added meta-chloroperoxybenzoic acid (980 mg, 2.85 mmol, 1.3 equiv., 50 % solution in H2O) at 0 °C. After 12 h the reaction was diluted with a saturated aqueous solution of sodium thiosulfate/sodium carbonate (30 mL, 1:1), and the aqueous layer extracted with dichloromethane (4 × 10 mL). The combined organic layers were dried over anhydrous sodium sulfate, concentrated under reduced pressure, and purified by column chromatography to yield N-oxide 27 (341 mg, 64 %). – m.p.: 138–140 °C. – 1H NMR (500 MHz): 4.09 (3 H, s), 7.32–7.36 (1 H, m), 7.75 (1 H, s), 8.02 (1 H, d, J = 8.5 Hz), 8.48 (1 H, d, J = 8.5 Hz) ppm. – 13C NMR (125 MHz): 63.1, 121.5, 122.1, 127.3, 129.8, 130.0, 130.1, 138.9, 139.1, 148.4 ppm. – IR: 1098, 1225, 1392, 1558, 2880, 2938, 3012 cm−1. – MS (ESI): 244.0, HRMS: 243.9987, calcd: 243.9927 [M+H+].
8-(tert-Butoxy)-5,7-dichloro-2-(trifluoromethyl)quinoline (28)
According to GP1, to a stirred solution of 27 (49 mg, 0.20 mmol), 3Å molecular sieves (50 mg), and trimethyl(trifluoromethyl)silane (45 μL, 0.30 mmol, 1.5 equiv.) in tetrahydrofuran (1 mL) at − 20°C was added potassium tert-butoxide (66 mg, 0.60 mmol, 3 equiv.) in 3 portions over 30 min. After 3 h the reaction was worked up and the crude product purified by column chromatography to yield quinoline 28 (37 mg, 54 %). – 1H NMR (500 MHz): 1.57 (9 H, s), 7.80 (1 H, s), 7.82 (1 H, d, J = 8.5 Hz), 8.70 (1 H, d, J = 8.5 Hz) ppm. – 13C NMR (125 MHz): 29.7, 86.3, 102.5, 117.5, 122.5, 126.7, 130.1, 130.7, 135.7, 145.0, 148.2 (d, J = 2 Hz), 150.1 ppm. – 19F NMR (282 MHz): −67.4 ppm. – IR: 1102, 1189, 1262, 1339, 1443, 2953, 2988, 3028 cm−1. – MS (ESI): 376.0, HRMS: 375.9874, calcd: 375.9880 [M+K+].
8-(tert-Butoxy)-5,7-dichloro-2-(perfluoroethyl)quinoline (29)
According to GP1, to a stirred solution of 27 (49 mg, 0.20 mmol), 3Å molecular sieves (50 mg), and trimethyl(pentafluoroethyl)silane (53 μL, 0.30 mmol, 1.5 equiv.) in tetrahydrofuran (1 mL) was added potassium tert-butoxide (67 mg, 0.6 mmol, 3 equiv.) in 3 portions over 30 min at −20 °C. After 3 h the reaction was worked up and the crude product was purified by column chromatography to yield quinoline 29 (34 mg, 44 %). – 1H NMR (500 MHz): 1.57 (9 H, s), 7.80 (1 H, s), 7.82 (1 H, d, J = 8.5 Hz), 8.70 (1 H, d, J = 8.5 Hz) ppm. – 13C NMR (125 MHz): 29.6, 86.4, 110.0, 111.3 (m), 113.7, 125.5, 126.6, 130.2, 130.8, 135.5, 145.3, 147.9, 150.1 ppm. – 19F NMR (282 MHz): −82.9, −116.9 ppm. – IR: 1011, 1158, 1262, 1300, 1393, 1440, 1585, 2934, 2980, 3035 cm−1. – MS (ESI): 388.0, HRMS: 388.0566, calcd: 388.0289 [M+H+].
5,7-Dichloro-2-(trifluoromethyl)quinolin-8-ol (31)
To a solution of 28 (12 mg, 0.04 mmol) in dichloromethane (4 mL) at 23 °C was added trifluoromethanesulfonic acid (36 mg,0.24 mmol, 6 equiv.). After 12 h the reaction was diluted with a saturated aqueous solution of sodium bicarbonate (10 mL) and the aqueous layer extracted with dichloromethane (3 × 10 mL). The combined organic layers were dried over anhydrous sodium sulfate and concentrated under reduced pressure to yield chlorquinaldol analogue 31 (10 mg, 88 %). – 1H NMR (500 MHz): 7.76 (1 H, s), 7.92 (1 H, d, J = 9 Hz), 8.74 (1 H, d, J = 9 Hz) ppm. – 13C NMR (125 MHz): 104.5, 110.0, 117.2, 118.3, 121.0, 125.9, 130.6, 136.2, 137.6, 148.0 (m) ppm. – 19F NMR (282 MHz): −67.4 ppm. – IR: 908, 1104, 1187, 1316, 1351, 1465, 2991, 3031, 3359 cm−1. – MS (ESI): 282.0, HRMS: 281.9666, calcd: 281.9695 [M+H+].
5,7-Dichloro-2-(perfluoroethyl)quinolin-8-ol (32)
To a solution of 29 (15 mg, 0.04 mmol) in dichloromethane (4 mL) at 23 °C was added trifluoromethanesulfonic acid (36 mg, 0.24 mmol, 6 equiv.). After 12 h the reaction was diluted with a saturated aqueous solution of sodium bicarbonate (10 mL) and the aqueous layer extracted with dichloromethane (3 × 10 mL). The combined organic layers were dried over anhydrous sodium sulfate and concentrated under reduced pressure to yield chlorquinaldol analogue 32 (12 mg, 90 %). – 1H NMR (500 MHz): 7.76 (1 H, s), 7.92 (1 H, d, J = 9 Hz), 8.74 (1 H, d, J = 9 Hz) ppm. – 13C NMR (125 MHz): 104.5, 109.89, 117.2, 118.4, 121.0, 125.8, 130.6, 136.1, 137.5, 147.8 (m) ppm. – 19F NMR (282 MHz): −82.7, −116.6 ppm. – IR: 960, 1121, 1163, 1331, 1460, 2925, 2963, 3033, 3442 cm−1. – MS (ESI): 331.9, HRMS: 331.9659, calcd: 331.9663 [M+H+].
Supplementary Material
Acknowledgments
Financial support by the Welch Foundation (AX-1788), the National Institute of General Medical Sciences (SC3GM105579), the Max and Minnie Tomerlin Voelcker Fund, NIH MBRS-RISE (GM060655), and the University of Texas at San Antonio is gratefully acknowledged. Mass spectroscopic analysis was supported by a grant from the National Institute on Minority Health and Health Disparities (G12MD007591).
Footnotes
Electronic Supplementary Information (ESI) available: Experimental procedures and spectroscopic data. See DOI: 10.1039/b000000x/
References
- 1.(a) Müller K, Faeh C, Diederich F. Science. 2007;317:1881–1886. doi: 10.1126/science.1131943. [DOI] [PubMed] [Google Scholar]; (b) Purser S, Moore PP, Swallow S, Gouverneur V. Chem Soc Rev. 2008;37:320–330. doi: 10.1039/b610213c. [DOI] [PubMed] [Google Scholar]; (c) O’Hagan D. Chem Soc Rev. 2008;37:308–319. doi: 10.1039/b711844a. [DOI] [PubMed] [Google Scholar]; (d) Wang J, Sánchez-Roselló, Luis Aceña J, del Pozo C, Sorochinsky AE, Fustero S, Soloshonok VA, Liu H. Chem Rev. 2014;114:2432–2506. doi: 10.1021/cr4002879. [DOI] [PubMed] [Google Scholar]
- 2.(a) Nakajima T, Groult H, editors. Fluorinated Materials for Energy Conversion. Elsevier; 2005. [Google Scholar]; (b) Pagliaro M, Ciriminna R. J Mater Chem. 2005;15:4981–4991. [Google Scholar]
- 3.(a) Merino E, Nevado C. Chem Soc Rev. 2014 doi: 10.1039/c4cs00025k. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Liu T, Shen Q. Eur J Org Chem. 2012:6679–6687. [Google Scholar]; (c) Tomashenko OA, Grushin VV. Chem Rev. 2011;111:4475–4521. doi: 10.1021/cr1004293. [DOI] [PubMed] [Google Scholar]; (d) Wu XF, Neumann H, Beller M. Chem Asian J. 2012;7:1744–1754. doi: 10.1002/asia.201200211. [DOI] [PubMed] [Google Scholar]; (e) Roy S, Gregg BT, Gribble GW, Le VD, Roy S. Tetrahedron. 2011;67:2161–2195. [Google Scholar]
- 4.(a) Nagib DA, MacMillan DW. Nature. 2011;480:224–228. doi: 10.1038/nature10647. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ji Y, Brueckl T, Baxter RD, Fujiwara Y, Seiple IB, Su S, Blackmond DG, Baran PS. Proc Natl Acad Sci USA. 2011;108:14411–14415. doi: 10.1073/pnas.1109059108. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Chu L, Qing FL. J Am Chem Soc. 2012;134:1298–304. doi: 10.1021/ja209992w. [DOI] [PubMed] [Google Scholar]
- 5.a) Caron S, Do NM, Sieser JE. Tetrahedron Lett. 2000;41:2299. [Google Scholar]; (b) Balicki R, Golinski J. Synth Commun. 2000;30:1529. [Google Scholar]; (c) Rosenau T, Potthast A, Kosma P. Synlett. 1999:1972. [Google Scholar]; (d) Larionov OV, Stephens D, Mfuh AM, Arman HD, Naumova AS, Chavez G, Skenderi B. Org Biomol Chem. 2014;12:3026. doi: 10.1039/c4ob00115j. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Coperet C, Adolfsson H, Khuong TAW, Yudin AK, Sharpless KB. J Org Chem. 1998;63:1740. [Google Scholar]
- 6.(a) Campeau LC, Rousseaux S, Fagnou K. J Am Chem Soc. 2005;127:18020. doi: 10.1021/ja056800x. [DOI] [PubMed] [Google Scholar]; (b) Kanyiva KS, Nakao Y, Hiyama T. Angew Chem Int Ed. 2007;46:8872. doi: 10.1002/anie.200703758. [DOI] [PubMed] [Google Scholar]; (c) Cho SH, Hwang SJ, Chang S. J Am Chem Soc. 2008;130:9254–9256. doi: 10.1021/ja8026295. [DOI] [PubMed] [Google Scholar]; (d) Wu J, Cui X, Chen L, Jiang G, Wu Y. J Am Chem Soc. 2009;131:13888–13889. doi: 10.1021/ja902762a. [DOI] [PubMed] [Google Scholar]; (e) Xiao B, Liu ZJ, Liu L, Fu Y. J Am Chem Soc. 2013;135:616–619. doi: 10.1021/ja3113752. [DOI] [PubMed] [Google Scholar]; (f) Larionov OV, Stephens D, Mfuh A, Chavez G. Org Lett. 2014;16:864–867. doi: 10.1021/ol403631k. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Zhu C, Yi M, Wei D, Chen X, Wu Y, Cui X. Org Lett. 2014;16:1840–1843. doi: 10.1021/ol500183w. [DOI] [PubMed] [Google Scholar]
- 7.(a) Wengryniuk SE, Weickgenannt A, Reiher C, Strotman NA, Chen K, Eastgate MD, Baran PS. Org Lett. 2013;15:792. doi: 10.1021/ol3034675. [DOI] [PubMed] [Google Scholar]; (b) Paudler WW, Jovanovic MV. J Org Chem. 1983;48:1064. [Google Scholar]
- 8.(a) Londregan AT, Jennings S, Wei L. Org Lett. 2011;13:1840. doi: 10.1021/ol200352g. [DOI] [PubMed] [Google Scholar]; (b) Keith JM. J Org Chem. 2010;75:2722. doi: 10.1021/jo1001017. [DOI] [PubMed] [Google Scholar]; (c) Londregan AT, Jennings S, Wei LQ. Org Lett. 2010;12:5254. doi: 10.1021/ol102301u. [DOI] [PubMed] [Google Scholar]; (d) Storz T, Bartberger MD, Sukits S, Wilde C, Soukup T. Synthesis. 2008:201. [Google Scholar]; (e) Yin J, Xiang B, Huffman MA, Raab CE, Davies IW. J Org Chem. 2007;72:4554. doi: 10.1021/jo070189y. [DOI] [PubMed] [Google Scholar]
- 9.(a) Liu X, Xu C, Wang M, Liu Q. Chem Rev. 2014 doi: 10.1021/cr400473a. [DOI] [PubMed] [Google Scholar]; (b) Prakash GKS, Mandal M. J Fluorine Chem. 2001;112:123–131. [Google Scholar]; (c) Ma JA, Cahard D. J Fluorine Chem. 2007;128:975–996. [Google Scholar]; (d) Prakash GKS, Krishnamurti R, Olah GA. J Am Chem Soc. 1989;111:393. [Google Scholar]; (e) Ruppert I, Schlich K, Volbach W. Tetrahedron Lett. 1984;25:2195. [Google Scholar]
- 10.(a) Prakash GKS, Krishnamoorthy S, Ganesh SK, Kulkarni A, Haiges R, Olah GA. Org Lett. 2014;16:54–57. doi: 10.1021/ol403007j. [DOI] [PubMed] [Google Scholar]; (b) Prakash GKS, Yudin AK. Chem Rev. 1997;97:757–786. doi: 10.1021/cr9408991. [DOI] [PubMed] [Google Scholar]
- 11.(a) 2007133637. World Pat, WO. 2007; (b) 2008059370. World Pat, WO. 2008; (c) 2009064449. World Pat, WO. 2009
- 12.Nishida T, Ida H, Kuninobu Y, Kanai M. Nature Comm. 2014 doi: 10.1038/ncomms4387. [DOI] [PubMed] [Google Scholar]
- 13.Loska R, Majcher M, Makosza M. J Org Chem. 2007;72:5574–5580. doi: 10.1021/jo0703552. [DOI] [PubMed] [Google Scholar]
- 14.(a) Pino P, Piccolo O, Straub B, Consiglio G, Dich CT. Helv Chim Acta. 1982;65:2102–2109. [Google Scholar]; (b) Wendeborn S, Winkler T, Foisy I. Tetrahedron Lett. 2000;41:6387–6391. [Google Scholar]; (c) Matsumoto K, Kannami M, Inokuchi D, Kurata H, Kawase T, Oda M. Org Lett. 2007;9:2903–2906. doi: 10.1021/ol071189n. [DOI] [PubMed] [Google Scholar]
- 15.(a) Shabalin AY, Adonin NY, Bardin VV, Prikhod’ko SA, Timofeeva MN, Bykova MV, Parmon VN. J Fluorine Chem. 2013;149:82–87. [Google Scholar]; (b) Yu D, Lu L, Shen Q. Org Lett. 2013;15:940–943. doi: 10.1021/ol303567t. [DOI] [PubMed] [Google Scholar]; (c) Liu C, Cao L, Yin X, Xu H, Zhang B. J Fluorine Chem. 2013;156:51–60. [Google Scholar]; (d) Filler R, Ayyangar NR, Gustowski W, Kang HH. J Org Chem. 1969;34:534–538. [Google Scholar]
- 16.Terenin VI, Gerasimenko VA, Nikishova NG, Kurts AL, Afanas’eva TA, Bundel YG. Zh Org Khim. 1990;26:2191–2198. [Google Scholar]
- 17.Tobisu M, Yasutome A, Yamakawa K, Shimasaki T, Chatani N. Tetrahedron. 2012;68:5157–5161. [Google Scholar]
- 18.(a) Kolomeitsev A, Rusanov E, Bissky G, Lork E, Röschenthaler GV, Kirsch P. Chem Commun. 1999:1017–1018. [Google Scholar]; (b) Suzuki Y, Bakar A, Muramatsu MDK, Sato M. Tetrahedron. 2006;62:4227–4231. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.