

Abstract
The blood-brain barrier (BBB) is the main entry route for chemicals into the mammalian central nervous system (CNS). Two transmembrane transporters of the ATP-binding cassette (ABC) family – Breast Cancer Resistance Protein (ABCG2 in humans, Abcg2 in rodents) and P-glycoprotein (ABCB1 in humans, Abcb1 in rodents) – play a key role in mediating this process. Pharmacological and genetic evidence suggests that Abcg2 prevents CNS access to a group of highly potent and selective O-arylcarbamate fatty-acid amidohydrolase (FAAH) inhibitors, which include the compound URB937 (cyclohexylcarbamic acid 3′-carbamoyl-6-hydroxybiphenyl-3-yl ester). To define structure-activity relationships of the interaction of these molecules with Abcg2, in the present study we tested various peripherally restricted and non-restricted O-arylcarbamate FAAH inhibitors for their ability to serve as transport substrates in monolayer cultures of Madin-Darby Canine Kidney-II (MDCKII) cells over-expressing Abcg2. Surprisingly, we found that the majority of compounds tested – even those able to enter the CNS in vivo – were substrates for Abcg2 in vitro. Additional experiments in MDCKII cells overexpressing ABCB1 revealed that only those compounds that were dual substrates for ABCB1 and Abcg2 in vitro were also peripherally restricted in vivo. The extent of such restriction seems to depend upon other physicochemical features of the compounds, in particular the polar surface area. Consistent with these in vitro results, we found that URB937 readily enters the brain in dual knockout mice lacking both Abcg2 and Abcb1, whereas it is either partially or completely excluded from the brain of mice lacking either transporter alone. The results suggest that Abcg2 and Abcb1 act together to restrict the access of URB937 to the CNS.
Keywords: Fatty-acid amidohydrolase (FAAH), FAAH inhibitor, Blood-brain barrier, ATP-binding cassette transporters, Abcb1/P-gp, Abcg2/Bcrp
1. Introduction
The blood-brain barrier (BBB) is the main entry route for molecules into the brain and spinal cord. This unique barrier system is composed of endothelial cells and associated perivascular elements, including astrocytic end-feet processes, perivascular neurons and pericytes [1]. Endothelial cells of the BBB are distinctive in that, while forming complex tight junctions that effectively seal the paracellular pathway, contain numerous membrane transporters that regulate the transcellular traffic of essential molecules between brain and blood, as well as the efflux of potentially harmful substances and waste products [1]. The efflux systems of the BBB are localized to the apical membrane of endothelial cells, and are primarily composed of ATP-binding cassette (ABC) transporters, such as P-glycoprotein (ABCB1 in humans, Abcb1 in rodents) and Breast Cancer Resistance Protein (ABCG2 in humans, Abcg2 in rodents) [2,3]. These two transporters, in particular, are highly expressed in a variety of rodent and human tissues (e.g., small intestine, liver, kidney, brain endothelium and placenta) and contribute in important ways to the absorption, elimination and distribution of xenobiotics [4]. They also play a critical role in the induction of multidrug resistance (MDR) in tumor cells [5]. Significant drug discovery efforts have been aimed at developing inhibitors for ABC transporters [6,7], but little is still known about their substrate preference. While recent structural advances have helped close knowledge gaps about the substrate polyspecificity of ABCB1/Abcb1 [8], the lack of detailed structural information about ABCG2/Abcg2 renders the pharmacophore characterization of its substrates more problematic. Recent attempts to elucidate the substrate-binding properties of ABCG2 have taken advantage of a combination of molecular docking with homology models [9] and mutational analysis [10]. Human ABCG2 and its rodent orthologue Abcg2 recognize a wide range of molecules, including chemotherapeutic agents (e.g. mitoxantrone, methotrexate) and non-chemotherapeutic agents (e.g. nitrofurantoin, cimetidine), as well as non-pharmaceutical compounds such as dietary flavonoids, porphyrins and estrone 3-sulfate [10,11]. Genetic and pharmacological evidence indicates that Abcg2 is also required for the extrusion of the highly potent and selective fatty-acid amidohydrolase (FAAH) inhibitor, URB937 (Fig. 1A), from brain and spinal cord [12,13] URB937 produces profound antinociceptive effects in mice and rats, which depend on the peripheral blockade of the FAAH-mediated deactivation of anandamide, an endogenous agonist of type-1 cannabinoid (CB1) receptors [12]. Two chemical moieties in the O-arylcarbamate scaffold of URB937 - the hydroxy group in para on the proximal phenyl ring and the amide group in meta on the distal phenyl ring (Fig. 1A) - are important for the exclusion of this compound from the central nervous system (CNS) [12–15]; The role of these two moieties is consistent with the results of ligand-based experiments showing that hydroxyl or amine groups on the outer ring of camptothecin analogs, imidazoacridinones, mitoxantrone and urolithins are essential for substrate recognition and efflux by ABCG2/Abcg2 [9,16–18].
Figure 1.
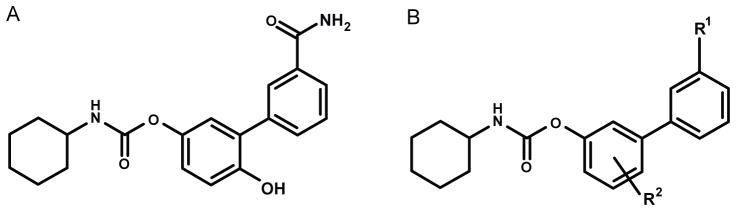
Chemical structures of A) URB937 and B) congeners.
Given the considerable overlap in substrate preference among different members of the ABC family, the distribution of molecules across tissue barriers can be rarely attributed to a single transporter [2,19,20]. Additionally, other factors such as polar surface area (PSA) or membrane lipid composition also affect the passage of chemicals through the BBB [21,22]. Therefore, to gain a better understanding of the mechanism(s) through which URB937 is excluded from the CNS, in the present study we investigated a select group of URB937 derivatives for their ability to serve as substrates for Abcg2 and ABCB1 in vitro, and assessed the access of URB937 to the CNS of mutant mice lacking Abcg2, Abcb1 or both. Our results provide novel structural insight on the substrate selectivity of Abcg2 and Abcb1 and suggest that the combined action of these two transporters restricts the entry of URB937 and its peripheral analogs to the CNS.
2. Material and methods
2.1 Chemicals
Ko143 was purchased from Tocris (Bristol, United Kingdom). PSC-833 (Valspodar) was from Sequoia Research Products (Pangbourne, UK). URB937 and analogs 1–7,9 were synthesized as previously described [12,15]. Compound 8 (cyclohexylcarbamic acid 3′-carbamoyl-6-methylbiphenyl-3-yl ester) was prepared as reported [12]. 8: White crystals. Mp: 165–166 °C (EtOH). MS (ESI): 353 (M+H+). 1H NMR (200 MHz, DMSO-d6): δ = 8.07 (s, 1H), 7.83–7.90 (m, 2H), 7.70–7.74 (m, 1H), 7.43–7.57 (m, 3H), 7.27–7.31 (m, 1H), 6.95–7.05 (m, 2H), 3.28–3.30 (m, 1H), 2.20 (s, 3H), 1.05–1.83 (m, 10H) ppm. IR (Nujol): ν = 3484, 3293, 3133, 1706 cm−1. All the other chemicals were of analytical grade and were available from commercial sources.
2.2 Animals
Adult (9-week) male wild-type FVB, Mdr1a/b−/−, Bcrp−/− and Mdr1a/b−/−-Bcrp−/− mice were obtained from Taconic Farms Inc (Hudson, NY) and kept in a temperature-controlled environment with a 12-h light/12-h dark cycle. Animals received standard chow and water ad libitum. All procedures were approved by the Institutional Animal Care and Use Committee of the University of California, Irvine.
2.3 Cell cultures
Madin-Darby Canine Kidney-II (MDCKII) and their Abcg2- and ABCB1-transduced subclones were a kind gift from Dr. A.H. Schinkel. Culture conditions were those previously described [23,24]. Cells were cultured at 37°C in the presence of 5% CO2 in Dulbecco-modified Eagles’s medium (DMEM) supplemented with Glutamax (Life Technologies, Inc., Carlsbad, CA, USA), penicillin (50 units/mL), streptomycin (50 μg/mL), and 10% (v/v) fetal calf serum (MP Biomedicals, Solon, OH, USA). Cells were trypsinized every 3–4 days for subculturing.
2.4 Transport studies
Transepithelial transport assays were carried out as previously described [25] with minor modifications. Cells were seeded on microporous polycarbonate membrane filters (3.0 μm pore size, 24 mm diameter; Transwell 3414; Costar, Corning, NY, USA) at a density of 1.0×106 cells per well. Cells were grown for 3 days, and the medium was replaced every day. Before starting the experiment transepithelial resistance was measured in each well using a Millicell ERS ohmmeter (Millipore, Bedford, MA); wells registering a resistance of 150 Ω or greater, after correcting for the resistance obtained in blank control wells, were used in the experiments. The measurements were repeated at the end of each experiment to assess the tightness of the monolayer. Experiments were performed using Optimem medium, a reduced serum medium that is a modification of Eagle’s minimum essential medium, buffered with HEPES and sodium bicarbonate. Two h before starting the experiment, medium on both sides of the monolayer was replaced with 2 mL of Optimem medium (Life Technologies, Inc., Carlsbad, CA, USA), without serum, either with or without Ko143 (1μM) or PSC-833 (5 μM). The experiment was then started (t = 0) by replacing the medium in either the apical or basolateral compartment with fresh Optimem medium, either with or without Ko143 (1 μM) or PSC-833 (5 μM), and containing 5 μM of each of the test compounds, except for compound 8, where the concentration used was 1 μM due to limited water solubility. Samples (0.1 mL) were removed from the acceptor compartment at t = 2h and 4h, and stored at −20°C until analysis. The appearance of the compound in the acceptor compartment is shown as fraction of total compound added to the donor compartment at the beginning of the experiment. Active transport was expressed by the relative transport ratio (r), defined as the percent of apically directed transport divided by the percent of basolaterally directed transport, after 4 h [26].
Concentration of tested compounds was quantified by LC-MS/MS. A nine-point calibration curve (1 nM to 10 μM) was prepared for each molecule by serial dilution in Optimem buffer containing 20% acetonitrile to ensure full solubilization. Samples were allowed to reach room temperature and vortexed for 30 s. Samples and calibrators were then transferred (0.1 ml) into 96-well plates and an equal volume of acetonitrile was added. After mixing and centrifugation, supernatant solutions (3 μL) were loaded on a Xevo-TQ UPLC-MS/MS system (Waters Inc., Milford, MA, USA) equipped with a 2.1X50 mm BEH reversed-phase column. Analytes were eluted with a linear gradient of acetonitrile in water (both containing formic acid 0.1%). Detection and quantification were performed in positive ion tandem-mass mode, comparing the multiple reaction monitoring (MRM) peak areas of the samples with those of the corresponding standard curve.
2.5 Experimental design of in vivo study
URB937 was dissolved in saline/PEG400/Tween-80 (18:1:1, v/v/v) and injected intraperitoneally at the volume of 10 mL/kg. Mice were sacrificed by decapitation under slight anesthesia with isoflurane, and brain and spinal cord were removed and snap frozen in liquid nitrogen. Blood was collected through a left cardioventricular puncture and centrifuged at 2000 × g for 20 min to obtain plasma. Brain samples were weighed and homogenized in ice-cold Tris–HCl buffer (50 mM, 5–9 vol., pH 7.5) containing 0.32 M sucrose. Homogenates were centrifuged at 1000 × g for 10 min at 4°C. Supernatants were collected (0.25 mL) and protein concentration determined using a bicinchoninic acid (BCA) assay kit (Pierce, Rockford, IL, USA). Remaining supernatant and pellet were further extracted with methanol/chloroform for URB937 analysis.
2.6 FAAH activity
FAAH activity was measured at 37°C for 30 min in 0.5mL of Tris-HCl buffer (50 mM, pH 7.5) containing fatty acid-free bovine serum albumin (BSA) (0.05%, w/v), tissue homogenates (S1 fraction, 50 μg), 10 μM anandamide, and anandamide-[ethanolamine-3H] (10,000 cpm, specific activity 60 Ci/mmol; American Radiolabeled Chemicals). The reactions were stopped with chloroform/methanol (1:1, 1 mL) and radioactivity was measured in the aqueous layers by liquid scintillation counting.
2.7 URB937 quantification by LC/MS
Tissue and plasma levels of URB937 were determined as previously described [12] with minor modifications. In brief, tissue homogenates and plasma samples were extracted with methanol/chloroform (1:2) containing N-cyclohexyl biphenyl-3-ylacetamide as internal standard. Organic phases were evaporated under nitrogen and reconstituted in 0.1 mL of methanol. Samples were analyzed using an 1100-LC system coupled to a 1946A-MS detector (Agilent Technologies, Inc., Palo Alto, CA) equipped with an electrospray ionization interface. URB937 and N-cyclohexyl biphenyl-3-ylacetamide (mass-to-charge ratio, m/z = 377 and 294 respectively) were eluted on an XDB Eclipse C18 column (50×4.6mm inner diameter, 1.8 μm, Zorbax) using a linear gradient of 60% to 100% of A in B over 3 min at a flow rate of 1.0 mL/min. Mobile phase A consisted of methanol containing 0.25% acetic acid and 5 mM ammonium acetate; mobile phase B consisted of water containing 0.25% acetic acid and 5 mM ammonium acetate.
2.8 Statistical analyses
Results are expressed as mean ± standard error of the mean (SEM) or standard deviation (SD) and the significance of differences was determined using one-way or two-way analysis of variance (ANOVA) followed by a Dunett’s test as post hoc. Differences were considered significant if P<0.05. Statistical analyses were conducted using GraphPad Prism Version 4.0 (San Diego, CA, USA).
3. Results
3.1 Transport of O-arylcarbamate FAAH inhibitors in wild-type MDCKII cells
We first measured the transepithelial transport of URB937 and a select group of O-arylcarbamate FAAH inhibitors across polarized monolayers of wild-type MDCKII cells. Results for the transport in the basolateral-to-apical and apical-to-basolateral directions, as well as the relative transport ratios, are reported in Table 1. URB937 and its analogs 1, 5 and 6 showed a moderate apically directed transport, relative to basolaterally directed transport (transport ratios ≥ 2) (Table 1). This asymmetric distribution was likely due to endogenous expression of native canine Abcb1, because it was abolished by incubation with the selective Abcb1 inhibitor, PSC833, but not by the Abcg2 inhibitor Ko143 (Supplementary Figure 1). Such directed translocation was not observed for compounds 3, 7 and 8, which displayed BA/AB ratios close to 1 (Table 1), thus suggesting that they are not substrates for endogenously expressed Abcb1. By contrast, compounds 4 and 9, initially designed to evaluate the H-bonding capacity of the distal amide residue, and the putative sulfate conjugation of URB937 that might occur in vivo respectively, showed minimal translocation in either the apical or basolateral direction. As this result was likely due to the presence in their structure of chemical groups that were ionized at physiological pH, the compounds were not further tested.
Table 1.
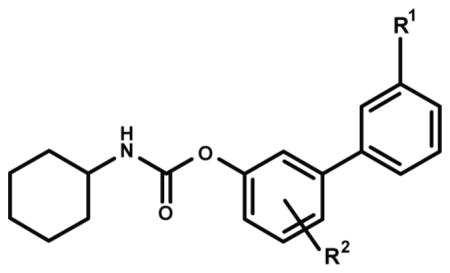
Transepithelial transport of URB937 and other O-arylcarbamate FAAH inhibitors in monolayers of wild-type MDCKII cells.
| |||||||
|---|---|---|---|---|---|---|---|
| Compound | R1 | R2 | PSAa | Brain ID50 (mg/kg)a | B to A transportb | A to B Transportb | Ratio BA/AB |
| URB937 | CONH2 | p-OH | 83 | 40 | 44.3 ± 5.35 | 17.78 ± 2.12 | 2.54 ± 0.49 |
| 1 | CONHCH3 | p-OH | 73 | 15 | 48.1 ± 2.33 | 21.7 ± 0.96 | 2.22 ± 0.17 |
| 2 | CON(CH3)2 | p-OH | 65 | 5 | 43.1 ± 4.48 | 25.6 ± 1.96 | 1.69 ± 0.22 |
| 3 | COCH3 | p-OH | 62 | <1 | 46.8 ± 2.73 | 39.4 ± 0.54 | 1.16 ± 0.42 |
| 4 | COOH | p-OH | 77 | 12 | 5.21 ± 0.42 | 4.93 ± 0.88 | 1.08 ± 0.17 |
| 5 | CONH2 | m-OH | 84 | 75 | 43.2 ± 4.21 | 19.6 ± 4.89 | 2.25 ± 0.37 |
| 6 | CONH2 | p-CH2OH | 83 | 15 | 43.0 ± 2.34 | 15.5 ± 0.57 | 2.76 ± 0.06 |
| 7 | CONH2 | p-OCH3 | 74 | <1 | 37.1 ± 1.48 | 29.3 ± 0.44 | 1.26 ± 0.03 |
| 8 | CONH2 | p-CH3 | 66 | <1 | 27.3 ± 3.8 | 26.0 ± 1.52 | 1.06 ± 0.15 |
| 9 | CONH2 | p-OSO3NH4 | 117 | >75 | 7.49 ± 0.39 | 7.92 ± 0.41 | 0.94 ± 0.04 |
The experiment was started with the addition of each compound to the apical (A) or basolateral (B) compartment. Drug concentration was 5 μM in all cases except for compound 8, which was tested at 1 μM due to solubility limitations.
Data obtained from (Moreno-Sanz et al., 2013) included in the table for clarity.
Percentage of drug appearing in the opposite compartment after 4h. PSA, Polar surface area (Å2).
3.2 Transport of O-arylcarbamate FAAH inhibitors in MDCKII cells monolayers overexpressing Abcg2
We next assessed transport of URB937 and its congeners in MDCKII cells engineered to overexpress murine Abcg2. Previous studies have shown that human and murine ABCG2 have similar substrate preferences [27]. The majority of test compounds (URB937, 1, 2, 5, 6, 7 and 8) displayed a substantial basolateral-to-apical translocation, and a diminished apical-to-basolateral translocation (Table 2). The observed relative transport ratios, which were in all cases ≥10, identify the compounds as excellent substrates for Abcg2 in vitro. Compounds bearing a primary or substituted amide in the R1 position and a hydroxy or a hydroxy-containing group in R2 (URB937, 5 and 6)(Fig. 1), although having relative transport ratios similar to those of other test compounds, displayed significantly smaller apical-to-basolateral transport, which correlates with a higher PSA value (≥80Å) and the low brain penetration previously reported for these inhibitors in vivo [15]. Surprisingly, compounds 7 and 8 were effectively transported by Abcg2 (Table 2), even though they were previously shown to readily enter the brain when administered to mice [15]. In the small set of molecules tested here, only compound 3 did not behave as a transport substrate for Abcg2. The translocation of all compounds was blocked by addition of the Abcg2 inhibitor, Ko143 (Supplementary Figure 2A).
Table 2.
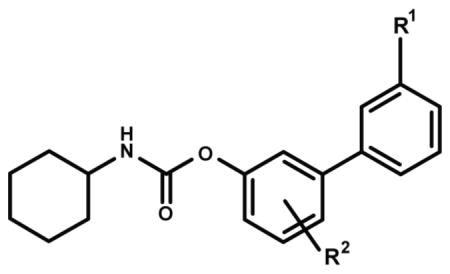
Transepithelial transport of URB937 and other O-arylcarbamate FAAH inhibitors in monolayers of MDCKII cells over-expressing Abcg2.
| |||||||
|---|---|---|---|---|---|---|---|
| Compound | R1 | R2 | PSAa | Brain ID50 (mg/kg)a | B to A transportb | A to B transportb | Ratio BA/AB |
| URB937 | CONH2 | p-OH | 83 | 40 | 25.9 ± 1.44 | 1.68 ± 0.12 | 15.4 ± 0.24 |
| 1 | CONHCH3 | p-OH | 73 | 15 | 69.8 ± 16.9 | 6.99 ± 0.99*** | 9.90 ± 1.0 |
| 2 | CON(CH3)2 | p-OH | 65 | 5 | 91.0 ± 12.7 | 6.95 ± 0.42*** | 13.1 ± 2.0 |
| 3 | COCH3 | p-OH | 62 | <1 | 30.5 ± 0.75 | 24.5 ± 2.39*** | 1.25 ± 0.13 |
| 5 | CONH2 | m-OH | 84 | 75 | 48.4 ± 1.16 | 2.79 ± 0.42 | 17.6 ± 2.7 |
| 6 | CONH2 | p-CH2OH | 83 | 15 | 56.2 ± 1.54 | 1.79 ± 0.17 | 31.4 ± 3.4 |
| 7 | CONH2 | p-OCH3 | 74 | <1 | 98.8 ± 16.6 | 7.29 ± 1.30*** | 13.08 ± 5.1 |
| 8 | CONH2 | p-CH3 | 66 | <1 | 64.8 ± 4.87 | 5.59 ± 0.58*** | 11.6 ± 0.92 |
The experiment started with the addition of each compound to the apical or basolateral compartment. Drug concentration was 5 μM in all cases except for compound 8, which was tested at 1 μM due to solubility limitations.
Data obtained from (Moreno-Sanz et al., 2013) included in the table for clarity.
Percentage of drug appearing in the opposite compartment after 4h. PSA, Polar surface area (Å2).
P< 0.001 vs. URB937; 1-way ANOVA with Dunnett’s post hoc test;
3.3 Transport of O-arylcarbamate FAAH inhibitors in MDCKII cells monolayers overexpressing ABCB1
We also asked whether URB937 and other O-arylcarbamate FAAH inhibitors are transported across monolayers of MDCKII cells transduced with human ABCB1 (Table 3). In this case, no significant vectorial transport was observed for analogs 3, 7 and 8. This indicates that these compounds are not ABCB1 substrates in vitro and suggests that both the m-amide group in the distal phenyl ring and the p-hydroxy group in the proximal phenyl ring of URB937 are required for its substrate recognition by ABCB1. Other test molecules displayed relative transport ratios >6, which shows that they were effectively translocated by ABCB1. In contrast to what observed in Abcg2 overexpressing cells, no significant difference was found among the apical-to-basolateral translocation, independently of the PSA value (Table 3). Inclusion of the selective ABCB1 inhibitor, PSC883, prevented transport, confirming that ABCB1 activity mediates this process (Supplementary Figure 2B).
Table 3.
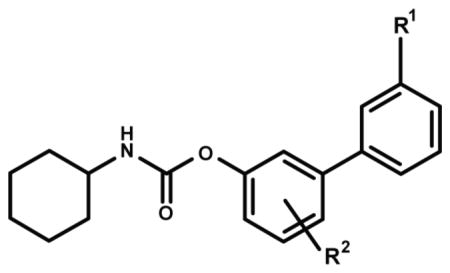
Transepithelial transport of URB937 and other O-arylcarbamate FAAH inhibitors in monolayers of MDCKII cells over-expressing ABCB1.
| |||||||
|---|---|---|---|---|---|---|---|
| Compound | R1 | R2 | PSAa | Brain ID50 (mg/kg)a | B to A transportb | A to B Transportb | Ratio BA/AB |
| URB937 | CONH2 | p-OH | 83 | 40 | 50.2 ± 4.02 | 5.89 ± 1.33 | 8.76 ± 1.4 |
| 1 | CONHCH3 | p-OH | 73 | 15 | 67.4 ± 14.5 | 8.55 ± 1.79 | 7.89 ± 0.58 |
| 2 | CON(CH3)2 | p-OH | 65 | 5 | 58.6 ± 3.85 | 8.86 ± 0.95 | 6.65 ± 0.36 |
| 3 | COCH3 | p-OH | 62 | < 1 | 57.6 ± 7.90 | 39.8 ± 1.88 | 1.44 ± 0.13 |
| 5 | CONH2 | m-OH | 84 | 75 | 46.2 ± 6.68 | 5.68 ± 0.63 | 8.35 ± 2.4 |
| 6 | CONH2 | p-CH2OH | 83 | 15 | 50.9 ± 14.0 | 6.07 ± 0.25 | 8.42 ± 2.6 |
| 7 | CONH2 | p-OCH3 | 74 | < 1 | 45.1 ± 3.61 | 21.6 ± 0.55 | 2.08 ± 0.11 |
| 8 | CONH2 | p-CH3 | 66 | < 1 | 35.8 ± 0.43 | 20.7 ± 1.00 | 1.73 ± 0.14 |
The experiment was started with the addition of each compound to the apical (A) or basolateral (B) compartment. Drug concentration was 5 μM in all cases except for compound 8, which was tested at 1 μM due to solubility limitations.
Data obtained from (Moreno-Sanz et al., 2013) included in the table for clarity.
Percentage of drug appearing in the opposite compartment after 4h. PSA, Polar surface area (Å2).
3.4 Brain penetration of URB937 in mice lacking Abcg2, Abcb1 or both
Previous studies have shown that URB937 inhibits FAAH activity in the brain with a median effective dose (ID50) of 40 mg/kg, which is 400 times greater than the dose needed to inhibit FAAH activity in peripheral organs such as the liver (ID50=0.1 mg/kg) [12]. A dose-exploration study of URB937 (3–15 mg/kg, intraperitoneal, i.p.) in mutant mice lacking Abcg2 (Bcrp−/− mice) yielded an IC50 of brain FAAH activity of 10 mg/kg (Fig. 2A), which suggested that Abcg2 is only partly responsible for the restricted access of URB937 to the brain. We selected the dose of 5 mg/kg, which inhibited brain FAAH by only 26%, to study the effect of removing Abcg2, Abcb1 or both. As expected, administration of URB937 (5 mg/kg, i.p.) did not affect FAAH activity in the brain of wild-type mice (Fig. 2B), but completely blocked FAAH activity in the liver (Supplementary Figure 3A). A similar result was obtained in Abcb1-deficient animals (Fig. 2B), where the brain-to-plasma ratio of URB937 was not significantly different from controls (Fig. 2C). These results indicate that URB937 is unable to enter the brain when Abcg2 is present, even in the absence of Abcb1. On the other hand, brain FAAH activity was drastically inhibited, and brain-to-plasma ratio of URB937 significantly increased, in mice lacking both Abcb1 and Abcg2 (Mdr1a/b−/−-Bcrp−/− mice) (Fig. 2B, C). Similar results were obtained in the spinal cord (Supplementary Figure 3B).
Figure 2. Abcg2 and Abcb1 cooperate to restrict entry of URB937 into mouse brain in vivo.
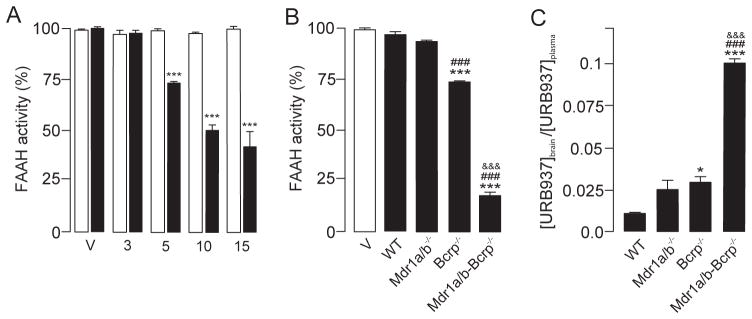
A) Dose-dependent inhibition of FAAH activity in the brain of mutant mice lacking Abcg2 (Bcrp−/− mice) by URB937 (3–15 mg/kg, i.p.). Mice were sacrificed 1 h after drug injection. *** P<0.001 vs. Vehicle; 2-way ANOVA with Dunnett’s post hoc test. B) Effect of URB937 (5 mg/kg, i.p) on brain FAAH activity in wild-type (WT) mice and in mice lacking Abcb1 (Mdr1a/b−/−), Abcg2 (Bcrp−/−) or Abcb1 plus Abcg2 (Mdr1a/b-Bcrp−/−). C) Brain-to-plasma ratio of URB937 1 h after administration of the drug (5 mg/kg, i.p.) to wild-type (WT) mice or to mice lacking Abcb1 (Mdr1a/b−/−), Abcg2 (Bcrp−/−) or both (Mdr1a/b-Bcrp−/−). * P<0.05, *** P<0.001 vs. wild-type (WT); ### P<0.001 vs. Mdr1a/b−/− mice; &&&P <0.001 vs. Bcrp−/−; n = 3–4. Results are expressed as mean ± SEM.
4. Discussion
The discovery of URB937, the first peripherally restricted FAAH inhibitor, revealed an unexpected role for the endocannabinoid anandamide, a FAAH substrate, in the control of pain initiation outside the central nervous system [12]. Pharmacological and genetic investigations in the mechanism underlying the peripheral distribution of URB937 have shown that the ABC transporter, Abcg2, plays an essential role in limiting the access of this compound to the CNS [12,13].
Recent efforts aimed at elucidating the structural determinants responsible for the peripheral segregation of URB937 in vivo yielded a small set of brain-impermeant FAAH inhibitors, which share certain common structural features. These include (i) a primary, secondary or tertiary amide in the meta position of the distal phenyl ring; and (ii) a hydroxy or a hydroxy-containing group in the meta or para position of the proximal phenyl ring (Fig. 1) [15]. To determine whether similar or different features underlie the recognition of these compounds by Abcg2, in the present study we tested a select group of O-arylcarbamate FAAH inhibitors in MDCKII cells overexpressing this transporter. The same set of molecules was also tested in MDCKII cells overexpressing ABCB1, another member of the ABC family of transporters. The results of this focused analysis suggest that the amide residue in the distal ring is essential for recognition by both Abcg2 and ABCB1, because its substitution with a ketone group (as in compound 3) completely prevented directional transport. This result is in agreement with previous studies showing that compound 3 readily enters the brain following systemic administration in mice [15]. In the same study, progressive alkylation of the m-amide group in URB937 was found to increase the access of analogs 1 and 2 to the brain. However, this finding does not match our present results indicating that both Abcg2 and ABCB1 translocate compounds 1 and 2 in vitro. A plausible explanation for this discrepancy is that replacing hydrogen atoms with methyl groups within the amide moiety of URB937 reduces the compounds’ polar surface area (PSA) and increases their tendency to diffuse passively through the BBB. Indeed, polarity may play an important role in the substrate specificity of Abcg2, as compounds with the greatest PSA (≥75Å2, URB937, 5 and 6) also showed significantly lower apical-to-basolateral translocation in Abcg2-, but not ABCB1-, overexpressing cells, and had the most restricted access to the CNS in vivo [15]. This is in agreement with the guiding principle that establishes 75 Å2 as the threshold value for PSA that, if exceeded, will dramatically increase the chances of transporter involvement, whereas lipophilic compounds below 75 Å2 are likely to be cleared by metabolism [28].
Surprisingly, substituting the p-hydroxyl group in the proximal phenyl ring (R2) with a methoxy (7) or methyl group (8), a modification that drastically increases brain penetration of these molecules in vivo [15], did not prevent Abcg2-mediated transport in vitro. On the other hand, neither 7 nor 8 were substrates for ABCB1, which suggests that the hydroxyl substituent in R2 is crucial for substrate preference by this transporter. Because 7 and 8 readily enter the brain after systemic administration in mice (ID50 ≤ 1 mg/kg, i.p.), it is reasonable to conclude that, among the set of molecules investigated in the present study, only those that act as dual substrates for Abcg2 and Abcb1 have impaired access to the CNS in vivo. Further, our results demonstrate that compounds such as 1 and 7, with identical polarity (PSA=73), lipophilicity (cLogP=3.9) and molecular weigh (MW=368 Da), can largely differ in their systemic distribution due to selective transport recognition.
Previous experiments with the non-selective inhibitor, verapamil, failed to reveal a significant role for Abcb1 in the peripheral segregation of URB937 [12]. We reexamined this issue using mutant mice that lack either Abcb1 (Mdr1a/b−/−), Abcg2 (Bcrp−/−), or both (Mdr1a/b-Bcrp−/−). Our results concord with those obtained by others for drugs – such as sorafenib [29] and dantrolene [30] – that have significantly higher affinity for Abcg2 than Abcb1. In those cases too, a role for Abcb1 was unmasked in triple Mdr1a/b-Bcrp−/− knockout mice. Strikingly, however, while the action of Abcg2 alone is sufficient to prevent the entry of URB937 into the brain of Mdr1a/b−/− mice, structural analogs of this compound lacking affinity for Abcb1 such as 7 and 8 can readily access the CNS. This observation, together with the substantial effect of dual Mdr1a/b-Bcrp−/− removal on the brain penetration of URB937, suggests the possibility of a functional cooperation between these two transporters. However, based on theoretical pharmacokinetic models, it has been noted that there is no need to postulate a direct synergistic interaction between Abcg2 and Abcb1 to account for the disproportionate brain accumulation observed for dual substrates in Mdr1a/b-Bcrp−/− compared to Mdr1a/b−/− and Bcrp−/− knockout mice [20,30]. Rather, such accumulation could be explained by the fact that the intrinsic efflux transport mediated by Abcg2 and Abcb1 is considerably larger than the remaining, most likely passive, efflux transport at the BBB in the absence of both transporters [2]. Unfortunately, we were not able to fit these kinetic equations to our observed data because our experiments were not carried out under steady-state conditions [30]. In the case of 7 and 8, the substitution of the hydroxyl moiety on the proximal ring by a methoxy or methyl group was accompanied by a reduction in PSA, which, according to the present results, may be important for the substrate recognition by Abcg2. Also, the substitution renders these compounds more capable of passively diffusing through biological membranes, compared to URB937.
Another ABC transporter, Abcc4, has been shown to collaborate with Abcg2 and Abcb1 in the extrusion of camptothecin analogs from the brain [31]. Although our present results cannot exclude a role for Abcc4 or any other transporter in the extrusion of URB937 from the brain, the extent of FAAH inhibition found in the CNS of Mdr1a/b-Bcrp−/− knockout mice appears to be inconsistent with the participation of other transporters.
5. Conclusion
The present results indicate that the peripheral FAAH inhibitor, URB937, and a small number of its congeners are substrates for both Abcg2 and Abcb1 in vitro, and identify two different structural features within the O-arylcarbamate scaffold that are critical for such recognition. This does not, however, exclude the possibility that other residues, outside of the regions studied here, might also be involved in the drug-protein interaction. Our data further suggest that, among this set of compounds, only those that are dual substrates for both transporters display a restricted access to the brain and spinal cord in vivo, the extent of which may vary depending upon other physicochemical properties (e.g. PSA). These findings could be useful on the design and development of novel therapeutic agents, by exploiting ABC transporter recognition to selectively control systemic distribution.
Supplementary Material
Acknowledgments
The authors thank Dr. A.H. Schinkel (The Netherlands Cancer Institute, Amsterdam, The Netherlands) who provided MDCKII cells and their transduced cell lines. This study was supported by grants from the National Institutes on Drug Abuse (RO1-DA-012413 to D.P.) and by research projects AGL2009-11730 and AGL2012-31116 from the Spanish Ministry of Economy and Competitiveness and the European Regional Development Fund (to G.M.) and by a predoctoral grant (FPU) from the Spanish Ministry of Education (to B.B.). The contribution of the Agilent Technologies/ UCI Analytical Discovery Facility is gratefully acknowledged.
Abbreviations
- ABC
ATP-binding cassette
- BBB
Blood-brain barrier
- Abcg2/Bcrp
Breast cancer resistance protein
- CNS
Central nervous system
- DMEM
Dulbecco-modified Eagles’s medium
- ESI
Electrospray Ionization
- FAAH
Fatty-acid amidohydrolase
- LC-MS
Liquid chromatography–mass spectrometry
- MDCKII
Madin-Darby Canine Kidney-II
- ID50
Median effective dose
- MDR
Multidrug resistance
- MRM
Multiple reaction monitoring
- Abcb1/P-gp
P-glycoprotein
- PSA
Polar surface area
- CB1
Type-1 cannabinoid receptor
- UPLC
Ultra performance liquid chromatography
Footnotes
Conflict of interest statement
The following conflict of interest is declared: GMS, TB, AD, GT, and DP are inventors in patent applications, filed by the University of California Irvine and the Istituto Italiano di Tecnologia, which protect URB937 and other O-arylcarbamate FAAH inhibitors.
Chemical compounds studied in this article
HEPES (PubChem CID: 23831); Ko143 (PubChem CID: 10322450); Polyethylene glycol (PubChem CID: 24762); PSC833 (PubChem CID: 5281884); Tween-80 (PubChem CID: 6364656); URB937 (PubChem CID: 53394762)
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Cecchelli R, Berezowski V, Lundquist S, Culot M, Renftel M, Dehouck MP, et al. Modelling of the blood-brain barrier in drug discovery and development. Nat Rev Drug Discov. 2007;6(8):650–61. doi: 10.1038/nrd2368. [DOI] [PubMed] [Google Scholar]
- 2.Agarwal S, Hartz AM, Elmquist WF, Bauer B. Breast cancer resistance protein and P-glycoprotein in brain cancer: two gatekeepers team up. Curr Pharm Des. 2011;17(26):2793–802. doi: 10.2174/138161211797440186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chaves C, Shawahna R, Jacob A, Scherrmann JM, Decleves X. Human ABC Transporters at blood-CNS Interfaces as Determinants of CNS Drug Penetration. Curr Pharm Des. 2014;20(10):1450–62. doi: 10.2174/13816128113199990466. [DOI] [PubMed] [Google Scholar]
- 4.Konig J, Muller F, Fromm MF. Transporters and drug-drug interactions: important determinants of drug disposition and effects. Pharmacol Rev. 2013;65(3):944–66. doi: 10.1124/pr.113.007518. [DOI] [PubMed] [Google Scholar]
- 5.Sharom FJ. ABC multidrug transporters: structure, function and role in chemoresistance. Pharmacogenomics. 2008;9(1):105–27. doi: 10.2217/14622416.9.1.105. [DOI] [PubMed] [Google Scholar]
- 6.Lecerf-Schmidt F, Peres B, Valdameri G, Gauthier C, Winter E, Payen L, et al. ABCG2: recent discovery of potent and highly selective inhibitors. Future Med Chem. 2013;5(9):1037–45. doi: 10.4155/fmc.13.71. [DOI] [PubMed] [Google Scholar]
- 7.Falasca M, Linton KJ. Investigational ABC transporter inhibitors. Expert Opin Investig Drugs. 2012;21(5):657–66. doi: 10.1517/13543784.2012.679339. [DOI] [PubMed] [Google Scholar]
- 8.Gutmann DA, Ward A, Urbatsch IL, Chang G, van Veen HW. Understanding polyspecificity of multidrug ABC transporters: closing in on the gaps in ABCB1. Trends Biochem Sci. 2010;35(1):36–42. doi: 10.1016/j.tibs.2009.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rosenberg MF, Bikadi Z, Chan J, Liu X, Ni Z, Cai X, et al. The human breast cancer resistance protein (BCRP/ABCG2) shows conformational changes with mitoxantrone. Structure. 2010;18(4):482–93. doi: 10.1016/j.str.2010.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ni Z, Bikadi Z, Rosenberg MF, Mao Q. Structure and function of the human breast cancer resistance protein (BCRP/ABCG2) Curr Drug Metab. 2010;11(7):603–17. doi: 10.2174/138920010792927325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Alvarez AI, Perez M, Prieto JG, Molina AJ, Real R, Merino G. Fluoroquinolone efflux mediated by ABC transporters. J Pharm Sci. 2008;97(9):3483–93. doi: 10.1002/jps.21233. [DOI] [PubMed] [Google Scholar]
- 12.Clapper JR, Moreno-Sanz G, Russo R, Guijarro A, Vacondio F, Duranti A, et al. Anandamide suppresses pain initiation through a peripheral endocannabinoid mechanism. Nat Neurosci. 2010;13(10):1265–70. doi: 10.1038/nn.2632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Moreno-Sanz G, Barrera B, Guijarro A, d’Elia I, Otero JA, Alvarez AI, et al. The ABC membrane transporter ABCG2 prevents access of FAAH inhibitor URB937 to the central nervous system. Pharmacol Res. 2011;64(4):359–63. doi: 10.1016/j.phrs.2011.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Clapper JR, Vacondio F, King AR, Duranti A, Tontini A, Silva C, et al. A second generation of carbamate-based fatty acid amide hydrolase inhibitors with improved activity in vivo. ChemMedChem. 2009;4(9):1505–13. doi: 10.1002/cmdc.200900210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Moreno-Sanz G, Duranti A, Melzig L, Fiorelli C, Ruda GF, Colombano G, et al. Synthesis and Structure-Activity Relationship Studies of O-Biphenyl-3-yl Carbamates as Peripherally Restricted Fatty Acid Amide Hydrolase Inhibitors. J Med Chem. 2013;56(14):5917–30. doi: 10.1021/jm4007017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yoshikawa M, Ikegami Y, Hayasaka S, Ishii K, Ito A, Sano K, et al. Novel camptothecin analogues that circumvent ABCG2-associated drug resistance in human tumor cells. Int J Cancer. 2004;110(6):921–7. doi: 10.1002/ijc.20216. [DOI] [PubMed] [Google Scholar]
- 17.Bram EE, Adar Y, Mesika N, Sabisz M, Skladanowski A, Assaraf YG. Structural determinants of imidazoacridinones facilitating antitumor activity are crucial for substrate recognition by ABCG2. Mol Pharmacol. 2009;75(5):1149–59. doi: 10.1124/mol.109.054791. [DOI] [PubMed] [Google Scholar]
- 18.Gonzalez-Sarrias A, Miguel V, Merino G, Lucas R, Morales JC, Tomas-Barberan F, et al. The gut microbiota ellagic acid-derived metabolite urolithin A and its sulfate conjugate are substrates for the drug efflux transporter breast cancer resistance protein (ABCG2/BCRP) J Agric Food Chem. 2013;61(18):4352–9. doi: 10.1021/jf4007505. [DOI] [PubMed] [Google Scholar]
- 19.Poller B, Iusuf D, Sparidans RW, Wagenaar E, Beijnen JH, Schinkel AH. Differential impact of P-glycoprotein (ABCB1) and breast cancer resistance protein (ABCG2) on axitinib brain accumulation and oral plasma pharmacokinetics. Drug Metab Dispos. 2011;39(5):729–35. doi: 10.1124/dmd.110.037317. [DOI] [PubMed] [Google Scholar]
- 20.Tang SC, de Vries N, Sparidans RW, Wagenaar E, Beijnen JH, Schinkel AH. Impact of P-glycoprotein (ABCB1) and breast cancer resistance protein (ABCG2) gene dosage on plasma pharmacokinetics and brain accumulation of dasatinib, sorafenib, and sunitinib. J Pharmacol Exp Ther. 2013;346(3):486–94. doi: 10.1124/jpet.113.205583. [DOI] [PubMed] [Google Scholar]
- 21.Shityakov S, Neuhaus W, Dandekar T, Forster C. Analysing molecular polar surface descriptors to predict blood-brain barrier permeation. Int J Comput Biol Drug Des. 2013;6(1–2):146–56. doi: 10.1504/IJCBDD.2013.052195. [DOI] [PubMed] [Google Scholar]
- 22.Pal A, Mehn D, Molnar E, Gedey S, Meszaros P, Nagy T, et al. Cholesterol potentiates ABCG2 activity in a heterologous expression system: improved in vitro model to study function of human ABCG2. J Pharmacol Exp Ther. 2007;321(3):1085–94. doi: 10.1124/jpet.106.119289. [DOI] [PubMed] [Google Scholar]
- 23.Jonker JW, Smit JW, Brinkhuis RF, Maliepaard M, Beijnen JH, Schellens JH, et al. Role of breast cancer resistance protein in the bioavailability and fetal penetration of topotecan. J Natl Cancer Inst. 2000;92(20):1651–6. doi: 10.1093/jnci/92.20.1651. [DOI] [PubMed] [Google Scholar]
- 24.Pavek P, Merino G, Wagenaar E, Bolscher E, Novotna M, Jonker JW, et al. Human breast cancer resistance protein: interactions with steroid drugs, hormones, the dietary carcinogen 2-amino-1-methyl-6-phenylimidazo(4,5-b)pyridine, and transport of cimetidine. J Pharmacol Exp Ther. 2005;312(1):144–52. doi: 10.1124/jpet.104.073916. [DOI] [PubMed] [Google Scholar]
- 25.Merino G, Jonker JW, Wagenaar E, Pulido MM, Molina AJ, Alvarez AI, et al. Transport of anthelmintic benzimidazole drugs by breast cancer resistance protein (BCRP/ABCG2) Drug Metab Dispos. 2005;33(5):614–8. doi: 10.1124/dmd.104.003319. [DOI] [PubMed] [Google Scholar]
- 26.Huisman MT, Chhatta AA, van Tellingen O, Beijnen JH, Schinkel AH. MRP2 (ABCC2) transports taxanes and confers paclitaxel resistance and both processes are stimulated by probenecid. Int J Cancer. 2005;116(5):824–9. doi: 10.1002/ijc.21013. [DOI] [PubMed] [Google Scholar]
- 27.Bakhsheshian J, Hall MD, Robey RW, Herrmann MA, Chen JQ, Bates SE, et al. Overlapping substrate and inhibitor specificity of human and murine ABCG2. Drug Metab Dispos. 2013;41(10):1805–12. doi: 10.1124/dmd.113.053140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Smith DA. Evolution of ADME science: where else can modeling and simulation contribute? Mol Pharm. 2013;10(4):1162–70. doi: 10.1021/mp3005319. [DOI] [PubMed] [Google Scholar]
- 29.Agarwal S, Elmquist WF. Insight into the cooperation of P-glycoprotein (ABCB1) and breast cancer resistance protein (ABCG2) at the blood-brain barrier: a case study examining sorafenib efflux clearance. Mol Pharm. 2012;9(3):678–84. doi: 10.1021/mp200465c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kodaira H, Kusuhara H, Ushiki J, Fuse E, Sugiyama Y. Kinetic analysis of the cooperation of P-glycoprotein (P-gp/Abcb1) and breast cancer resistance protein (Bcrp/Abcg2) in limiting the brain and testis penetration of erlotinib, flavopiridol, and mitoxantrone. J Pharmacol Exp Ther. 2010;333(3):788–96. doi: 10.1124/jpet.109.162321. [DOI] [PubMed] [Google Scholar]
- 31.Lin F, Marchetti S, Pluim D, Iusuf D, Mazzanti R, Schellens JH, et al. Abcc4 together with abcb1 and abcg2 form a robust cooperative drug efflux system that restricts the brain entry of camptothecin analogues. Clin Cancer Res. 2013;19(8):2084–95. doi: 10.1158/1078-0432.CCR-12-3105. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.