

Abstract
Podocyte number is significantly reduced in diabetic patients and animal models, but the mechanism remains unclear. In the present study, we found that high glucose induced apoptosis in control podocytes which express transient receptor potential canonical 6 (TRPC6) channels, but not in TRPC6 knockdown podocytes in which TRPC6 was knocked down by TRPC6 silencing short hairpin RNA (shRNA). This effect was reproduced by treatment of podocytes with the reactive oxygen species (ROS), hydrogen peroxide (H2O2). Single-channel data from cell-attached, patch-clamp experiments showed that both high glucose and H2O2 activated the TRPC6 channel in control podocytes, but not in TRPC6 knockdown podocytes. Confocal microscopy showed that high glucose elevated ROS in podocytes and that H2O2 reduced the membrane potential of podocytes and elevated intracellular Ca2+ via activation of TRPC6. Since intracellular Ca2+ overload induces apoptosis, H2O2-induced apoptosis may result from TRPC6-mediated elevation of intracellular Ca2+. These data together suggest that high glucose induces apoptosis in podocytes by stimulating TRPC6 via elevation of ROS.
Keywords: ROS, Patch-clamp technique, Confocal microscopy, Hydrogen peroxide, Oxidative stress, Intracellular Ca2+
1. Introduction
Podocyte injury or the so-called podocytopathy includes changes in either the morphology or the number of podocytes in the glomerulus [1]. Loss of podocytes represents an early change in diabetic patients [17,20] and animal models [24]. Hyperglycemia induces podocyte loss via apoptosis by elevating intracellular ROS [25]. These studies suggest that the loss of podocytes due to apoptosis may account for glomerular hyperfiltration at the early stages of diabetes. Since depletion of podocytes results in focal segmental glomerulosclerosis [1,26], the loss of podocyte due to apoptosis may also account for the pathogenesis of glomerulosclerosis at the late stages of diabetes. Therefore, podocyte apoptosis may play an important role in promoting the development of diabetic nephropathy from the early glomerular hyperfiltration to the late glomerulosclerosis. Since the genetic form of glomerulosclerosis is caused by a mutation in the TRPC6 gene [29], altered TRPC6 channel activity may account for the development of diabetic glomerulosclerosis. TRPC6 is a Ca2+-permeable channel [12] which is highly expressed in podocytes [21]. TRPC6-mediated increases of intracellular Ca2+ mediate podocyte apoptosis induced by angiotensin II treatment [31], albumin overload [3], and sustained activation of atypical N-methyl-D-aspartate (NMDA) receptors [14]. It is known that high glucose induces podocyte apoptosis by elevating intracellular ROS [25] and that H2O2 activates TRPC6 and promotes TRPC6 trafficking to the plasma membrane [10]. These lines of indirect evidence suggest that TRPC6 may mediate high glucose-induced podocyte apoptosis.
However, it appears that the role of TRPC6 in regulating cell fate depends on cell types. In brain, inhibition of TRPC6 degradation protects neurons against ischemic damage [7]. In vascular smooth muscle cells, H2O2 elevates intracellular Ca2+ via TRPC6, but instead of causing apoptosis, it induces vessel contraction [6]. In glomerular mesangial cells of diabetic rats, the expression of TRPC6 is decreased [11]. Since mesangial cells are smooth muscle-like cells, decreased TRPC6 expression may reduce mesangial cell contraction and, together with loss of podocytes, account for the glomerular hyperfiltration found in early diabetes mellitus. In cancer cells, activation of TRPC6 participates in malignant cell proliferation [2,4]. In the lung, TRPC6 plays a role opposite to those in neurons, mesangial cells, and cancer cells; activation of TRPC6 actually participates in ischemia-reperfusion induced edema [28]. Although high glucose-induced oxidative stress increases TRPC6 expression in human monocytes [30], instead of inducing apoptosis, it causes activation of monocytes. Although ROS activate TRPC6 in a heterologous expression system [10], due to the cell-type dependence of TRPC6, it is necessary to examine whether ROS activate TRPC6 in podocytes and whether activation of TRPC6 accounts for high glucose-induced apoptosis of podocytes.
In the present study, using the cell-attached patch-clamp technique combined with confocal microscopy analysis, we show that high glucose induces apoptosis in podocytes by increasing TRPC6 channel open probability through elevation of intracellular ROS.
2. Materials and methods
2.1. Cell culture
An immortalized human podocyte cell line was provided by Dr. Moin Saleem at Children’s Renal Unit and Academic Renal Unit, University of Bristol, Southmead Hospital, Bristol, UK. The cells were cultured as described previously [22]. Briefly, cells were cultured in RPMI-1640 medium supplemented with 10% fetal bovine serum and 100 U/ml penicillin–streptomycin, in humidified 5% CO2 incubators and first incubated at 33 °C. After the cells became 70% confluent, they were incubated at 37 °C for 2 weeks before any experiment manipulations. To knock down TRPC6 expression, podocytes were stably transfected with TRPC6 silencing short hairpin RNA (shRNA) carried by a lentiviral vector (Santa Cruz Biotechnology). The podocytes with reduced TRPC6 expression (TRPC6 knockdown podocytes)were further selected in the presence of G418. Before the TRPC6 knockdown podocytes were used, the reduction of TRPC6 expression was confirmed by Western blot experiments. All the experiments in this study were performed at room temperature.
2.2. Patch-clamp recordings
Cell-attached patch-clamp recordings of TRPC6 single-channel currents from podocytes were carried out using an Axopatch 200B amplifier (Molecular Devices, Sunnyvale, CA). Prior to the experiments, podocytes cultured on 35 mm petri dish were thoroughly washed with NaCl bath solution containing (in mM): 145 NaCl, 3.4 KCl, 1 CaCl2, 1 MgCl2, and 10 HEPES; pH was adjusted to 7.4 with NaOH. The glass micropipette was filled with the NaCl bath solution (the pipette resistance is 7–10 MΩ). Most single-channel currents were obtained with an applied pipette potential of 20 mV (−Vpipette = −20 mV). For detecting the current–voltage relationship, several applied pipette potentials were used. The current traces were filtered at 1 kHz, and sampled every 50 μs with Clampex 10 software. All experiments were conducted at room temperature. The total numbers of functional channels in the patch were estimated according to the number of peaks detected on the current amplitude histograms using at least 5 min record. The open probability (PO) of TRPC6 before (3-minute record) and 5 min after each experimental manipulation (3-minute record) was calculated using Clampfit 10.
2.3. Confocal microscopy imaging
To evaluate apoptosis, podocytes were stained with both FITC-conjugated annexin V (AV) and propidium iodide (PI). Confocal microscopy experiments were performed, as we previously describe [27], to determine how glucose induces apoptosis in podocytes. The cell membrane of apoptotic cells was stained with AV because phosphatidylserine, a lipid which has a high binding affinity to AV, is externalized in apoptotic cells. The nuclei of apoptotic cells were stained with PI because the nuclear membrane of apoptotic cells becomes permeable to PI. AV was excited with 488 nm laser and visualized through a 515 nm emission filter, shown in green. PI was excited with 488 nm laser and visualized through a 590 nm emission filter, shown in red. To detect the levels of intracellular reactive oxygen species (ROS), podocytes were incubated with 25 μM 2′,7′-dichlorodihydrofluorescein diacetate a membrane-permeable, ROS-sensitive, fluorescent probe, for 15 min. To determine the membrane potential, podocytes were incubated with 10 μM bis-(1,3- dibutylbarbituric acid) trimethine oxonol, a fluorescent voltage-sensitive dye, for 30 min. Prior to the confocal microscopy experiments, cells were washed twice with NaCl solution. To determine intracellular Ca2+, podocytes were incubated with 5 μM fluo-4 AM, a fluorescent Ca2+ indicator for 30 min. Confocal microscopy XY scanning of podocytes was accomplished within 5–15 min. In each set of experiments, images were taken using the same parameter settings.
2.4. Western blotting
Either control podocytes or TRPC6 knockdown podocytes were cultured as described above. Cell lysates (100 μg) were loaded and electrophoresed on 10% SDS-PAGE gels for 60 to 90 min. Gels were blotted onto polyvinylidene fluoride (PVDF) membranes for 1 h at 90 volts. After 1 h blocking with 5% BSA-PBST buffer, PVDF membranes were incubated with primary antibody (1:1000 dilution) of rabbit polyclonal antibody to TRPC6 (Sigma, Lot # 8831P1) overnight at 4 °C, and then incubated with horseradish peroxidase (HRP)-conjugated sheep anti-rabbit IgG secondary antibody (1:5000 dilution, GE healthcare) for 1 h after 4 vigorous washes. Finally, blots were visualized with chemiluminescence using ECL Plus Western Blotting Detection System (GE healthcare).
2.5. Chemicals
Most chemicals were purchased from Sigma-Aldrich (St. Louis, MO). H2O2 was purchased from Fisher Scientific. H2O2 was diluted with NaCl solution before it was added to the bath. All the solutions were either premade and stored in −20 °C freezer or freshly made before the experiments. All the concentrations listed in this study represent the final concentrations.
2.6. Statistical analysis
Data are reported as mean values ± SD. Statistical analysis was performed with SigmaPlot and SigmaStat software (Jandel Scientific, CA). Paired t-test was used for comparisons between pre and post treatment activities. Z-test and chi-squared test were used for comparisons between the changes in percentage. Results were considered significant if p < 0.05.
3. Results
3.1. High glucose induces apoptosis of podocytes via TRPC6
High glucose causes apoptosis of podocytes [25], but the mechanism remains largely unknown. Using confocal microscopy to analyze apoptotic podocytes stained with annexin V-FITC and propidium iodide, we found that treatment of podocytes with 33 mM glucose for 7 days (high glucose) induced significant apoptosis in control podocytes. After the treatment, the percentage of apoptotic podocytes was increased from 1 ± 2% to 24 ± 10% (n = 15, p < 0.001), but remained at a low level (7 ± 5%) when the culture medium contained 10 μM 1-[2-(4-methoxyphenyl)-2-[3-(4-methoxyphenyl) propoxy]ethyl]imidazole (SKF-96365), a generic blocker of TRPC channels (Fig. 1, A, C). To determine whether TRPC6 is involved in this process, podocytes were stably transfected with TRPC6 silencing short hairpin RNA (shRNA) to knock down TRPC6 expression in podocytes. Western blotting data showed that TRPC6 expression was significantly reduced in these podocytes (Fig. 1D). In these TRPC6 knockdown podocytes, high glucose did not induce significant apoptosis; the percentage of apoptotic podocytes tended to be increased, from 2 ± 3% to 5 ± 5%, but it was not statistically significant (n = 15, p > 0.05) (Fig. 1, B, C). These data suggest that high glucose induces apoptosis via a TRPC6-dependent pathway.
Fig. 1.

High glucose induces apoptosis of podocytes via TRPC6. A, representative confocal microscopy data showing control podocytes in either control conditions (left) or treated with high glucose (33 mM D-glucose for all the experiments in this study) for 7 days in the absence (middle) or presence of 10 μM SKF-96365 (right), a generic blocker of TRPC channels. B shows that high glucose did not induce significant apoptosis in TRPC6 knockdown podocytes in which TRPC6 was knocked down by its shRNA. C, percent apoptotic cells in each experimental condition (calculated from 15 microscopy field of four separate experiments). Apoptotic cells were stained with FITC-conjugated Annexin V (AV, shown in green) due to phosphatidylserine externalization and propidium iodide (PI, shown in red) due to permeable nuclear membrane. DIC images were taken to show total podocytes in each microscopy field. Reduced TRPC6 expression was confirmed by Western blotting experiments (D).
3.2. High glucose activates TRPC6 in podocytes
To determine how high glucose regulates TRPC6 at the single-channel level, we performed cell-attached patch-clamp experiments both in control podocytes and TRPC6 knockdown podocytes. The data showed that high glucose induced a channel activity in a cell-attached patch established on a control podocyte (Fig. 2A). The single-channel current events versus the potentials applied to the patch pipette were plotted in Fig. 2B. The current–voltage relationship of this channel was summarized from 6 cell-attached patches and was shown in Fig. 2C. It has a linear conductance of 21 pS, but slightly outwardly rectified (the chord conductance between 40 and 60 mV = 29 pS). In contrast, in another cell-attached patch established on a TRPC6 knockdown podocyte, high glucose did not induce such a channel activity (Fig. 2D), indicating that the channel induced by high glucose is TRPC6. Fig. 2E shows that high glucose significantly increased open probability (PO) of this channel from 0.02 ± 0.04 to 0.33 ± 0.16 in control podocytes (n = 6, p < 0.01). In untreated TRPC6 knockdown podocytes, we were unable to record any channel activity in 6 cell-attached patches. After the treatment with high glucose, only 1 out of 6 cell-attached patches contained channel activity; the mean PO is 0.02 ± 0.04. There is no difference in PO between untreated podocytes and the podocytes treated with high glucose (n = 6, p > 0.05). These data together suggest that high glucose activates TRPC6 in podocytes.
Fig. 2.

High glucose induces TRPC6 channel activity in podocytes. A, representative single-channel records from two cell-attached patches when 20 mV was applied to the patch pipette (−Vpipette = −20 mV): one from an untreated podocyte (upper); the other from a podocyte treated with high glucose for 3 days (lower). Patch pipettes were filled with NaCl bath solution (see Materials and methods). B, single-channel current traces at different applied pipette potentials from the same patch shown in A after the podocyte was treated with high glucose. C, summary plot of current–voltage relationship of this channel recorded from 5 glucose-treated podocytes, showing that the channel has a linear conductance of 21 pS and is slightly outwardly voltage-rectified. D shows that high glucose failed to induce such a channel in a cell-attached patch formed on a TRPC6 knockdown podocyte. E, summary plots of open probability (PO) of 6 cell-attached patches in each experimental condition. “-C” or “C-” shows the baseline when channel is closed. Downward events show inward currents.
3.3. High glucose causes oxidative stress in podocytes
Previous studies showed that high glucose gradually elevates intracellular ROS in immortalized mouse podocytes during the period from 30 min to 24 h [25]. To test whether high glucose also elevates ROS in human podocytes, cultured human podocytes were stained with a membrane-permeable, ROS-sensitive, fluorescent probe, 2′,7′-dichlorodihydrofluorescein diacetate. Confocal microscopy data showed that treatment of human podocytes with 33 mM glucose for 3 days caused more than 3-fold increase of the fluorescent intensity (Fig. 3, A, B) (n = 6, p < 0.001). These data suggest that high glucose elevates intracellular ROS in human podocytes and the elevation is maintained for a long period.
Fig. 3.
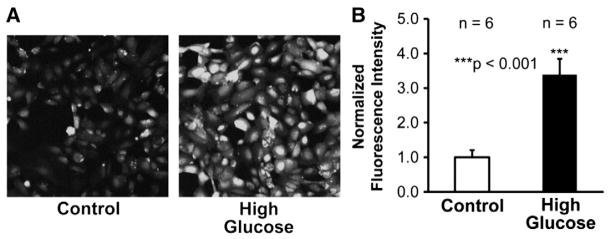
High glucose causes intracellular oxidative stress. A, representative confocal microscopy data from podocytes either in control conditions (left) or treated with high glucose for 3 days (right). Podocytes were stained with 2′,7′-dichlorodihydrofluorescein diacetate, a membrane-permeable fluorescent probe detecting intracellular ROS. These confocal microscopy data represent consistent results from six separate experiments in each group. B, summary plots of fluorescence intensity of podocytes either under control conditions (white bar) or treated with high glucose for 3 days (black bar).
3.4. H2O2 induces apoptosis of podocytes via TRPC6
The above experiments showed that high glucose elevated intracellular ROS and induced apoptosis. To determine whether high glucose induces apoptosis by elevating ROS in podocytes, we used H2O2 as a tool to manipulate the levels of intracellular ROS. Confocal microscopy experiments similar to those in Fig. 1 were carried out, except that in these experiments podocytes were treated with 5 mM H2O2 for 1 h. Fig. 4 shows that treatment with 5 mM H2O2 for 1 h induced apoptosis in control podocytes; the percentage of apoptotic podocytes was increased, from 1 ± 2% to 14 ± 8% (n = 15, p < 0.001), but remained at a low level (2 ± 3%) when culture medium contained 10 μM SKF-96365, a generic blocker of TRPC channels (Fig. 4, A–C). In contrast, H2O2 applied to the cells also for 1 h at the same concentration (5 mM) did not induce apoptosis in TRPC6 knockdown podocytes. The percentage of apoptotic podocytes tended to increase from 1 ± 2% to 3 ± 5%, but the increase was not statistically significant (n = 15, p > 0.05) (Fig. 4, B, C). We noticed that a very high concentration of H2O2 was required to induce the effect. This is not surprising because in the hippocampus, the IC50 for extracellular H2O2 to affect postsynaptic potentials is nearly 6 mM [18]. Our recent studies also show that a high concentration of exogenous H2O2 is required to finally elevate intracellular ROS and regulate the epithelial sodium channel in A6 distal nephron cells [15]. These data together suggest that H2O2 at 5 mM did not cause a nonspecific podocyte injury, but a TRPC6-mediated apoptosis.
Fig. 4.

H2O2 also induces apoptosis of podocytes via TRPC6. A, representative confocal microscopy data showing podocytes either in control conditions (left) or treated with 5 mM H2O2 for 1 h in the absence (middle) or presence of 10 μM SKF-96365 (right). B shows that H2O2 failed to induce apoptosis in TRPC6 knockdown podocytes. C, percent apoptotic cells in each experimental condition, as indicated (calculated from 15 microscopy fields from three separate experiments). Apoptotic cells were stained as described in Fig. 1.
3.5. H2O2 activates TRPC6 in podocytes
In Figs. 2 and 3, we showed that high glucose elevated intracellular ROS and activated TRPC6 in podocytes. Although it has been shown in a heterologous system that H2O2 stimulates TRPC6 [10], it remains unknown whether H2O2 could stimulate TRPC6 in podocytes. Therefore, cell-attached patch-clamp experiments were carried out in both control and TRPC6 knockdown podocytes. We found that application of 5 mM H2O2 to the bath activated a channel in a cell-attached patch formed on a control podocyte (Fig. 5A). The H2O2-induced channel activity was blocked by 10 μM SKF-96365 in the patch pipette (Fig. 5B). The PO of this channel was significantly increased from 0.02 ± 0.04 to 0.46 ± 0.15 (n = 6, p < 0.01) when the patch pipette contained NaCl bath solution alone, but remained unchanged (0.01 ± 0.02 to 0.04 ± 0.05; n = 6, p > 0.05) when the patch pipette contained NaCl bath solution plus 10 μM SKF-96365. To confirm that the channel we observed is TRPC6, a total of 6 cell-attached patches were established on TRPC6 knockdown podocytes. None of these patches contained any H2O2-induced channel activity, even when the cells were exposed to 5 mM H2O2 (n = 6, PO = 0) (Fig. 5E), indicating that the channel activated by H2O2 is TRPC6. To characterize the current– voltage relationship, H2O2-induced single-channel current events versus the potentials applied to the patch pipette were plotted in Fig. 6A. The current–voltage relationship of this channel was summarized from 6 cell-attached patches and was shown in Fig. 6C. Similar to the channel activated by high glucose, the channel activated by H2O2 also has a linear conductance of 21 pS. It appears that, for some unknown reason, the outward rectification appears to become apparent (the chord conductance between 40 and 60 mV = 42 pS). These data suggest that H2O2 can mimic the effect of high glucose on TRPC6 channel activity.
Fig. 5.

H2O2 also induces TRPC6 channel activity in podocytes. A, application of 5 mM H2O2 to the bath induced a channel in a cell-attached patch from a podocyte when patch pipettes contained NaCl bath solution. Such channel activity was barely observed when patch pipettes contained 10 μM SKF-96365 (B). C and D, summary plots of PO before and after application of 5 mM H2O2 to the bath; patch pipettes contained NaCl bath solution either without (C) or with 10 μM SKF-96365 (D) (note: in D, the data are from a total of 6 cell-attached patches; the POs in three cell-attached patches are overlapped because the value is zero even after addition of H2O2 to the bath). E shows that 5 mM H2O2 did not induce the same type of channel activity in TRPC6 knockdown podocytes in any of 6 cell-attached patches.
Fig. 6.

Blockade of TRPC6 channels outside the patch pipette shifts the reversal potential of TRPC6 channel recorded in the patch. A and B, H2O2-induced single-channel currents versus potentials applied to the patch pipette in a cell-attached patch before (A) and after addition of 10 μM SKF-96365 to the bath to block all the TRPC6 channels outside the patch pipette across the whole cell membrane (B). C, summary plots of current–voltage relationship of H2O2-induced single-channel currents versus pipette potentials before (open circles) and after addition of 10 μM SKF-96365 to the bath (filled circles).
3.6. Activation of TRPC6 reduces the membrane potential and elevates intracellular Ca2+
In Fig. 2, we noticed that the reversal potential of high glucose-induced TRPC6 single-channel current was near zero. Similarly, the reversal potential of H2O2-induced TRPC6 single-channel current was also near zero (Fig. 6C). Since TRPC6 is a non-selective cation channel [12], theoretically, activation of TRPC6 should depolarize the membrane potential. If H2O2 could cause the depolarization by activation of TRPC6, blockade of TRPC6 should abolish the depolarization and shift the reversal potential. Indeed, the data showed that application of 10 μM SKF-96365 to the bath to block the TRPC6 channels outside the patch pipette shifted the reversal potential from near zero to 27 mV (Fig. 6, B, C) implying that the membrane potential was hyperpolarized by 27 mV. To confirm the patch-clamp data, podocytes were stained with a fluorescent voltage-sensitive dye, bis-(1,3-dibutylbarbituric acid) trimethine oxonol. At 5 min after exposure of podocytes to 5 mM H2O2, the fluorescence intensity was reduced by about 40%, indicating that the membrane potential was depolarized. The depolarization was abolished by 10 μM SKF-96365, even though 5 mM H2O2 is still in the bath (Fig. 7, A, B). These data suggest that activation of TRPC6 depolarizes podocyte membrane potential.
Fig. 7.
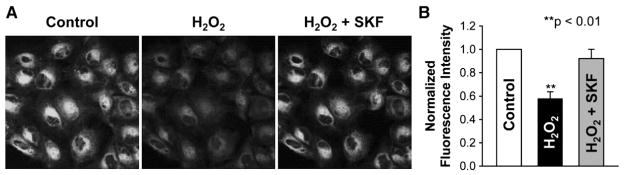
Activation of TRPC6 by H2O2 depolarizes podocyte membrane potential. A, representative confocal microscopy images of podocytes stained with 10 μM bis- (1,3-dibutylbarbituric acid) trimethine oxonol, a voltage-sensitive fluorescent probe detecting the membrane potential. B, summary plots of normalized fluorescence intensity from podocytes either under control condition (white bar), after addition of 5 mMH2O2 to the bath (black bar), or after addition of 10 μMSKF-96365 to the bath in the presence of H2O2.
Since TRPC6 is a Ca2+-permeable cation channel [12], activation of TRPC6 should result in elevation of intracellular Ca2+. However, in a heterologous expression system, TRPC6 channel appears to have limited Ca2+ permeability [8]. To test whether activation of native TRPC6 channel could elevate intracellular Ca2+, both control and TRPC6 knockdown podocytes were incubated with Fluo-4, a Ca2+ probe. The data showed application of 5 mMH2O2 to the cells remarkably elevated intracellular Ca2+ in untreated control podocytes (Fig. 8A), but Ca2+ was not increased in either the podocytes pretreated with 10 μM SKF-96365 (Fig. 8B) or in TRPC6 knockdown podocytes (Fig. 8C). Fig. 8D shows that there is about four-fold increase of the fluorescence intensity at 5 min after application of 5 mM H2O2 to the bath in control podocytes; but H2O2 did not alter the fluorescence intensity either in podocytes pretreated with 10 μM SKF-96365 or in TRPC6 knockdown podocytes. These data suggest that activation of TRPC6 by H2O2 results in significant increases in intracellular Ca2+.
Fig. 8.

H2O2 elevates intracellular Ca2+ in podocytes via TRPC6. A and B show representative confocal microscopy data from control podocytes before (the first image) and after addition of 5 mM H2O2 to the bath either in control conditions (A) or pretreated with 10 μM SKF-96365 (B). C shows representative confocal microscopy data from TRPC6 knockdown podocytes before (the first image) and after addition of 5 mM H2O2 to the bath. D shows summary plots of relative fluorescence intensity.
4. Discussion
The role of TRPC6 in podocyte pathology has been highlighted by recent discoveries that TRPC6 is a channel in glomerular slit diaphragm [21] and that a mutation in the TRPC6 gene causes familial focal segmental glomerulosclerosis [29]. Since overexpression of TRPC6 by in vivo gene delivery causes proteinuria [16], which is one of the characteristic symptoms of glomerulosclerosis, the abnormalities in TRPC6 for causing familiar focal segmental glomerulosclerosis are likely to be gain-of-function mutations [19]. However, it is not clear whether TRPC6 is active in podocytes from normal subjects and whether it can be activated in diabetic conditions. Using a cultured human podocyte cell line, we show that TRPC6 is inactive in control conditions, indicating that basal TRPC6 activity may not be required for glomerular function. This argument is consistent with the evidence that TRPC6-deficient mice do not show any obvious renal phenotype [5]. However, our data indicate that the silent TRPC6 channel in human podocytes can be strongly activated by high glucose-induced oxidative stress. For the first time, our single-channel data clearly demonstrate that ROS activation of TRPC6 channels mediates high glucose-induced apoptosis of podocytes by causing a massive elevation of intracellular Ca2+. Since podocyte injury is known to play an important role in the development and progression of diabetic glomerulosclerosis [9,13], the present study suggests that elevated TRPC6 channel activity may account for diabetic glomerulosclerosis. Further studies may suggest the clinical use of antioxidants or TRPC6 channel blockers for preventing glomerular damage in diabetic patients.
It has long been suggested that intracellular Ca2+ regulates both cell survival and cell death [23]. Since TRPC6 is a Ca2+-permeable channel [12], we argue that TRPC6 regulates both cell survival and cell death depending on its activity. Moderate activation of TRPC6 should result in a moderate elevation of intracellular Ca2+ which should promote cell survival. Since TRPC6 promotes malignant proliferation [2,4], we argue that TRPC6 may be moderately activated in cancer cells to mediate cell proliferation. In contrast, excess activation of TRPC6 should result in a massive increase in intracellular Ca2+ which causes apoptosis. The present study suggests that high glucose induces apoptosis of podocytes by causing intracellular Ca2+ overload through ROS activation of TRPC6. Therefore, full characterization of TRPC6 channel gating may provide convincing explanation for the contrary roles of TRPC6 in cell proliferation and apoptosis.
In addition to TRPC6 channel gating, the downstream pathway for ROS to strongly activate TRPC6 also requires further investigation. First, it is not clear which type of ROS in the cells plays a role in regulating TRPC6 (H2O2, superoxide, or both). It seems that strong oxidants may be necessary to activate TRPC6 because in order to activate the channel a very high concentration of H2O2 (a relatively mild oxidant) is required. However, it remains to be further determined which type of ROS (superoxide, H2O2, or both) is the native activator of TRPC6. Second, it remains obscure how ROS regulate the channel. Since we applied H2O2 to the bath outside the patch pipette, it is unlikely that H2O2 could directly regulate TRPC6 by oxidizing the extracellular domains of the channel. Previous studies also showed that H2O2 failed to activate TRPC6 in inside-out patches [10], indicating that H2O2 does not directly regulate TRPC6. These topics will serve as interesting directions for our future studies.
Acknowledgments
This research was supported by Department of Health and Human Services (National Institutes of Health Grant 5R01-DK067110 to He-Ping Ma).
References
- 1.Barisoni L, Schnaper HW, Kopp JB. Advances in the biology and genetics of the podocytopathies: implications for diagnosis and therapy. Arch Pathol Lab Med. 2009;133:201–216. doi: 10.1043/1543-2165-133.2.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cai R, Ding X, Zhou K, Shi Y, Ge R, Ren G, Jin Y, Wang Y. Blockade of TRPC6 channels induced G2/M phase arrest and suppressed growth in human gastric cancer cells. Int J Cancer. 2009;125:2281–2287. doi: 10.1002/ijc.24551. [DOI] [PubMed] [Google Scholar]
- 3.Chen S, He FF, Wang H, Fang Z, Shao N, Tian XJ, Liu JS, Zhu ZH, Wang YM, Wang S, Huang K, Zhang C. Calcium entry via TRPC6 mediates albumin overload-induced endoplasmic reticulum stress and apoptosis in podocytes. Cell Calcium. 2011;50:523–529. doi: 10.1016/j.ceca.2011.08.008. [DOI] [PubMed] [Google Scholar]
- 4.Chigurupati S, Venkataraman R, Barrera D, Naganathan A, Madan M, Paul L, Pattisapu JV, Kyriazis GA, Sugaya K, Bushnev S, Lathia JD, Rich JN, Chan SL. Receptor channel TRPC6 is a key mediator of Notch-driven glioblastoma growth and invasiveness. Cancer Res. 2010;70:418–427. doi: 10.1158/0008-5472.CAN-09-2654. [DOI] [PubMed] [Google Scholar]
- 5.Dietrich A, Mederos YS, Gollasch M, Gross V, Storch U, Dubrovska G, Obst M, Yildirim E, Salanova B, Kalwa H, Essin K, Pinkenburg O, Luft FC, Gudermann T, Birnbaumer L. Increased vascular smooth muscle contractility in TRPC6−/− mice. Mol Cell Biol. 2005;25:6980–6989. doi: 10.1128/MCB.25.16.6980-6989.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ding Y, Winters A, Ding M, Graham S, Akopova I, Muallem S, Wang Y, Hong JH, Gryczynski Z, Yang SH, Birnbaumer L, Ma R. Reactive oxygen species-mediated TRPC6 protein activation in vascular myocytes, a mechanism for vasoconstrictor-regulated vascular tone. J Biol Chem. 2011;286:31799–31809. doi: 10.1074/jbc.M111.248344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Du W, Huang J, Yao H, Zhou K, Duan B, Wang Y. Inhibition of TRPC6 degradation suppresses ischemic brain damage in rats. J Clin Invest. 2010;120:3480–3492. doi: 10.1172/JCI43165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Estacion M, Sinkins WG, Jones SW, Applegate MA, Schilling WP. Human TRPC6 expressed in HEK 293 cells forms non-selective cation channels with limited Ca2+ permeability. J Physiol. 2006;572:359–377. doi: 10.1113/jphysiol.2005.103143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gassler N, Elger M, Kranzlin B, Kriz W, Gretz N, Hahnel B, Hosser H, Hartmann I. Podocyte injury underlies the progression of focal segmental glomerulosclerosis in the fa/fa Zucker rat. Kidney Int. 2001;60:106–116. doi: 10.1046/j.1523-1755.2001.00777.x. [DOI] [PubMed] [Google Scholar]
- 10.Graham S, Ding M, Ding Y, Sours-Brothers S, Luchowski R, Gryczynski Z, Yorio T, Ma H, Ma R. Canonical transient receptor potential 6 (TRPC6), a redox-regulated cation channel. J Biol Chem. 2010;285:23466–23476. doi: 10.1074/jbc.M109.093500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Graham S, Gorin Y, Abboud HE, Ding M, Lee DY, Shi H, Ding Y, Ma R. Abundance of TRPC6 protein in glomerular mesangial cells is decreased by ROS and PKC in diabetes. Am J Physiol Cell Physiol. 2011;301:C304–C315. doi: 10.1152/ajpcell.00014.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hofmann T, Obukhov AG, Schaefer M, Harteneck C, Gudermann T, Schultz G. Direct activation of human TRPC6 and TRPC3 channels by diacylglycerol. Nature. 1999;397:259–263. doi: 10.1038/16711. [DOI] [PubMed] [Google Scholar]
- 13.Hoshi S, Shu Y, Yoshida F, Inagaki T, Sonoda J, Watanabe T, Nomoto K, Nagata M. Podocyte injury promotes progressive nephropathy in zucker diabetic fatty rats. Lab Invest. 2002;82:25–35. doi: 10.1038/labinvest.3780392. [DOI] [PubMed] [Google Scholar]
- 14.Kim EY, Anderson M, Dryer SE. Sustained activation of N-methyl-D-aspartate receptors in podocytes leads to oxidative stress, mobilization of transient receptor potential canonical 6 channels, nuclear factor of activated T cells activation, and apoptotic cell death. Mol Pharmacol. 2012;82:728–737. doi: 10.1124/mol.112.079376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ma HP. Hydrogen peroxide stimulates the epithelial sodium channel through a phosphatidylinositide 3-kinase-dependent pathway. J Biol Chem. 2011;286:32444–32453. doi: 10.1074/jbc.M111.254102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Moller CC, Wei C, Altintas MM, Li J, Greka A, Ohse T, Pippin JW, Rastaldi MP, Wawersik S, Schiavi S, Henger A, Kretzler M, Shankland SJ, Reiser J. Induction of TRPC6 channel in acquired forms of proteinuric kidney disease. J Am Soc Nephrol. 2007;18:29–36. doi: 10.1681/ASN.2006091010. [DOI] [PubMed] [Google Scholar]
- 17.Nakamura T, Ushiyama C, Suzuki S, Hara M, Shimada N, Ebihara I, Koide H. Urinary excretion of podocytes in patients with diabetic nephropathy. Nephrol Dial Transplant. 2000;15:1379–1383. doi: 10.1093/ndt/15.9.1379. [DOI] [PubMed] [Google Scholar]
- 18.Nistico R, Piccirilli S, Cucchiaroni ML, Armogida M, Guatteo E, Giampa C, Fusco FR, Bernardi G, Nistico G, Mercuri NB. Neuroprotective effect of hydrogen peroxide on an in vitro model of brain ischaemia. Br J Pharmacol. 2008;153:1022–1029. doi: 10.1038/sj.bjp.0707587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Patrakka J, Tryggvason K. New insights into the role of podocytes in proteinuria. Nat Rev Nephrol. 2009;5:463–468. doi: 10.1038/nrneph.2009.108. [DOI] [PubMed] [Google Scholar]
- 20.Reidy K, Susztak K. Epithelial-mesenchymal transition and podocyte loss in diabetic kidney disease. Am J Kidney Dis. 2009;54:590–593. doi: 10.1053/j.ajkd.2009.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Reiser J, Polu KR, Moller CC, Kenlan P, Altintas MM, Wei C, Faul C, Herbert S, Villegas I, Avila-Casado C, McGee M, Sugimoto H, Brown D, Kalluri R, Mundel P, Smith PL, Clapham DE, Pollak MR. TRPC6 is a glomerular slit diaphragm-associated channel required for normal renal function. Nat Genet. 2005;37:739–744. doi: 10.1038/ng1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Saleem MA, O’Hare MJ, Reiser J, Coward RJ, Inward CD, Farren T, Xing CY, Ni L, Mathieson PW, Mundel P. A conditionally immortalized human podocyte cell line demonstrating nephrin and podocin expression. J Am Soc Nephrol. 2002;13:630–638. doi: 10.1681/ASN.V133630. [DOI] [PubMed] [Google Scholar]
- 23.Shapovalov G, Lehen’kyi V, Skryma R, Prevarskaya N. TRP channels in cell survival and cell death in normal and transformed cells. Cell Calcium. 2011;50:295–302. doi: 10.1016/j.ceca.2011.05.006. [DOI] [PubMed] [Google Scholar]
- 24.Siu B, Saha J, Smoyer WE, Sullivan KA, Brosius FC., III Reduction in podocyte density as a pathologic feature in early diabetic nephropathy in rodents: prevention by lipoic acid treatment. BMC Nephrol. 2006;7:6. doi: 10.1186/1471-2369-7-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Susztak K, Raff AC, Schiffer M, Bottinger EP. Glucose-induced reactive oxygen species cause apoptosis of podocytes and podocyte depletion at the onset of diabetic nephropathy. Diabetes. 2006;55:225–233. [PubMed] [Google Scholar]
- 26.Wang L, Tang Y, Howell DN, Ruiz P, Spurney RF. A novel mouse model of podocyte depletion. Nephron Exp Nephrol. 2012;121:e10–e22. doi: 10.1159/000342369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang LH, Wang N, Lu XY, Liu BC, Yanda MK, Song JZ, Dai HM, Sun YL, Bao HF, Eaton DC, Ma HP. Rituximab inhibits Kv1.3 channels in human B lymphoma cells via activation of FcgammaRIIB receptors. Biochim Biophys Acta. 2011;1823:505–513. doi: 10.1016/j.bbamcr.2011.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Weissmann N, Sydykov A, Kalwa H, Storch U, Fuchs B, Schnitzler M, Brandes RP, Grimminger F, Meissner M, Freichel M, Offermanns S, Veit F, Pak O, Krause KH, Schermuly RT, Brewer AC, Schmidt HH, Seeger W, Shah AM, Gudermann T, Ghofrani HA, Dietrich A. Activation of TRPC6 channels is essential for lung ischaemia-reperfusion induced oedema in mice. Nat Commun. 2012;3:649. doi: 10.1038/ncomms1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Winn MP, Conlon PJ, Lynn KL, Farrington MK, Creazzo T, Hawkins AF, Daskalakis N, Kwan SY, Ebersviller S, Burchette JL, Pericak-Vance MA, Howell DN, Vance JM, Rosenberg PB. A mutation in the TRPC6 cation channel causes familial focal segmental glomerulosclerosis. Science. 2005;308:1801–1804. doi: 10.1126/science.1106215. [DOI] [PubMed] [Google Scholar]
- 30.Wuensch T, Thilo F, Krueger K, Scholze A, Ristow M, Tepel M. High glucose-induced oxidative stress increases transient receptor potential channel expression in human monocytes. Diabetes. 2010;59:844–849. doi: 10.2337/db09-1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang H, Ding J, Fan Q, Liu S. TRPC6 up-regulation in Ang II-induced podocyte apoptosis might result from ERK activation and NF-kappaB translocation. Exp Biol Med (Maywood) 2009;234:1029–1036. doi: 10.3181/0901-RM-11. [DOI] [PubMed] [Google Scholar]