

Summary
Phagocytosis of apoptotic cells (efferocytosis) is essential for regulation of immune responses and tissue homeostasis, and is mediated by phagocytic receptors. In this study we found that urokinase plasminogen activator receptor (uPAR) plays an important role in internalization of apoptotic cells, and also characterized the underlying mechanisms. In a flow cytometry-based phagocytic assay, uPAR-deficient (uPAR−/−) macrophages displayed significant defect in internalization but not tethering of apoptotic cells. When uPAR−/− mice were challenged with apoptotic cells, they exhibited pronounced splenomegaly resulting from accumulation of abundant apoptotic cells in spleen. Overexpression of uPAR in HEK-293 cells enhanced efferocytosis, which was inhibited by annexin V and phosphatidylserine (PS) liposome, suggesting that uPAR-mediated efferocytosis is dependent on PS. In serum lacking high-molecular-weight kininogen (HK), a uPAR ligand, uPAR-mediated efferocytosis was significantly attenuated, which was rescued by replenishment of HK. As detected by flow cytometry, HK selectively bound to apoptotic cells, but not viable cells. In purified systems, HK was specifically associated with PS liposome. HK binding to apoptotic cells induced its rapid cleavage to two-chain HKa and bradykinin. Both heavy chain and light chain of HKa were associated with PS liposome and apoptotic cells. HKa has higher binding affinity than HK to uPAR. Overexpression of Rac1/N17 cDNA inhibited uPAR-mediated efferocytosis. HK plus PS liposome stimulated a complex formation of CrkII with p130Cas and Dock-180, and Rac1 activation in uPAR-293 cells, but not in control HEK-293 cells. Thus, uPAR mediates efferocytosis through HK interaction with PS on apoptotic cells and activation of Rac1 pathway.
Introduction
Efficient clearance of apoptotic cells by phagocytes (efferocytosis) is an essential mechanism for maintenance of normal tissue homeostasis and regulation of immune responses (1–2). Phagocytes, such as macrophages, utilize a variety of receptors to recognize and internalize apoptotic cells. After engulfing an apoptotic cell, macrophages produce anti-inflammatory cytokines, such as interleukin-10 and transforming growth factor-β, which prevent inflammation and tissue damage. However, if apoptotic cells are not rapidly cleared, they will become secondary necrotic, causing the release of toxic intracellular antigens that induce tissue damage and production of proinflammatory cytokines. Dysfunctional efferocytosis is often associated with chronic inflammation, with a variety of pathological sequelae including autoimmune diseases and development of necrotic core in atherosclerotic plaque (3–4). Thus, elucidation of efferocytosis process is critical for understanding tissue homeostasis and inflammation resolution.
Phagocytic receptors that interact with apoptotic cells must recognize specific ‘eat-me’ signals on the apoptotic cells. The omnipresent exposure of phosphatidylserine (PS) on a variety of apoptotic cells suggests that PS is a general ‘eat-me’ signal (1–2). Phagocytes may recognize PS directly through the PS receptors such as T cell immunoglobulin and mucin domain (TIM) 1/4 and Stablin-2 (5–6). Another pool of phagocytic receptors, such as αv integrins and receptor tyrosine kinase Mer, interact with PS via opsonins, including milk fat globule-EGF factor 8 protein and growth arrest-specific 6, which bind to PS (1, 7). However, so far, it is still surprising that so little is known about how the single PS stimulus directly and indirectly transmits the ‘eat-me’ signal to multiple PS receptors, and how a phagocyte reaps meaningful information from such a general signal. Elucidation of these important issues relies on more comprehensive understanding of key players involved in this process.
Urokinase plasminogen activator receptor (uPAR, CD87) is a multidomain glycosylphosphatidylinositol (GPI)-anchored protein, implicated in many cellular processes, ranging from cell proliferation, motility, angiogenesis, wound repair, inflammation, tumor invasion, to metastasis (8–9). Moreover, the altered expression and function of uPAR are closely related with inflammatory conditions and autoimmune disorders such as lupus and atherosclerosis (10–14) We and others have reported that uPAR facilitates clearance of not only bacteria and parasites (15–16), but also apoptotic cells (17–18). Given that efferocytosis is critical for resolution of inflammation and uPAR expression and function are modulated in many pathological processes, understanding the mechanisms for uPAR-mediated efferocytosis is very important. On the other hand, the study by Park et al. has proposed uPAR as a negative regulator of phagocytosis of viable cells (17). These studies suggest that uPAR plays a differential role in engulfment of apoptotic cells and viable cells and its function is complex in these processes (19). Therefore, it is necessary to further investigate the role of uPAR in efferocytosis and the underlying mechanisms.
In this study, by using uPAR-deficient mouse model we present evidence that uPAR is required for internalization of apoptotic cells, which is mediated by a PS-dependent pathway. High-molecular-weight kininogen (HK), a known uPAR ligand in plasma, physically binds to PS and serves as an opsonin bridging the interaction of uPAR with PS. Since HK is a critical component of plasma kallikrein-kinin system (also named as plasma contact activation system), its involvement in phagocytosis of apoptotic cells reveals a novel role for this system in regulation of innate immunity and apoptotic cells-associated procoagulant activity.
Materials and Methods
Mice and reagents
Breeding pairs of uPAR knockout (uPAR−/−) mice on C57BL/6 background were purchased from the Jackson Laboratory and housed in a specific pathogen-free facility. Eight to ten week-old mice were used in experiments, and wild-type littermates were used control. All of the animal protocols have been reviewed and approved by Institutional Animal Care and Use Committee of Soochow University and Temple University. Reagents were purchased form Sigma unless otherwise specified.
Preparation of recombinant proteins and synthetic peptides
The recombinant soluble human uPAR (suPAR) protein was generated and purified as previously described (20). Recombinant protein of human HK heavy chain (HC, aa 18–380) and light chain (aa 390–644) was generated as previously described (21). The synthetic peptides GKD25(GKDFVQPPTKICVGCPRDIPTNSPE), GCP28(GCPRDIPTNSPELEETLTHTITKLNAEN), NAT26(NATFYFKIDNVKKARVQVVAGKKYFI),KKY30(KKYFIDFVARETTCSKESNEELTESCETKK), RET27(RETTCSKESNEELTESCETKKLGQSLD), LDC27(LDCNAEVYVVPWEKKIYPTVNCQPLGM), VSP21(VSPPHTSMAPAQDEERDSGKE), GKE19(GKEQGHTRRHDWGHEKQRK), HNL21(HNLGHGHKHERDQGHGHQRGH), GHG19(GHGLGHGHEQQHGLGHGHK), FKL20(FKLDDDLEHQGGHVLDHGHK), and HKH20 (HKHGHGHGKHKNKGKKNGKH) were based on sequences in HK as described previously (22–23).
Cell preparation and transfection
Human full-length uPAR cDNA was subcloned into pIRES2-EGFP at the XhoI and SalI sites (18). Peritoneal macrophages were elicited from mice as previously described (24). Culture of HEK293 cells and transfection using Lipofectamine™ 2000 were performed as previously described (18).
Preparation of liposomes and phospholipid-coated beads
Phosphatidylcholine (PC), PS, 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) and 1-oleoyl-2-{6-[(7-nitro-2-1,3-benzoxadiazol-4-yl)amino]hexanoyl}-sn-glycero-3- phosphocholine (NBD-PC) were from Avanti-Polar Lipids. To prepare liposome containing 100% PC (PC liposome) or PC/PS (80:20 mol%, PS liposome), the appropriate amounts of phospholipids were dissolved in chloroform in a glass tube. After dried under nitrogen, phospholipids were suspended in PBS and vortexed (25–26). To prepare phospholipids-coated beads, 6.5 mg/mL of Nucleosil 120–3 C18 beads (Macherey-Nagel, Inc) in 1 mL of chloroform were incubated with 2.5 mg/mL DOPC (PC beads) or mixed DOPC:PS (80:20 mol%, PS beads). After dried under nitrogen, the beads were resuspended in PBS and sonicated, followed by labeling with 2.5 μM NBD-PC at 37°C for 20 minutes. After centrifugation at 13,000 × g for 10 minutes, the beads were washed with ice-cold PBS twice to remove remaining NBD-PC, and resuspended in PBS.
In vitro and in vivo phagocytosis assay
Apoptotic neutrophils or PS-coated beads were used as phagocytic targets. Studies using human blood were performed after approval by the Soochow University or Temple University Institution Review Boards with informed consent in accordance with the Declaration of Helsinki. Human neutrophils were collected and purified from peripheral blood as previously described (27). Mouse neutrophils were purified from bone marrow cells using negative selection kit (Miltenyi Biotec Inc.). Apoptosis was induced by heating at 42°C for 60 minutes and incubation at 37°C for 3 hours. Apoptosis was evaluated by an Apoptosis Detection Kit (BD Biosciences) and flow cytometry, and verified as positive for annexin V and less than 10% positive to PI staining. Apoptotic cells were purified by magnetic annexin V microbeads (Miltenyi Biotec, Inc.). Phagocytosis assay by flow cytometry was performed as previously described (7).
The in vivo phagocytosis assay was performed according to the method described by Morelli et al. with slight modification (28). A total of 4×107 NBD-labeled PS beads were injected intravenously into mice. After 12 hours, the mice were euthanized, spleen was collected and digested with 400 U/ml type IV collagenase at 37°C for 30 minutes. Splenic mononuclear cells were isolated by density gradient centrifugation at 300g for 30 minutes, and labeled with Biotin-conjugated anti-F4/80 antibody (eBioscience). F4/80-positive macrophages were collected by anti-Biotin magnetic microbeads (Miltenyi Biotec, Inc.). Macrophages engulfing NBD-PS beads were measured by flow cytometry.
Spleen and thymus index and in situ identification of nuclear DNA fragmentation
Apoptotic cells (2×107 per mouse) were intravenously injected in to mice every day for one week. After mice were sacrificed, spleen and thymus were removed, blotted dry, freed of blood clots. Both of body weight and spleen or thymus weight were weighed for calculation of spleen/thymus index (%) [= spleen/thymus weight (mg)/body weight(g)×100%](29).
For in situ calculation of apoptotic cells accumulated in spleen, spleen was immersion-fixed in 10% buffered formalin and embedded in paraffin. After tissue was sectioned and deparaffinized, it was stained using TdT-mediated dUTP nick-end labeling (TUNEL) method (ApopTag fluorescein in situ apoptosis detection kit, Millipore). Nuclei were counterstained with DAPI to aid in visualization. The total DAPI positive cells and cells with FITC-labeled apoptotic nuclei was numerated under fluorescent microscope and analyzed with KS400 Image Analysis System (KS400; Zeiss, Heidelberg, Germany).
Amnis ImageStream Data Analysis
As we previously described (18), ImageStream data for the uptake of NBD-PC labeled PS beads or apoptotic cells by PE-F4/80 labeled macrophages were acquired using the Amnis ImageStream Analyzer instrument equipped with the Amnis INSPIRE software. This software can distinguish the binding and internalization of targets. The beads or apoptotic cells that overlapped with two peripheral pixels of phagocytes was considered as bound but not internalized, the rest was considered as internalized. The percentage of cells of binding and internalization were analyzed with the Amnis IDEAS software.
Measurement of the binding of plasma proteins to apoptotic cells and liposomes
Single chain human HK (Enzyme Research Laboratories), uPA and Vitronectin (R&D Systems) was biotinylated using EZ-Link® Sulfo-NHS-LC-Biotinylation Kit (Thermo Scientific) (30). Binding of biotinylated-protein to cell surface was measured by flow cytometry. Binding of HK to liposomes was analyzed using Western blot.
Surface placsmon resonance (SPR) assay
The SPR analysis was carried out with the Biacore X system (GE Healthcare, Pittsburgh, PA). PBS-Zn buffer (16mM Na2HPO4, 3mM NaH2PO4, 0.15M NaCl, 50μM ZnCl2, pH 7.4) was used as running buffer, and 7.4μg/ml HK protein was diluted in 10mM acetate (pH4.0) and immobilized onto flow cell 2 (FL2) of CM5 sensor chip (GE Healthcare). The remaining activated sites on the chip surfaces were blocked with a 35 μl injection of an ethanolamine hydrochloride solution (1 M at pH 8.5), followed by a 60 s wash with 2 M NaCl to remove any nonspecifically adsorbed materials. Approximately 3000 RU of the immobilized HK protein were obtained. For all subsequent measurements a flow rate of 30 μl/min was used. PS liposome (PS: PC = 1:9) or PC diluted with running buffer were allowed to bind to HK protein for 3 min to reach equilibrium, followed by 20 min of dissociation. The response curves were obtained by subtraction of the signals over the reference surface from the binding response to HK immobilized surface, and analyzed using BIAevaluation software, version 2.0 (GE Healthcare).
Measurement of bradykinin production
Bradykinin was measured using an ELISA kit (Enzo Life Sciences, Farmingdale, NY, USA).
Immunoprecipitation and immunoblotting
Immunoprecipitation and immunoblotting were performed as previously described (7).
Assay for Rac1 activation
GTP-loaded Rac1 was precipitated with GST-fusion protein expressing human PAK1 (GST-PAK1, aa56–272), and detected by immunoblotting with anti-Rac1 MoAb (Millipore) (7).
Statistical analysis
The results are expressed as means ± SD. In One-way analysis of variance (ANOVA, for multiple groups) or Student’s t test (for comparisons between 2 groups), a p value <0.05 was considered statistically significant. Unless stated otherwise, the data are from a single experiment representative of at least three separate experiments.
Results
uPAR is required for the internalization of apoptotic cells by macrophages
Our previous study using uPAR-overexpression cell lines have shown that uPAR plays an important role in phagocytosis of apoptotic cells (18). Since uPAR overexpression failed to stimulate engulfment of viable cells, uPAR recognizes determinant signal on the surface of apoptotic cells (18). Our initial motivation was to verify the function of uPAR using a genetic model and determine the underlying mechanism. Tethering (binding) and internalization of apoptotic cells by phagocytes are two key steps of efferocytosis. To verify the function of uPAR in efferocytosis, the binding and internalization of apoptotic cells was compared between wild type (WT) and uPAR−/− macrophages. As shown in Figure 1A, the uPAR protein was absent in uPAR−/− peritoneal macrophages (upper panel), and its membrane expression was deficient on uPAR−/− macrophages (lower panel). In the flow cytometry-based phagocytosis assay, there was no remarkable difference in the association, including binding and ingestion, of apoptotic cells between WT and uPAR−/− macrophages (Figure 1 Bi). To distinguish the role of uPAR in binding and internalization of apoptotic cells, macrophages were treated with trypan blue (TB), which completely quenches fluorescence of the dye that stains unengulfed cells (Figure 1B, ii). Although when treated with TB wild-type macrophages had similar internalization with those without TB treatment, uPAR−/− macrophages displayed significantly lower internalization of apoptotic cells than WT macrophages by 60% (TB:+, Figure 1B, i). This result suggests that uPAR is selectively required for internalization of apoptotic cells, but not the tethering. About 80% wild-type macrophage internalized more than 1 cells, the percentage of phagocytic cells was of no difference before and after TB treatment. It has been suggested that uPAR on both phagocytes and target cells regulates phagocytosis (17). Thus, we used this TB treatment method to examine the specific contribution of uPAR expressed on both macrophages and apoptotic cells to internalization of apoptotic cells. As shown in Figure 1C, when viable neutrophils were used as target, both WT and uPAR−/− macrophages had low internalization capacity, and uPAR−/− macrophages had a significantly stronger ingestion than WT macrophages. However, when incubated with apoptotic neutrophils, uPAR−/− macrophages displayed a dramatic reduction in internalization of apoptotic cells, regardless of the presence or absence of uPAR on apoptotic cells (Figure 1C). This result indicates that uPAR is a bona fide receptor for internalization of apoptotic cells. Similarly to uPAR−/− macrophages, when uPAR expression of human monocyte cell line THP-1 cells was downregulated by siRNA, their internalization of apoptotic cells was significantly reduced (Supplemental Figure 1). As shown in Figure 1C, uPAR−/− macrophages had greater phagocytosis of viable cells than wild type macrophages and wild type macrophages have greater phagocytosis of viable uPAR−/− cells than that of viable wild type cells, which is consistent with the observations of Park et al (17). The results from these two studies suggest that uPAR plays a distinct role in phagocytosis of viable cells and phagocytosis of apoptotic cells.
Figure 1. uPAR is required for the internalization of apoptotic cells in vitro.
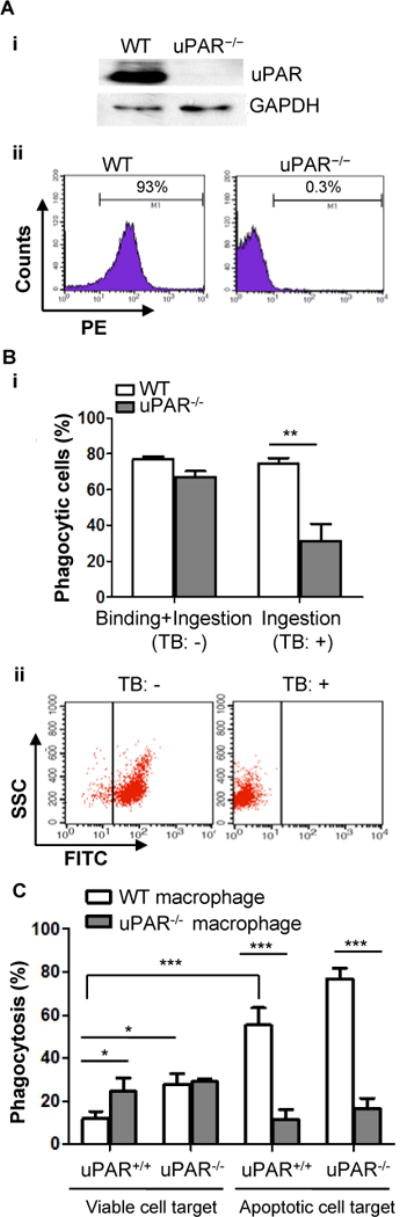
(A) Absence of uPAR expression in uPAR-deficient macrophages. Macrophages were isolated from WT and uPAR−/− mice (n=3) and pooled together, respectively, their expression of uPAR was measured by Western blot analysis (upper panel) and flow cytometry (lower panel). The data are representative of three experiments.(B) Internalization of apoptotic cells. Peritoneal macrophages from WT and uPAR−/− mice were incubated with apoptotic cells at 37°C for 15 minutes, followed by treatment with or without 0.04% trypan blue (TB) to quench the fluorescence of unengulfed apoptotic cells. Phagocytosis of apoptotic cells was analyzed by flow cytometry. Association is indicated in percentage as binding and ingestion (TB:-) and ingestion (TB:+). **, p<0.01(i). Quench of fluorescence signal of CFDA-SE-stained apoptotic cells by TB was confirmed by flow cytometry (ii). (C) Viable or apoptotic neutrophils (5×105) from WT or uPAR−/− mice were incubated with adherent WT or uPAR−/− macrophages (5×104) on 96-well plate at 37°C for 15 minutes. After macrophages were treated with TB, phagocytosis was analyzed by flow cytometry. *, p<0.05; ***, p<0.001.
Amnis cellular imaging technology is able to distinguish the binding and internalization of phagocytic targets by visualizing interactions between phagocytes and target cells with confocal accuracy, and is capable of quantitating phagocytes with the bound and internalized targets. PS is major “eat-me signal” on apoptotic cells. We used this technique and PS beads as a second approach to verify the role of uPAR in PS-dependent internalization. Shown in Figure 2A(i) are representative panels of peritoneal macrophages binding and engulfing NBD-labeled PS beads. Analysis by Amnis INSPIRE software indicates that uPAR−/− macrophages exhibited higher capacity of binding PS-beads than WT macrophages (58.2±5.7% versus 32.5 ±4.6%, p<0.01, Figure 2A, ii). However, uPAR−/− macrophages had significantly reduced internalization of PS-beads (28.9±3.9%versus 60.8±8.4 %, p<0.01, Figure 2A, ii). When apoptotic cells were used as target, uPAR−/− macrophages also exhibited a significant decrease in internalization (78.9±3.4% versus 30.2±7.3 %, p<0.01), and higher capacity of binding than WT macrophages (22.1±2.9% versus 68.8 ±7.2%, p<0.01) (Figure 2B). These results serve as additional evidence showing that uPAR is indeed required for internalization of apoptotic cells. These data also suggest that apoptotic cells and PS-expressing beads may bind to uPAR through HK and to other PS receptors simultaneously, the PS-mediated crosslinking of uPAR with other receptors is required for the internalization. In the absence of uPAR, macrophages have defect in internalization, but the targets are still bound to other receptors and thus tethered to the surface of macrophages.
Figure 2. uPAR is required for internalization but not the binding of PS beads.
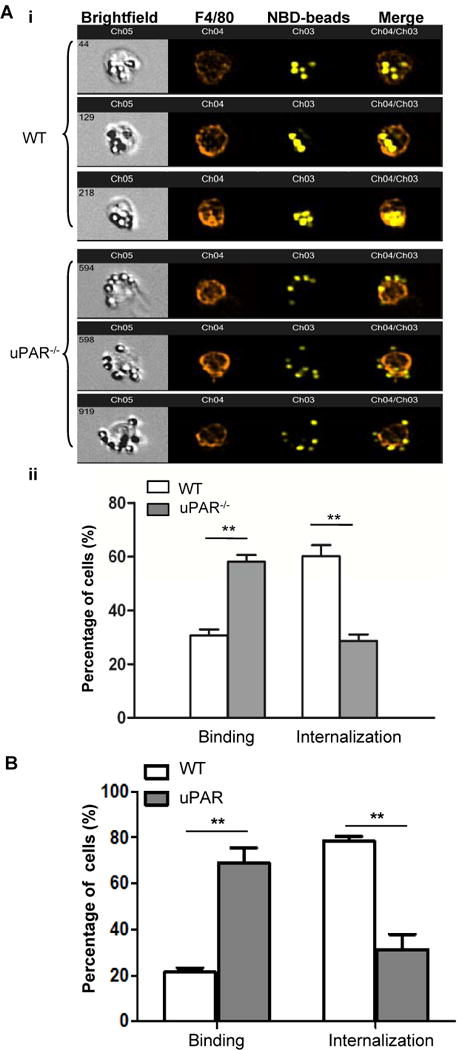
(A) Amnis ImageStream Data Analysis. Peritoneal macrophages from WT and uPAR−/− mice were incubated with NBD-labeled PS beads at 37°C for 20 minutes. After washing with PBS, macrophages were labeled with PE-F4/80 antibody and evaluated using Amnis ImageStream Data Analysis, as described in the Materials and Methods. Shown are the representative output images after Amnis acquisition (i). ImageStream data were acquired using the Amnis ImageStream Analyzer and the percentage of macrophages that bound (binding) and internalized (internalization) PS-beads were quantitated with the Amnis IDEAS software. (**, p<0.01, ii). (B) As described above, apoptotic cells were used as target instead of PS-beads, peritoneal macrophages from WT and uPAR−/− mice were labeled with CFDA-SE prior to incubation with PKH26-labeled apoptotic cells at 37°C for 20 minutes. The percentage of macrophages binding (Binding) and internalizing (Internalization) apoptotic cells were analyzed with the Amnis IDEAS software (**, p<0.01).
Deficiency of uPAR leads to accumulation of apoptotic cells in mouse spleen and decreases splenic macrophage efferocytosis in vivo
We next examined the in vivo function of uPAR in efferocytosis. After mice were challenged with intravenous injection of apoptotic cells, uPAR-deficient mice but not WT mice exhibited pronounced splenomegaly (Figure 3A, i and ii), although WT and uPAR-deficient mice without injection of apoptotic cells showed no significant difference in mean spleen index at baseline (Figure 3A, ii). After mice received injection of apoptotic cells, the size of thymus (mean thymus index) of uPAR-deficient mice was not significantly different with that of WT (1.52±0.52 versus 1.67±0.33, p>0.05), suggesting that spleen is probably the major site where uPAR is needed for the clearance of injected apoptotic cells. As measured by a TUNEL assay, the accumulation of apoptotic cells in uPAR−/− spleens was increased by about 10 fold than WT spleens (p<0.01, Figure 3B, i and ii). Moreover, as shown in Figure 3C, in an in vivo phagocytosis assay, uPAR−/− macrophages displayed a lower internalization of PS beads (i) or apoptotic cells (ii) by about 50% than WT macrophages, which is consistent with the in vitro observations (Figure 1C). Because the exposure of a large number of apoptotic cells causes autoantibody production in mice, especially anti-PS antibodies, autoantibodies in sera were measured with an ELISA assay. In sera from uPAR−/− mice, the level of anti-PS immmunoglobulins was significantly higher than that in WT mice (Supplemental Figure 2), suggesting that the impaired phagocytosis of apoptotic cells in uPAR−/− mice causes autoantibody production in vivo.
Figure 3. uPAR-deficient mice exhibit impaired clearance of apoptotic cells.
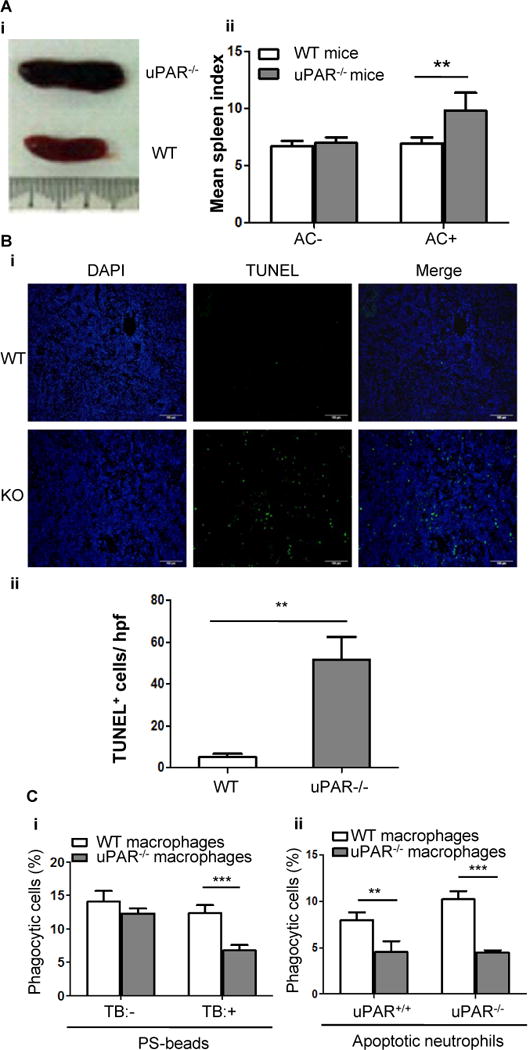
(A) Apoptotic cells (2×107 per mouse) were intravenously injected into WT and uPAR−/− mice (n=7) every day for 7 days. Shown is the representative gross appearance of spleens on day 8 (i). (ii) The mean spleen index of WT and uPAR−/− mice that received injection of PBS (AC−) and apoptotic cells (AC+) is shown.**, p<0.01. (B) Spleens of the above experiments were further processed for paraffin-embedded sections. Apoptotic cells were detected by TUNEL method. Nuclei were counterstained with DAPI. Representative images (20×) were shown (i, bars =100 μm). (ii) TUNEL-positive cells were enumerated in 10 randomly chosen high-power fields (hpf). **, p<0.01. (C) Internalization of PS-coated beads or apoptotic neutrophils in vivo. As described in the Materials and Methods, 4×107 NBD-PC-labeled PS-beads (i) or apoptotic neutrophils isolated from WT or uPAR−/− mice (ii) were intravenously injected into WT and uPAR−/− mice (n=5). Six hours after injection, spleens were harvested and homogenized, and F4/80-positive macrophages were purified. After treatment with or without TB, internalization of PS-coated beads was analyzed by flow cytometry (i). After treatment with TB, internalization of uPAR+/+ and uPAR−/− apoptotic neutrophils was analyzed by flow cytometry (ii). **, p<0.01, ***, p<0.001.
uPAR mediates internalization of apoptotic cells through recognition of PS
The decrease in internalization of PS beads by uPAR−/− macrophages suggest that uPAR mediates efferocytosis is through interaction with PS. To further dissect the mechanism underlying the role of uPAR in efferocytosis, we examined uPAR-dependent events using a cell line. We generated HEK293 cells overexpressing uPAR and EGFP (uPAR-293 cells) using a bicistronic IRES vector, the expression of uPAR on cell surface was confirmed by flow cytometry (Figure 4A). Compared with EGFP-293 cells that were transfected with empty IRES vector expressing EGFP alone (control mock), uPAR-293 cells exhibited significantly higher capacity in internalization of apoptotic cells (Figure 4B). However, the expression of uPAR on uPAR-293 cells did not increase the internalization of viable cells (Figure 4B). Treatment of these cells with soluble form of uPAR (suPAR) significantly inhibited internalization of apoptotic cells (Figure 4C), suggesting that the gain of functional phagocytosis is conferred by surface expression of uPAR. Using this cell model, we determined whether uPAR-mediated internalization of apoptotic cells is dependent on PS. Annexin V is a 35 kDa phospholipid-binding protein and has a high binding affinity for PS in the presence of calcium (Ca2+). When apoptotic cells were preincubated with 50 μg/mL of unlabeled annexin V and Ca2+, the binding of FITC-labeled annexin V was completely blocked (Figure 4D, i), suggesting that the binding of annexin V at the concentration of 50 μg/mL to PS on apoptotic cells became saturated. At the same concentration, annexin V significantly inhibited the engulfment by uPAR-293 cells in the presence of Ca2+, but not the absence of Ca2+ (Figure 4D, ii). In addition, PS liposome, but not PC liposome, significantly inhibited uPAR-mediated engulfment of apoptotic cells (Figure 4E). These results suggest that uPAR-mediated internalization of apoptotic cells is dependent on PS signal.
Figure 4. Phosphatidylserine is necessary for uPAR-mediated internalization of apoptotic cells.
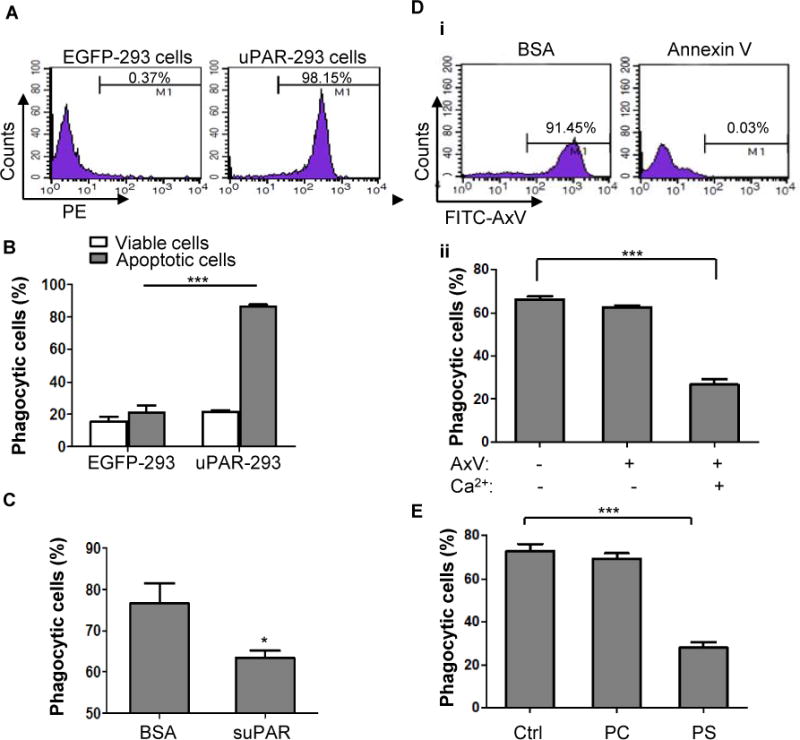
(A) HEK293 cells were transfected with pIRES2-EGFP plasmid (EGFP-293 cells) or IRES2-EGFP-uPAR plasmid (uPAR-293 cells), their expression of uPAR on membrane was measured by flow cytometry. (B) EGFP-293 cells and uPAR-293 cells were incubated with PKH26-labeled viable or apoptotic cells for 2 hours, respectively. Phagocytosis by green fluorescent 293 cells was evaluated after treatment with TB as described in the legend for Figure 1. ***, p<0.001. (C) suPAR inhibits phagocytosis of apoptotic cells in uPAR-293 cells. uPAR-293 cells were incubated with PKH26-labeled apoptotic cells in the presence of 1 μg/mL suPAR or BSA for 2 hours, which were subjected to the phagocytosis assay. *, p <0.05.(D) After preincubation with 50 μg/mL of unlabeled annexin V or BSA plus 2.5 mM CaCl2 for 20 minutes, apoptotic cells were labeled with FITC-conjugated annexin V (FITC-AxV), followed by flow cytometric analysis (i). After preincubation with 50 μg/mL of unlabeled annexin V in the presence or absence of 2.5 mM CaCl2 for 20 minutes, apoptotic cells were co-cultured with uPAR-293 cells, followed by treatment with TB and internalization analysis using flow cytometry (ii). ***, p < 0.001. (E) After preincubation with PBS (Ctrl), 50 nM PC liposome or PS liposome for 20 minutes, apoptotic cells were co-cultured with uPAR-293 cells, followed by treatment with TB and internalization analysis using flow cytometry.***, p < 0.001.
Plasma high molecular weight kininogen (HK) binds to PS and opsonizes uPAR-mediated internalization of apoptotic cells
To date, there are no data showing the direct interaction between uPAR and PS. In a cell-free system we failed to detect recombinant suPAR protein bound to PS liposome (data not shown). We presumed that uPAR interacts with PS via opsonin(s), and thus compared the internalization capacity of uPAR-293 cells in the presence and absence of serum. As shown in Figure 5A, the internalization of apoptotic cells by uPAR-293 cells was completely prevented in the absence of serum, suggesting that opsonin(s) in serum is necessary for uPAR-mediated efferocytosis. It has been known that uPAR has three ligands in plasma, uPA, vitronectin and HK(31–32). To test which protein(s) mediates uPAR efferocytosis, the assays were done using the serum depleted of each protein that was achieved by an immunoprecipitation method. Compared with normal serum, the internalization by uPAR-293 cells was markedly compromised in the serum depleted of HK [HK(−), p<0.01], which was reversed by replenishment of either HK or its cleaved form two chain HKa (Figure 5B). In contrast, when uPAR-293 cells were cocultured with apoptotic cells in uPA- or vitronectin-depleted serum, there was no reduction in engulfment (Supplemental Figure 3). Thus, HK is required for uPAR-mediated internalization of apoptotic cells.
Figure 5. HK binds to apoptotic cells and is required for uPAR-mediated internalization.
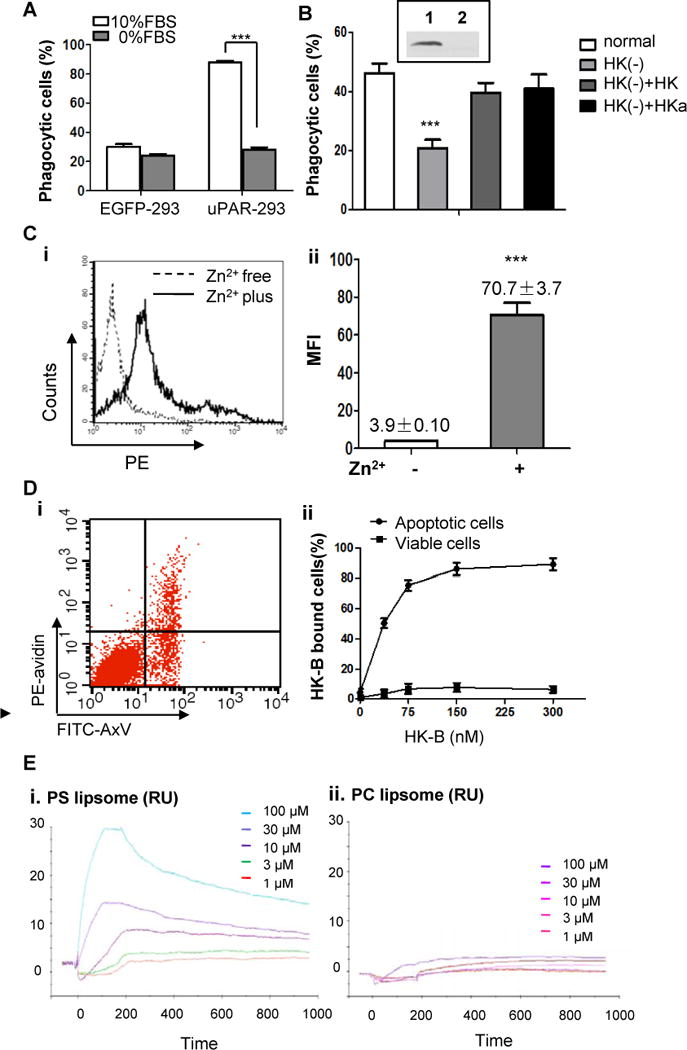
(A) EGFP-293 cells and uPAR-293 cells were co-cultured with apoptotic cells in the presence 10% FBS or 0% FBS for 2 hours. Internalization of apoptotic cells was analyzed by flow cytometry after treatment with TB as described in the legend for Figure 4. ***, p <0.001.(B) The levels of HK in normal serum (Lane 1, insert) and HK-depleted serum (lane 2, insert) were analyzed by immunoblotting. The uPAR-293 cells were incubated with apoptotic cells in presence of normal serum, HK-depleted serum [HK(−)], HK-depleted serum replenished with 50 nM HK [HK(−)+HK] or HKa [HK(−)+HKa], respectively. After treatment with TB, the internalization of apoptotic cells was evaluated by flow cytometry as described in the legend for Figure 4. ***, p< 0.001. (C) Apoptotic cells were incubated with 100 nM biotinylated HK (HK-B) in PBS with or without 50 μM ZnCl2 at 4°C for 15 minutes. After washing with PBS containing 0.35% BSA and 50 μM ZnCl2, the cells were stained with PE-avidin and the fluorescence intensity of was quantitated by flow cytometry. Representative data of HK bound to apoptotic cells in the presence (Zn2+ plus) or absence (Zn2+ free) of zinc ions (i) and the mean fluorescence intensity (MFI) from triplicate samples (ii) are shown. ***, p <0.001. (D) Mixed apoptotic cells and viable cells were incubated with 50 nM HK-B in Tyrode’s buffer containing 0.35% BSA and 50 μM ZnCl2 at 4°C for 15 minutes. After washing with PBS containing 50 μM ZnCl2, the cells were labeled with PE-avidin and non-saturated amounts of FITC-annexin V (AxV). The intensity of PE fluorescence for HK-B bound to apoptotic cells (FITC-AxV-positive) and viable cells (FITC-AxV-negative) was analyzed by flow cytometry (i). Shown in (ii) was the quantification of the percentage of PE-positive cells at the indicated concentrations of HK-B, representing HK-B bound cells (%). (E) SPR assay. PS liposome (i) or PC liposome (ii) at the indicated concentrations was allowed to bind to HK-immobilized CM5 sensor chip to reach equilibrium, followed by dissociation. The response curves were analyzed using BIAevaluation software.
We next studied whether and how HK binds to apoptotic cells. We have previously shown that HK binding to cell surface or negatively-charged molecules is dependent on ZnCl2 (Zn2+)(31, 33), we began with investigation of the binding of HK to apoptotic cells in the presence and absence of Zn2+. As shown in Figure 5C, in the absence of Zn2+, few HK-B bound to apoptotic cells. However, the addition of 50 μM Zn2+increased the binding of HK-B by about 18-fold (Figure 5C, i and ii). When 50 nM HK was incubated with mixed apoptotic cells and viable cells (Figure 5D, i), HK selectively bound to apoptotic cells that were annexin V-positive, but not to annexin-V negative viable cells, whereas the binding of HK at this concentration was not saturable (Figure 5D, ii). The binding of HK to apoptotic cells was concentration-dependent (Figure 5D, ii), and the Km was less than 30 nM. Thus, HK specifically binds to apoptotic cells. In contrast, vitronectin and uPA, bound to both apoptotic cells and viable cells (Supplemental Figure 4). SPR experiments were performed to investigate the binding of PS liposome to immobilized HK. Each concentration used were diluted and injected in triplicate and the three curves were averaged to one curve per concentration (as shown) and analyzed using the Biaevaluation program as described in Materials and Methods. The increase in response units in all five traces after the injection of liposomes indicates the binding of the PS liposomes, but not PC liposomes, to the surface of the chip (Figure 5E). Additional injections of PS liposomes (300 μM) did not increase the signal, indicating a complete coverage of the surface with PS (data not shown). These results demonstrate that HK is physically and specifically associated with PS, HK may recognize apoptotic cells by direct binding to PS.
HK has two functional domains, heavy chain (HC) and light chain (LC), which are produced by the cleavage. We further test which domain is involved in HK association with apoptotic cells. When 50 nM-Biotin-labeled HK was incubated with apoptotic cells in the presence of recombinant HC or LC, both of HC and LC inhibited the binding of Bio-HK to apoptotic cells (Figure 6A,i), suggesting that HK binds to apoptotic cells through both HC and LC. To examine whether HK directly binds to PS, HK was incubated with PC or PS liposome. As shown in Figure 6B(i), HK bound to PS liposome was much more abundant than that to PC liposome. Moreover, like HK, HC and LC bound to PS liposome directly (Figure 6B, ii). It has been shown that D3 (273~376 aa) in HC and D5 (384~498 aa) in LC are major surface binding regions, we thus used peptide inhibition method to map the binding domain of HK to apoptotic cells. As shown in Figure 6C, two D3 peptides LDC21 and RET27 and one D5 peptide HKH20 dose-dependently inhibited the binding of HK to apoptotic cells. However, a GKD25 and other D3 and D5 peptides had no inhibitory effect (Figure 6C and data not shown). These data suggest that HK binding to apoptotic cells through, at least, its D3 (324~375 aa) and D5 (479~498 aa).
Figure 6. Both of heavy chain and light chain are involved in HK binding to apoptotic cells and PS.
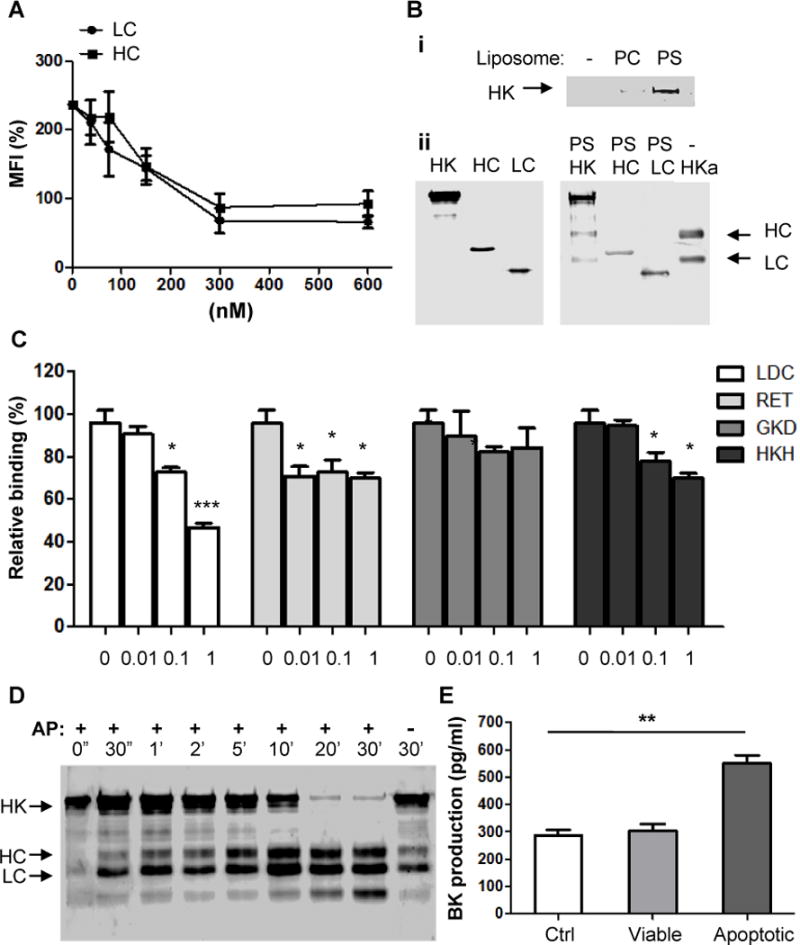
(A) HC and LC inhibit the binding of HK to apoptotic cells. Apoptotic cells were incubated with HC and LC at the indicated concentrations at 4°C for 20minutes, followed by addition of biotin-HK (100 nM) and PE-labeled avidin. The rest steps were as described in the legend for Figure 5(B). Shown are the measurements from 3 separate experiments. (B) HK binds to PS liposome via its heavy chain and light chain. (i) Five μ g of HK protein were incubated with PBS (−), 50 nM PC or PS liposomes in Tyrode’s buffer containing 0.35% BSA and 50 μM ZnCl2 at 4°C for 30 minutes. After ultracentrifugation at 55000 rpm, the pellets were washed twice and solubilized by sample buffer and analyzed by immunoblotting with polyclonal anti-HK antibody. (ii) Before (left panel) or after (left panel) five μg of HK, recombinant heavy chain (HC) or light chain (LC) of HK were incubated with 50 nM PS liposomes. After incubation with PS liposomes, the samples were ultracentrifuged and the pellets were collected. Samples were analyzed by immunoblotting with polyclonal anti-HK antibody. HKa serves as control for molecular weight. (C) In the presence of various peptides at the indicated concentrations, apoptotic cells were incubated with 100 nM biotinylated HK (HK-B), followed by staining with PE-avidin and quantitation by flow cytometry. (D) HK at 30 nM was incubated with apoptotic cells in the presence of prekallikrein (30 nM) at 37°C for the indicated time periods. Apoptotic cells were sonicated and centrifuged at 800 g for 5 minutes. Pellets containing membrane fraction was analyzed by Western blotting using anti-HK Ab. (E) Bradykinin production. Fresh human citrated cell-free plasma was incubated with 5×105 viable cells (Viable), apoptotic cells (Apoptotic) or equal volume of PBS (Ctrl) at 37°C for 30minutes, followed by measurement of bradykinin production. **, p <0.01.
Association with apoptotic cells induces the cleavage of HK and production of bradykinin
As a major component of plasma KKS, HK is responsible for binding to activation surfaces for assembly of KKS components, leading to kallikrein activation and its cleavage of HK to two-chain HKa and bradykinin. We thus examined whether the association of HK with apoptotic cells mediates its cleavage to HL can LC. As detected by Western blotting, incubation with apoptotic cells resulted in the rapid cleavage of HK as early as 30 seconds, and as a function of time (Figure 6D). In a reduced SDS gel, the cleavage products include three major fragments with molecular weight of 62, 56, and 45 kDa, respectively, which represent heavy chain (HC, aa 18–380, 62kDa) and light chain (LC, aa 390–644, 56 kDa), and a secondary 45 kDacleavage fragment of LC. Correspondingly, incubation of serum with apoptotic cells, but not with viable cells, significantly increased the production of bradykinin (Figure 6E). Thus, apoptotic cell membrane is a novel surface for activation of plasma KKS.
uPAR-mediates efferocytosis through p130Cas-CrkII-Dock-180-Rac1 pathway
Since uPAR is a GPI-anchored protein and is localized in the membrane rafts, we thus tested whether the integrity of rafts is involved in uPAR efferocytosis. As shown in Figure 7(A), treatment MβCD that depletes lipid in rafts significantly inhibited internalization of apoptotic cells by uPAR-293 cells, suggesting the dependence of uPAR-mediated efferocytosis on membrane rafts. It has been known that membrane rafts is associated with intracellular signaling molecules complex for Rac1 activation. Because Rac1 activation mediated by p130Cas-CrkII-Dock180 pathway is critical for internalization of apoptotic cells, and uPAR activates this pathway in other cells (34–35), we investigated whether Rac1 activation is required for uPAR efferocytosis. In uPAR-293 cells expressing dominant negative Rac1/N17 cDNA (Figure 7B,i), their internalization capacity was significantly reduced (p<0.01, Figure 7B, ii). Moreover, when uPAR-293 cells were stimulated with PS liposome plus HK, CrkII formed a complex with p130Cas and Dock-180, and active forma of Rac1 was increased (Figure 7C). However, HK or PS-liposome itself alone did not have such effect (Figure 7D). These data suggest that HK mediates PS–uPAR interaction and promotes clustering of uPAR in the plasma membrane, thereby enhancing the intracellular p130Cas-CrkII-Dock-180 pathway leading to Rac1 activation.
Figure 7. Integrity of lipid rafts and Rac1 activation are required for uPAR mediated-internalization of apoptotic cells.
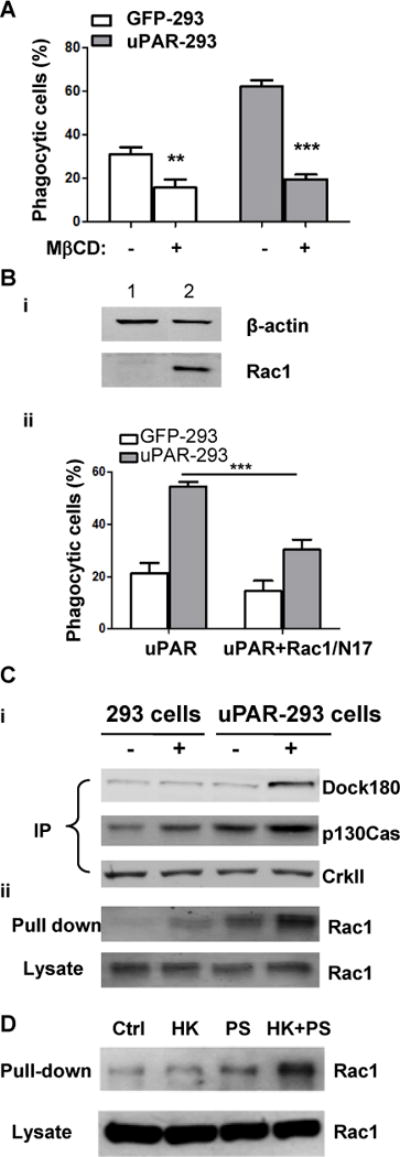
(A) After preincubation with or without 2.5 mM mβCD for 30 minutes, apoptotic cells were co-cultured with EGFP-293 or uPAR-293 cells. The internalization of apoptotic cells was analyzed by flow cytometry as described in the legend for Figure 4. ***, p< 0.001. (B) EGFP-293 or uPAR-293 cells were transfected with empty vector (lane 1) or Rac1/N17 (lane 2) cDNA for 48 hours, the overexpression of Rac1/N17 was detected by Western blotting using anti-Rac1 antibody with β-actin serving as a loading control (i). Internalization of apoptotic cells was analyzed (ii). **, p<0.01. (C) EGFP-293 cells and uPAR-293 cells were starved in DMEM plus 2% FBS for 8 hours, and incubated without (lanes 1 and 3) or with (lane 2 and 4) 1.0 μM PS liposome plus 600 nM HK for 1 hour in basal DMEM containing 0.35% BSA and 50 μM ZnCl2. Cell lysates were subjected to immunoprecipitation with anti-CrkII antibody (A) and Rac1 activity assay (B), respectively. The immunoprecipitates using anti-CrkII antibody were analyzed by immunoblotting with MoAb against p130Cas and polyclonal antibodies against CrkII and Dock180. Active GTP-loaded form of Rac1 and total Rac1 in cell lysates were detected by immunoblotting. (D) After starvation in DMEM plus 2% FBS for 8 hours, uPAR-293 cells were incubated with PBS (control, Ctrl), 600 nM HK (HK), 1.0 μM PS liposome (PS), or 600 nM HK plus 1.0 μM PS liposome (HK+PS) for 1 hour in DMEM containing 0.35% BSA and 50 μM ZnCl2. Cell lysates were incubated with GST-PAK1 conjugated with Glutathione Sepharose 4B beads and active GTP-loaded form of Rac1 (Pull-down) and total Rac1 in cell lysates (Lysate) were detected by immunoblotting.
Discussion
Our present study shows that uPAR plays an important role in internalization of apoptotic cells. The deficiency of uPAR causes splenomegaly resulting from impaired clearance of apoptotic cells. uPAR-mediated efferocytosis is dependent on PS pathway. Moreover, we show that HK is a novel PS-binding protein and serves as an opsonin bridging uPAR on macrophages and apoptotic cells. uPAR mediates internalization of apoptotic cells by activating p130Cas-CrkII-Dock180-Rac1 pathway. However, its role in tethering apoptotic cells is dispensable. Because uPAR is widely involved in a variety of vascular processes, its function in efferocytosis should provide a novel insight into its function in vascular biology and pathological disorders.
In our previous studies, we discovered that αvβ5 integrin is important in engulfment of apoptotic cells (1, 7, 18). We posited that integrins might dynamically form a cluster with its partner receptors in specialized membrane domains like membrane rafts that activate small GTPases. Because we have shown that uPAR, which is GPI-anchored protein localized in a lipid raft, is important for αv integrin function in endothelial cells (30). Initially we investigated whether uPAR association with αvβ5 or αvβ3 integrins in lipid rafts could be a requisite for engulfment. Although we succeeded in observing that uPAR is a novel phagocytic receptor, several lines of our data have indicated that uPAR’s function in efferocytosis is independent of integrins (18). Park et al reported that the loss of uPAR expression contributes to the enhanced clearance of viable cells (17). Thus, the role of uPAR in efferocytosis and the underlying mechanisms are complex. In this study, using trypan blue treatment (Figures 1 to 3) and the Amnis ImageStream Data Analysis (Figure 2), we found that uPAR is required for the internalization, but not for the tethering/binding of apoptotic cells. It is likely that the discrepancy on the role of uPAR in efferocytosis results from a fact that uPAR plays distinct roles in phagocytosis of apoptotic cells and phagocytosis of viable cells. It might serve as an unconventional “don’t eat me” to prevent the phagocytes to engulf healthy cells when the “eat-me” signals do not exist. When viable cells become apoptotic, uPAR may recognize “eat-me” signal PS and switch to be a phagocytic receptor. We speculate that phagocytes recognize and bind to PS on apoptotic cells via uPAR and its partner PS receptors, the PS-mediated crosslinking of uPAR with other receptors is required for the internalization. In the absence of uPAR, its partner PS receptors may still bind to apoptotic cells but have defect in internalization.
The redistribution of PS to external membrane surface is a key element of apoptotic cell recognition and is a molecular signature that dying cells can be distinguished from viable cells (1). In this study we provide evidence for the first time that uPAR-mediated efferocytosis is dependent on PS. Both of annexin V and PS liposome significantly inhibit uPAR-mediated phagocytosis of apoptotic cells (Figure 4). Unlike other receptors capable of directly recognizing PS, such as TIM-1/4(5), Stabilin-2(6), and brain angiogenesis inhibitor 1 (BAI1) (36), recombinant uPAR protein did not bind to PS liposome and apoptotic cells (data not shown). Correspondingly, in the absence of serum proteins, uPAR-mediated phagocytosis was completely prevented (Figure 5A). Thus, the role of uPAR in efferocytosis is dependent on opsonization of serum protein(s). Although three serum proteins uPA, vitronectin and HK serve as ligands for uPAR (8), we found that only HK binds to apoptotic cells and PS liposome, and the binding was in a concentration-dependent manner (Figure 5). In consistent, uPAR-mediated efferocytosis is inhibited in the serum lacking HK (Figure 5). Thus, HK serves as an opsonin for uPAR-mediated efferocytosis. This concept is supported by previous observations. First, using 2-D gel electrophoresis and mass spectrometry, Anderson et al. identified three proteins in enriched factors from serum for stimulating macrophage efferocytosis, which include albumin, protein S and HK (37). Although HK is abundant in serum, they could not observe that kininogen is involved in efferocytosis in their system (37). It has been known that HK binds to anionic surface is largely dependent of Zn2+, kininogen is devoid of prophagocytic activity in their assay is probably due to no addition of Zn2+. In this study, we found that HK binding to apoptotic cells is indeed Zn2+-dependent (Figure 5). Second, Sugi et al. found that the antibody purified from plasma of patients with anti-phospholipid syndrome recognizes HK in the presence of PS (38), suggesting that plasma HK forms a complex with PS in vivo which becomes immunogenic. Third, we and others have previously shown that HK binds to a variety of cells including leukocytes and endothelial cells via uPAR (30–31, 39–41). Fourth, our recent study using surface plasmon resonance has shown that compared to HK, the affinity of suPAR for HKa was 53-fold tighter (42). Since incubation with apoptotic cells induces HK cleavage to HKa (Figure 6), the binding of HK to apoptotic cells causes its cleavage, which may expose its binding site for uPAR. This conformational change in HK being cleaved to HKa dynamically facilitates its more stable interaction with uPAR on phagocytes, and increases the engulfment. Besides HK, plasma also contains low molecular weight kininogen which has exact the same heavy chain but no light chain. However, LK does not bind to suPAR in our Biacore assay (42), indicating that uPAR binds to light chain of HK. Consistently, in other cells such as endothelial cells, HKa bind to uPAR (43), moreover, HKa is associated with uPAR in lipid rafts via its light chain (Domain 5). Since HK is abundant in circulation at 660 nM, it readily serves as a sensor of apoptotic cells, especially when a large numbers of apoptotic cells persistently exist. Using domain protein and peptide inhibitory assay, we found that HK binds to apoptotic cells via its D3 and D5 regions.
It has been known that Rac1 activation is the main signaling pathway leading to phagocytosis of apoptotic cells (1–2), its activation is dependent on localization in lipid rafts. Because the dominant negative expression of Rac1/N17 inhibited uPAR-mediated engulfment (Figure 7), uPAR internalizes apoptotic cells by activating Rac1 pathway. Supportively, the challenge of uPAR by HK plus PS stimulated the complex formation of CrkII with p130Cas and Dock-180, and the downstream Rac1 activation (Figure 7). During the process of cell mobilization, Rac1 is mainly activated in the membrane rafts (44). Since uPAR is a GPI-anchored protein is localized in lipid rafts, it might play a key role in recruitment of other phagocytic receptors to membrane rafts, proximal to intracellular downstream signaling such as Rac1. Because uPAR lacks a trans-membrane domain, the signal transduction originating from this receptor, at least at the plasma membrane, requires lateral interactions with co-receptors (9). It awaits further investigation that which receptor(s) cooperates with uPAR and is responsible for mediating its signaling leading to Rac1 activation.
Our results propose a novel concept that uPAR is important in clearance of apoptotic cells and maintenance of vascular homeostasis. Given that uPAR is frequently cleaved in atherosclerotic plaque, systemic bacterial infection and cancer, the altered expression of uPAR may contribute to accumulation of apoptotic cells, leading to inflammatory and autoimmune disorders and tissue damage (9, 45–48). This is helpful to explain the increase in cleaved form of uPAR in plasma associated with development of atherosclerotic lesion and production of inflammatory cytokines in plasma and plaques (10, 49).
HK, as a major component of plasma KKS, possesses multiple functions and plays an important role in thrombosis, inflammation and fibrinolysis. Our current study reveals a novel function for the KKS system on its modulation of apoptotic cell clearance and “innate apoptotic immunity”. So far, it has been known that bacteria (50), misfolded proteins (51–52), collagen (52), and cellular releasate such as heparin and polyphosphate (53) can activate the plasma KKS in vivo. Because incubation with apoptotic cells induces HK cleavage and bradykinin production (Figure 6, D and E), apoptotic cell membrane could be a new surface for plasma KKS activation in vivo.
In summary, we have demonstrated that uPAR plays an important role in internalization of apoptotic cells, which is mediated by HK opsonization of uPAR interaction with PS. In the emerging picture, this study raises the intriguing question as to whether altered activation of the plasma KKS and uPA-uPAR system and their crosstalk contribute to the immune responses triggered by apoptotic cells. A further detailed molecular dissection of these issues could lead to the better understanding of innate immunity and the development of novel therapeutic strategies that modulate inflammatory response.
Supplementary Material
Acknowledgments
We thank Xuemei Zhu for recombinant protein preparation.
This study was supported in part by grants from the National Natural Science Foundation of China (30971491, 31201058, 81270592 and 81301534), the Priority Academic Program Development of Jiangsu Higher Education Institutions, and the National Institute of Health (AR057542 and AR063290).
Footnotes
Author Contributions: A.Y., J.D., Z.X. and Y.W. performed research, collected data, analyzed and interpreted data, and performed statistical analysis; J.D., Y.W. and Q.W. designed research. Q.W. and R.W.C. contributed vital reagents. Y.W. and R.B.B. wrote the paper.
Conflicts of Interest: The authors declare that they have no conflict of interest.
References
- 1.Wu Y, Tibrewal N, Birge RB. Phosphatidylserine recognition by phagocytes: a view to a kill. Trends Cell Biol. 2006;16:189–197. doi: 10.1016/j.tcb.2006.02.003. [DOI] [PubMed] [Google Scholar]
- 2.Ravichandran Kodi S. Beginnings of a Good Apoptotic Meal: The Find-Me and Eat-Me Signaling Pathways. Immunity. 2011;35:445–455. doi: 10.1016/j.immuni.2011.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kolodgie FD, Narula J, Guillo P, Virmani R. Apoptosis in human atherosclerotic plaques. Apoptosis. 1999;4:5–10. doi: 10.1023/a:1009645730270. [DOI] [PubMed] [Google Scholar]
- 4.Van Vre EA, Ait-Oufella H, Tedgui A, Mallat Z. Apoptotic cell death and efferocytosis in atherosclerosis. Arterioscler Thromb Vasc Biol. 2012;32:887–893. doi: 10.1161/ATVBAHA.111.224873. [DOI] [PubMed] [Google Scholar]
- 5.Kobayashi N, Karisola P, Pena-Cruz V, Dorfman DM, Jinushi M, Umetsu SE, Butte MJ, Nagumo H, Chernova I, Zhu B, Sharpe AH, Ito S, Dranoff G, Kaplan GG, Casasnovas JM, Umetsu DT, Dekruyff RH, Freeman GJ. TIM-1 and TIM-4 glycoproteins bind phosphatidylserine and mediate uptake of apoptotic cells. Immunity. 2007;27:927–940. doi: 10.1016/j.immuni.2007.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Park SY, Jung MY, Kim HJ, Lee SJ, Kim SY, Lee BH, Kwon TH, Park RW, Kim IS. Rapid cell corpse clearance by stabilin-2, a membrane phosphatidylserine receptor. Cell Death Differ. 2008;15:192–201. doi: 10.1038/sj.cdd.4402242. [DOI] [PubMed] [Google Scholar]
- 7.Wu Y, Singh S, Georgescu MM, Birge RB. A role for Mer tyrosine kinase in alphavbeta5 integrin-mediated phagocytosis of apoptotic cells. J Cell Sci. 2005;118:539–553. doi: 10.1242/jcs.01632. [DOI] [PubMed] [Google Scholar]
- 8.Mondino A, Blasi F. uPA and uPAR in fibrinolysis, immunity and pathology. Trends in Immunology. 2004;25:450–455. doi: 10.1016/j.it.2004.06.004. [DOI] [PubMed] [Google Scholar]
- 9.Smith HW, Marshall CJ. Regulation of cell signalling by uPAR. Nat Rev Mol Cell Biol. 2010;11:23–36. doi: 10.1038/nrm2821. [DOI] [PubMed] [Google Scholar]
- 10.Edsfeldt A, Nitulescu M, Grufman H, Gronberg C, Persson A, Nilsson M, Persson M, Bjorkbacka H, Goncalves I. Soluble urokinase plasminogen activator receptor is associated with inflammation in the vulnerable human atherosclerotic plaque. Stroke. 2012;43:3305–3312. doi: 10.1161/STROKEAHA.112.664094. [DOI] [PubMed] [Google Scholar]
- 11.Farris SD, Hu JH, Krishnan R, Emery I, Chu T, Du L, Kremen M, Dichek HL, Gold E, Ramsey SA, Dichek DA. Mechanisms of urokinase plasminogen activator (uPA)-mediated atherosclerosis: role of the uPA receptor and S100A8/A9 proteins. J Biol Chem. 2011;286:22665–22677. doi: 10.1074/jbc.M110.202135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fuhrman B. The urokinase system in the pathogenesis of atherosclerosis. Atherosclerosis. 2012;222:8–14. doi: 10.1016/j.atherosclerosis.2011.10.044. [DOI] [PubMed] [Google Scholar]
- 13.Lugano R, Pena E, Badimon L, Padro T. Aggregated low-density lipoprotein induce impairment of the cytoskeleton dynamics through urokinase-type plasminogen activator/urokinase-type plasminogen activator receptor in human vascular smooth muscle cell. J Thromb Haemost. 2012;10:2158–2167. doi: 10.1111/j.1538-7836.2012.04896.x. [DOI] [PubMed] [Google Scholar]
- 14.Svensson PA, Olson FJ, Hagg DA, Ryndel M, Wiklund O, Karlstrom L, Hulthe J, Carlsson LM, Fagerberg B. Urokinase-type plasminogen activator receptor is associated with macrophages and plaque rupture in symptomatic carotid atherosclerosis. Int J Mol Med. 2008;22:459–464. [PubMed] [Google Scholar]
- 15.Hovius JWR, Bijlsma MF, van der Windt GJW, Wiersinga WJ, Boukens BJD, Coumou J, Oei A, de Beer R, de Vos AF, Veer Cvt, van Dam AP, Wang P, Fikrig E, Levi MM, Roelofs JJTH, van der Poll T. The Urokinase Receptor (uPAR) Facilitates Clearance of Borrelia burgdorferi. PLoS Pathog. 2009;5:e1000447. doi: 10.1371/journal.ppat.1000447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wiersinga WJ, Kager LM, Hovius JWR, van der Windt GJW, de Vos AF, Meijers JCM, Roelofs JJ, Dondorp A, Levi M, Day NP, Peacock SJ, van der Poll T. Urokinase Receptor Is Necessary for Bacterial Defense against Pneumonia-Derived Septic Melioidosis by Facilitating Phagocytosis. The Journal of Immunology. 2010;184:3079–3086. doi: 10.4049/jimmunol.0901008. [DOI] [PubMed] [Google Scholar]
- 17.Park YJ, Liu G, Tsuruta Y, Lorne E, Abraham E. Participation of the urokinase receptor in neutrophil efferocytosis. Blood. 2009;114:860–870. doi: 10.1182/blood-2008-12-193524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.D’Mello V, Singh S, Wu Y, Birge RB. The urokinase plasminogen activator receptor promotes efferocytosis of apoptotic cells. J Biol Chem. 2009;284:17030–17038. doi: 10.1074/jbc.M109.010066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Blasi F, Sidenius N. Efferocytosis: another function of uPAR. Blood. 2009;114:752–753. doi: 10.1182/blood-2009-05-220657. [DOI] [PubMed] [Google Scholar]
- 20.Higazi AA, Mazar A, Wang J, Quan N, Griffin R, Reilly R, Henkin J, Cines DB. Soluble human urokinase receptor is composed of two active units. J Biol Chem. 1997;272:5348–5353. doi: 10.1074/jbc.272.8.5348. [DOI] [PubMed] [Google Scholar]
- 21.Kunapuli SP, DeLa Cadena RA, Colman RW. Deletion mutagenesis of high molecular weight kininogen light chain. Identification of two anionic surface binding subdomains. J Biol Chem. 1993;268:2486–2492. [PubMed] [Google Scholar]
- 22.Hasan AA, Cines DB, Herwald H, Schmaier AH, Muller-Esterl W. Mapping the cell binding site on high molecular weight kininogen domain 5. J Biol Chem. 1995;270:19256–19261. doi: 10.1074/jbc.270.33.19256. [DOI] [PubMed] [Google Scholar]
- 23.Herwald H, Hasan AA, Godovac-Zimmermann J, Schmaier AH, Muller-Esterl W. Identification of an endothelial cell binding site on kininogen domain D3. J Biol Chem. 1995;270:14634–14642. [PubMed] [Google Scholar]
- 24.Dai JH, Iwatani Y, Ishida T, Terunuma H, Kasai H, Iwakula Y, Fujiwara H, Ito M. Glycyrrhizin enhances interleukin-12 production in peritoneal macrophages. Immunology. 2001;103:235–243. doi: 10.1046/j.1365-2567.2001.01224.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yeung T, Gilbert GE, Shi J, Silvius J, Kapus A, Grinstein S. Membrane phosphatidylserine regulates surface charge and protein localization. Science. 2008;319:210–213. doi: 10.1126/science.1152066. [DOI] [PubMed] [Google Scholar]
- 26.Martinez J, Almendinger J, Oberst A, Ness R, Dillon CP, Fitzgerald P, Hengartner MO, Green DR. Microtubule-associated protein 1 light chain 3 alpha (LC3)-associated phagocytosis is required for the efficient clearance of dead cells. Proc Natl Acad Sci U S A. 2011;108:17396–17401. doi: 10.1073/pnas.1113421108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Khan MMH, Kunapuli SP, Lin Y, Majluf-Cruz A, Cadena RAD, Cooper SL, Colman RW. Three noncontiguous peptides comprise binding sites on high-molecular-weight kininogen to neutrophils. American Journal of Physiology – Heart and Circulatory Physiology. 1998;275:H145–H150. doi: 10.1152/ajpheart.1998.275.1.H145. [DOI] [PubMed] [Google Scholar]
- 28.Morelli AE, Larregina AT, Shufesky WJ, Zahorchak AF, Logar AJ, Papworth GD, Wang Z, Watkins SC, Falo LD, Thomson AW. Internalization of circulating apoptotic cells by splenic marginal zone dendritic cells: dependence on complement receptors and effect on cytokine production. Blood. 2003;101:611–620. doi: 10.1182/blood-2002-06-1769. [DOI] [PubMed] [Google Scholar]
- 29.Wang S, Dale GL, Song P, Viollet B, Zou M-h. AMPKα1 Deletion Shortens Erythrocyte Life Span in Mice: ROLE OF OXIDATIVE STRESS. Journal of Biological Chemistry. 2010;285:19976–19985. doi: 10.1074/jbc.M110.102467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wu Y, Rizzo V, Liu Y, Sainz IM, Schmuckler NG, Colman RW. Kininostatin associates with membrane rafts and inhibits alpha(v)beta3 integrin activation in human umbilical vein endothelial cells. Arterioscler Thromb Vasc Biol. 2007;27:1968–1975. doi: 10.1161/ATVBAHA.107.148759. [DOI] [PubMed] [Google Scholar]
- 31.Colman RW, Pixley RA, Najamunnisa S, Yan W, Wang J, Mazar A, McCrae KR. Binding of high molecular weight kininogen to human endothelial cells is mediated via a site within domains 2 and 3 of the urokinase receptor. J Clin Invest. 1997;100:1481–1487. doi: 10.1172/JCI119669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Blasi F, Sidenius N. The urokinase receptor: Focused cell surface proteolysis, cell adhesion and signaling. FEBS Letters. 2010;584:1923–1930. doi: 10.1016/j.febslet.2009.12.039. [DOI] [PubMed] [Google Scholar]
- 33.Pixley RA, Lin Y, Isordia-Salas I, Colman RW. Fine mapping of the sequences in domain 5 of high molecular weight kininogen (HK) interacting with heparin and zinc. J Thromb Haemost. 2003;1:1791–1798. doi: 10.1046/j.1538-7836.2003.00291.x. [DOI] [PubMed] [Google Scholar]
- 34.Kjoller L, Hall A. Rac mediates cytoskeletal rearrangements and increased cell motility induced by urokinase-type plasminogen activator receptor binding to vitronectin. J Cell Biol. 2001;152:1145–1157. doi: 10.1083/jcb.152.6.1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Smith HW, Marra P, Marshall CJ. uPAR promotes formation of the p130Cas-Crk complex to activate Rac through DOCK180. J Cell Biol. 2008;182:777–790. doi: 10.1083/jcb.200712050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Park D, Tosello-Trampont AC, Elliott MR, Lu M, Haney LB, Ma Z, Klibanov AL, Mandell JW, Ravichandran KS. BAI1 is an engulfment receptor for apoptotic cells upstream of the ELMO/Dock180/Rac module. Nature. 2007;450:430–434. doi: 10.1038/nature06329. [DOI] [PubMed] [Google Scholar]
- 37.Anderson HA, Maylock CA, Williams JA, Paweletz CP, Shu H, Shacter E. Serum-derived protein S binds to phosphatidylserine and stimulates the phagocytosis of apoptotic cells. Nat Immunol. 2003;4:87–91. doi: 10.1038/ni871. [DOI] [PubMed] [Google Scholar]
- 38.Sugi T, McIntyre JA. Autoantibodies to phosphatidylethanolamine (PE) recognize a kininogen-PE complex. Blood. 1995;86:3083–3089. [PubMed] [Google Scholar]
- 39.Barbasz A, Guevara-Lora I, Rapala-Kozik M, Kozik A. Kininogen binding to the surfaces of macrophages. Int Immunopharmacol. 2008;8:211–216. doi: 10.1016/j.intimp.2007.08.002. [DOI] [PubMed] [Google Scholar]
- 40.Chavakis T, Kanse SM, Pixley RA, May AE, Isordia-Salas I, Colman RW, Preissner KT. Regulation of leukocyte recruitment by polypeptides derived from high molecular weight kininogen. FASEB J. 2001;15:2365–2376. doi: 10.1096/fj.01-0201com. [DOI] [PubMed] [Google Scholar]
- 41.Khan MM, Bradford HN, Isordia-Salas I, Liu Y, Wu Y, Espinola RG, Ghebrehiwet B, Colman RW. High-molecular-weight kininogen fragments stimulate the secretion of cytokines and chemokines through uPAR, Mac-1, and gC1qR in monocytes. Arterioscler Thromb Vasc Biol. 2006;26:2260–2266. doi: 10.1161/01.ATV.0000240290.70852.c0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pixley RA, Espinola RG, Ghebrehiwet B, Joseph K, Kao A, Bdeir K, Cines DB, Colman RW. Interaction of high-molecular-weight kininogen with endothelial cell binding proteins suPAR, gC1qR and cytokeratin 1 determined by surface plasmon resonance (BiaCore) Thromb Haemost. 2011;105:1053–1059. doi: 10.1160/TH10-09-0591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tesfay L, Huhn AJ, Hatcher H, Torti FM, Torti SV. Ferritin blocks inhibitory effects of two-chain high molecular weight kininogen (HKa) on adhesion and survival signaling in endothelial cells. PLoS One. 2012;7:e40030. doi: 10.1371/journal.pone.0040030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.del Pozo MA, Alderson NB, Kiosses WB, Chiang HH, Anderson RG, Schwartz MA. Integrins regulate Rac targeting by internalization of membrane domains. Science. 2004;303:839–842. doi: 10.1126/science.1092571. [DOI] [PubMed] [Google Scholar]
- 45.Wei C, Moller CC, Altintas MM, Li J, Schwarz K, Zacchigna S, Xie L, Henger A, Schmid H, Rastaldi MP, Cowan P, Kretzler M, Parrilla R, Bendayan M, Gupta V, Nikolic B, Kalluri R, Carmeliet P, Mundel P, Reiser J. Modification of kidney barrier function by the urokinase receptor. Nat Med. 2008;14:55–63. doi: 10.1038/nm1696. [DOI] [PubMed] [Google Scholar]
- 46.Furlan F, Galbiati C, Jorgensen NR, Jensen JEB, Mrak E, Rubinacci A, Talotta F, Verde P, Blasi F. Urokinase Plasminogen Activator Receptor Affects Bone Homeostasis by Regulating Osteoblast and Osteoclast Function. Journal of Bone and Mineral Research. 2007;22:1387–1396. doi: 10.1359/jbmr.070516. [DOI] [PubMed] [Google Scholar]
- 47.Tjwa M, Sidenius N, Moura R, Jansen S, Theunissen K, Andolfo A, De Mol M, Dewerchin M, Moons L, Blasi F, Verfaillie C, Carmeliet P. Membrane-anchored uPAR regulates the proliferation, marrow pool size, engraftment, and mobilization of mouse hematopoietic stem/progenitor cells. The Journal of Clinical Investigation. 2009;119:1008–1018. doi: 10.1172/JCI36010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.May AE, Kanse SM, Lund LR, Gisler RH, Imhof BA, Preissner KT. Urokinase Receptor (CD87) Regulates Leukocyte Recruitment via β2 Integrins In Vivo. The Journal of Experimental Medicine. 1998;188:1029–1037. doi: 10.1084/jem.188.6.1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Olson FJ, Thurison T, Ryndel M, Hoyer-Hansen G, Fagerberg B. Soluble urokinase-type plasminogen activator receptor forms in plasma as markers of atherosclerotic plaque vulnerability. Clin Biochem. 2010;43:124–130. doi: 10.1016/j.clinbiochem.2009.09.028. [DOI] [PubMed] [Google Scholar]
- 50.Frick IM, Akesson P, Herwald H, Morgelin M, Malmsten M, Nagler DK, Bjorck L. The contact system–a novel branch of innate immunity generating antibacterial peptides. EMBO J. 2006;25:5569–5578. doi: 10.1038/sj.emboj.7601422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Maas C, Govers-Riemslag JW, Bouma B, Schiks B, Hazenberg BP, Lokhorst HM, Hammarstrom P, ten Cate H, de Groot PG, Bouma BN, Gebbink MF. Misfolded proteins activate factor XII in humans, leading to kallikrein formation without initiating coagulation. J Clin Invest. 2008;118:3208–3218. doi: 10.1172/JCI35424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.van der Meijden PE, I, Munnix C, Auger JM, Govers-Riemslag JW, Cosemans JM, Kuijpers MJ, Spronk HM, Watson SP, Renne T, Heemskerk JW. Dual role of collagen in factor XII-dependent thrombus formation. Blood. 2009;114:881–890. doi: 10.1182/blood-2008-07-171066. [DOI] [PubMed] [Google Scholar]
- 53.Muller F, Mutch NJ, Schenk WA, Smith SA, Esterl L, Spronk HM, Schmidbauer S, Gahl WA, Morrissey JH, Renne T. Platelet polyphosphates are proinflammatory and procoagulant mediators in vivo. Cell. 2009;139:1143–1156. doi: 10.1016/j.cell.2009.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.