

Abstract
Host innate-immune responses are tailored by cell-type to control and eradicate specific infectious agents. For example, an acute RNA virus infection can result in high-level expression of type 1 interferons (IFNs) by both conventional (cDCs) and plasmacytoid dendritic cells (pDCs), but while cDCs preferentially utilize RIG-I-like Receptor (RLR) signaling to produce type 1 IFNs, pDCs predominantly employ Toll-like Receptors (TLR) to induce these cytokines. We previously found that the IKKβ/NF-κB pathway regulates early IFN-β expression but not the magnitude of type 1 IFN expression following RLR engagement. In this study, we use IKKβ inhibition and mice deficient in IKKβ or canonical NF-κB subunits (p50, RelA/p65 and cRel) to demonstrate that the IKKβ/NF-κB axis is critically important for virus-induced type 1 IFN expression in pDCs, but not in cDCs. We also reveal a crucial and more general requirement for IKKβ/NF-κB in TLR - but not RLR- induced expression of type 1 IFNs and inflammatory cytokines. Together, these findings reveal a previously unappreciated specificity of the IKKβ/NF-κB signaling axis in regulation of anti-microbial responses by different classes of PRR, and therefore by individual cell-types reliant on particular PRRs for their innate-immune transcriptional responses.
Introduction
The mammalian immune system is exquisitely sensitive to the nature of invading infectious agents and tailors host responses that are best suited to eradicate specific agents (1, 2). Mammalian pattern recognition receptors (PRRs) such as Toll-Like Receptors (TLRs), RIG-I-like Receptors (RLRs), cGAS/STING and NOD-like receptors play key roles in protective innate and adaptive immune responses against microbial agents (3–8). Since different PRRs recognize distinct microbial components, and since individual immune cell types differ in the classes of PRR they express, host innate-immune responses to an infectious agent are specified at both the molecular and the cellular level. For example, infection of different dendritic cell (DC; see below) subsets with bacterial, viral or fungal pathogens triggers distinct and specialized transcriptional responses in each subset (1), the molecular basis for which remains an area of intense research.
DCs are a mobile subset of leucocytes specialized for regulating diverse host responses against microbes (9–12). They are broadly characterized as conventional (cDCs) and plasmacytoid DCs (pDCs). cDCs play a crucial role in T cell activation, and express high levels of co-stimulatory molecules, pro-inflammatory cytokines, and type 1 IFNs following direct infection by virus (13–15). pDCs (also known as IFN producing cells; IPC) are a unique subset of circulating DCs that, upon stimulation by microbial components, are specialized in the production of very high levels of type 1 IFN (15–19). Recent studies have revealed critical differences in the manner in which cDCs and pDCs produce type 1 IFNs following an acute RNA virus infection. While cDCs are critically reliant on the RLR pathway for induction of type 1 IFNs following infection, pDCs instead employ TLR-dependent sensing mechanisms to detect RNA viruses (20–22). In particular, TLR7 and TLR9 expression in pDCs detects viral ssRNA and DNA, respectively, leading to high type 1 IFN expression (8, 23).
Members of the Interferon Regulatory Factor (IRF) and NF-κB transcription factor families are activated following engagement of multiple PRRs (3, 23–31). Both IRF3 and IRF7 are crucial for inducing IFN-α/β (32, 33). However, while IRF3 contributes to IFN expression following RLR engagement in cDCs, IRF7 is essential for optimal IFN expression in pDCs (32–35). NF-κB activation by PRRs typically depends on IκB kinase β (IKKβ) (36). Our previous studies indicate that while IKKβ/NF-κB is required for early induction of IFN-β after virus infection, it does not regulate the magnitude of type 1 IFN expression following virus triggering of the RLR pathway in cDCs and MEFs (37, 38). In the present study, we examined the role of the NF-κB signaling axis in TLR signaling. We report that, in contrast to the RLR pathway, NF-κB is essential for the induction of type 1 IFNs downstream of TLRs. We show that induction of type 1 IFNs by pDCs is severely defective when IKKβ is inhibited, or when components of the IKKβ/canonical NF-κB signaling module are genetically ablated. We also reveal a broad requirement for NF-κB activity in TLR-driven expression of type 1 IFNs and other pro-inflammatory genes in multiple cell-types. Together with our previous findings, these results demonstrate that the IKKβ/NF-κB axis is essential for TLR - but not RLR- mediated induction of type 1 IFNs, indicative of a previously unappreciated specificity in transcription factor utilization by distinct cell-types and classes of PRRs.
Materials and Methods
Mice and materials
RelA−/−, p50−/− and cRel−/− mice have been described previously (39). IKKβ+/− mice (40) were kindly provided by Dr. Zhi-Wei Li (H. Lee Moffitt Cancer Center). E14 wild-type (WT) and RelA−/− fetal liver hematopoietic precursors (CD45.2) were adoptively transferred into lethally irradiated CD45.1 recipient mice as previously described (39). Recipient mice were typically used after 6–8 weeks. All experiments with mice were carried out in accordance with institutional guidelines. E. coli LPS 0127:B8 (Sigma) was used at 100ng/ml and R837 (imiquimod from Invitrogen) was used at 5μg/ml. The IKKβ inhibitor PS-1145 was used at 10μM and was obtained from Sigma. PS-1145 does not prevent phosphorylation (activation) of IKKβ but inhibits ability to phosphorylate substrates (41).
Cells
Bone marrow-derived conventional DCs (BMDCs/cDCs) were cultured from mouse bone marrow precursors as described (42). To obtain Flt-3 ligand-induced pDCs (BM-pDCs), bone marrow cells (2 ×106 cells/ml) were cultured in 24-well plates in DC medium (RPMI 1640, 5% of fetal bovine serum, 5 μM of 2-ME, antibiotics) in the presence of 200 ng/ml of Flt-3 ligand. On day 7, pDCs were purified using anti-B220 MicroBeads (Miltenyi) and re-suspended in fresh DC medium. Mouse splenic pDCs were purified using anti-mPDCA-1 magnetic beads or FACS sorted (B220+CD11cint population) and cultured in DC medium. Mouse embryonic fibroblasts (MEFs) were prepared from day 12.5 embryos.
SeV and NDV infections
Unless otherwise indicated, SeV was used at 200 HA units/ml and Newcastle disease virus (NDV) was used at a multiplicity of infection (MOI) of 2 in DCs and 10 in MEFs, as described previously (42). For infection with NDV in vivo, mice received intravenous injections of virus (1 × 107 pfu/mouse) or saline control. 8 hours after injection, splenic pDCs were purified using anti-mPDCA-1 MicroBeads (Miltenyi Biotec)
Real-time PCR, Ribonuclease Protection Assay, immnoblotting and transfection
Real-time PCR methods and primers were used as previously described (42). Whole cell extracts were prepared by lysing cells in RIPA buffer (25 mM Tris-HCl, pH7.9, 150 mM NaCl, 2 mM EDTA, 0.5% NP-40) supplemented with protease inhibitor cocktail (Roche) and phosphatase inhibitors (NaF and Na3VO4). The extracts were subjected to immunoblot analysis using antibodies against phospho-IKKα/β and total IKKβ (Cell Signaling). IRF-3 dimerization was detected by native-gel immunoblot analysis as described (43). Western blotting for IRF-3 was performed using anti-IRF-3 antibody (Zymed). Ribonuclease Protection Assay (RPA) was performed as previously described (44). Poly (I:C) (Invivogen) was used as dsRNA for transfection using electroporation as previously described (45). Briefly, 4 × 106 cDC cells were resuspended in 300μl RPMI medium with or without 3μg Poly(I:C) and pulsed once at 300V, 150mF (Bio-Rad Gene Pulser II). Cell suspensions were washed with 1.7ml DC media once, then seeded to low attachment 24-well plates for 6h after which cells were collected for RT-PCR.
ELISA and EMSA
After different treatments, cells were cultured for indicated time periods and supernatants were collected. Expression of IFN-α and IFN-β was determined by ELISA kits from PBL Biomedical Laboratories following the manufacturer’s recommendations. Readings below the first standard curve sample were considered undetected (U.D). For detection of NF-κB activation, nuclear extracts were prepared and EMSA was performed as previously described (46).
Statistical Analysis
All statistical analysis was performed using Student’s t test, with a p-value of less than 0.05 considered significant. P-values in figure legends are specified as *<0.05, **<0.01, ***<0.001.
Results
Crucial role of IKKβ/NF-κB in virus-induced type 1 IFN expression in pDCs
We previously found that the overall magnitude of type I IFN expression after virus infection of cDCs is not altered in the absence of NF-κB RelA, p50 or cRel subunits (38). However, RelA is uniquely required for basal and early virus-induced IFN-β expression in cDCs and MEFs (37, 38, 47). Type 1 IFN expression by pDCs is believed to be especially important for the host anti-virus response. To determine the potential role of IKKβ/NF-κB pathway in virus-induced type 1 IFN expression in pDCs, we utilized PS-1145, a highly specific IKKβ inhibitor (41, 48). Interestingly, Sendai virus (SeV)-induced type 1 IFN production in WT splenic pDCs as well as bone marrow-derived pDCs (BM-pDCs) was significantly reduced in the presence of PS-1145 (Fig. 1A). Similar results were obtained following Newcastle disease virus (NDV) infection of splenic and BM-pDCs (Fig. S1). In contrast, and consistent with our previous results indicating lack of essential requirement for IKKβ/NF-κB (37, 38), PS-1145 did not inhibit SeV-induced expression of IFN-α or IFN-β in cDCs (Fig. 1B). Furthermore, no significant effect of PS-1145 was noticed after 24h SeV infection while NDV-induced IFN-β was only slightly reduced (Fig. S2). Together, these results suggest that virus-induced type 1 IFN is selectively dependent on IKKβ in pDCs but not in cDCs.
Fig. 1. Differential effect of IKKβ inhibition on virus-induced type 1 IFN expression in cDCs and pDCs.

(A) Effect of IKKβ inhibitor PS-1145 on SeV infection-induced type 1 IFN production by splenic pDCs and BM-derived pDCs determined by ELISA. (B) Effect of PS-1145 on SeV infection-induced IFN-β and IFN-α protein expression in cDCs determined by ELISA. All virus infections were for 6h. SeV: sendai virus. PS: PS-1145. Sp_pDCs: splenic plasmacytoid dendritic cells. BM_pDCs: bone marrow-derived plasmacytoid dendritic cells generated by FLT-3 ligand. P-values are specified as *<0.05, **<0.01, ***<0.001. Representative results from 3 independent experiments are shown.
After SeV infection, IFN-β expression was slightly but significantly reduced by ~30% in RelA−/− cDCs while no reduction was noticed in p50−/−, cRel−/− and p50−/−cRel−/− cDCs (Fig. 2A). In contrast, IFN-β and IFN-α production was substantially impaired in pDCs derived from RelA−/−, p50−/−, cRel−/− and p50−/−cRel−/− knockout mice, with the most reduction in RelA−/− and p50−/−cRel−/− pDCs (Fig. 2B). While absence of RelA in cDCs reduced IFN-β expression by ~30%, a ~75% reduction in RelA absence was seen in pDCs (Fig. 2A and 2B). p50+cRel absence did not reduce IFN-β expression in cDCs but significantly reduced it in pDCs (Fig. 2A and 2B). Therefore, these NF-κB subunits are substantially more important for IFN-β expression after virus infection of pDCs versus cDCs.
Fig. 2. NF-κB deficiency severely impairs in vitro and in vivo virus-induced type 1 IFN expression in pDCs.

(A) ELISA analysis of IFN-β production induced by SeV infection in WT and NF-κB knockout cDCs. (B) ELISA analysis of type 1 IFN production induced by SeV infection in WT and NF-κB knockout splenic pDCs. (C) WT and p50−/−cRel−/− cDC were transfected with Poly (I:C) by electroporation. Poly (I:C) administration and transfection are indicated. Real-time PCR analysis of IFN-β mRNA expression is shown. (D) Real-time PCR analysis of IFN-β mRNA expression in splenic pDCs from WT and p50−/−cRel−/− mice infected with NDV. NDV: newcastle disease virus. All virus infections were for 6h. P-values are specified as *<0.05, **<0.01, ***<0.001. Representative results from 2 independent experiments are shown.
Since absence of p50+cRel does not impact mouse viability, unlike RelA absence, we utilized p50−/−cRel−/− mice for several additional studies. First, we sought to more directly test role of these NF-κB subunits in IFN-β expression after triggering of the RLR pathway. Transfection of double-stranded RNA (dsRNA) in cDCs is a powerful stimulus for IFN-β expression through activation of the RLR pathway (45). Importantly, p50−/−cRel−/− cDCs induced robust and similar expression of IFN-β compared to WT cDCs after dsRNA transfection (Fig. 2C). Second, unlike p50−/−cRel−/− pDC (Fig. 2B), cDCs from p50−/−cRel−/− spleens induced normal levels of IFN-β (Fig. S3). Third, we determined the in vivo role of p50+cRel in virus-induced type 1 IFN expression in pDCs. WT and p50−/−cRel−/− mice were infected with NDV (107pfu/mouse). 8 hours after infection, mRNA levels of IFN-β in splenic pDCs was determined. As shown in Fig 2D, IFN-β mRNA in pDCs from NDV-infected WT mice was robustly induced compared to the PBS control. In striking contrast, induction of IFN-β mRNA in pDCs from p50−/−cRel−/− mice was greatly reduced (Fig. 2D). These findings indicate a critical role for NF-κB p50 and cRel subunits in regulating virus-induced IFN-β expression in pDCs in vivo. On the other hand, our results indicate that these NF-κB subunits are not essential for RLR-induced IFN-β expression triggered by dsRNA transfection (Fig. 2C) or by virus infection of cDCs.
IKKβ inhibition suppresses TLR-induced type 1 IFN expression in cDCs
As mentioned above, pDCs specifically require TLR while cDCs require the RLR pathway for virus-induced type 1 IFN expression. An interesting possibility is that IKKβ/NF-κB dependence for virus-induced type 1 IFN expression in pDC is due to unique requirement for this signaling module in TLR signaling. To test this possibility, we determined whether TLR-induced type 1 IFN expression in cDCs was dependent on IKKβ/NF-κB. Importantly, unlike lack of effect after virus infection (Fig. 1B), TLR4 ligand LPS-induced IFN-β mRNA (not shown) and protein expression was substantially inhibited by PS-1145 in cDCs (Fig. 3A). TLR7 ligand R837 induced IFN-β expression in cDCs was also significantly inhibited by PS-1145 (Fig. 3A). As expected, PS-1145 significantly inhibited LPS-induced NF-κB activation (Fig. 3B) but had no appreciable effect on IRF-3 activation (as determined by activation-induced dimerization) in cDCs (Fig. 3C). These results therefore indicate that TLR-induced IKKβ/NF-κB activation in both pDCs (by virus infection) and cDCs (by LPS, R837) is required for type 1 IFN expression.
Fig. 3. IKKβ inhibition suppresses TLR-induced type 1 IFN expression in cDCs.

(A) Effect of PS-1145 on LPS- or R837-induced IFN-β protein expression (6h) determined by ELISA in cDCs. (B) Effect of PS-1145 on LPS-induced NF-κB activation (6h) in cDCs determined by EMSA. (C) Effect of PS-1145 on LPS-induced IRF-3 activation (dimerization) (6h) in cDCs determined by Native Gel Western Blotting. P-values are specified as *<0.05, **<0.01, ***<0.001. Representative results from 2 or 3 independent experiments are shown.
IKKβ is essential for TLR but not RLR induced type 1 IFN and inflammatory cytokine expression in MEFs
We wished to extend analysis of the role for IKKβ using IKKβ−/− cDCs. Since IKKβ deficiency results in embryonic day 12 lethality (40), we used IKKβ−/− fetal liver cells to generate radiation chimeras for obtaining IKKβ−/− cDCs. However, as we previously observed with p50−/−RelA−/− mice (39), spleen cDCs, pDCs or BM-derived cDCs were not obtained in the absence of IKKβ (data not shown). Similar to cDCs, virus-induced type 1 IFN expression is known to depend on RLR signaling in MEFs (20). Therefore, to further understand the role of IKKβ in TLR versus RLR-induced gene expression, we utilized early passage IKKβ−/− MEFs (40). Importantly, LPS-induced IFN-β expression was severely impaired in IKKβ−/− MEFs (Fig. 4A). In contrast, SeV or NDV infection induced slightly elevated expression of IFN-β in IKKβ−/− MEFs compared to WT MEFs (Fig. 4A). Similar findings were obtained when IFN-α4 and IFN-α (non4) mRNA expression was analyzed in IKKβ−/− MEFs after virus infection (Fig. 4A), while LPS did not induce detectable expression of IFN-α4 and IFN-α (non4) mRNA in MEFs or cDCs (data not shown). Overall, these findings indicate a key role for IKKβ in TLR-induced but not RLR-induced type 1 IFN expression in MEFs.
Fig. 4. IKKβ is essential for TLR but not RLR induced type 1 IFN and inflammatory cytokine expression in MEFs.

(A) IFN-β, IFN-α4, and IFN-α (non4) mRNA expression in IKKβ+/+ and IKKβ−/− MEFs induced by LPS, SeV or NDV infection as determined by real-time PCR. (B) TNFα mRNA expression in IKKβ+/+ and IKKβ−/− MEFs induced by LPS, SeV or NDV infection as determined by real-time PCR. (C) IL-6 mRNA expression in IKKβ+/+ and IKKβ−/− MEFs induced by LPS, SeV or NDV infection as determined by real-time PCR. P-values are specified as *<0.05, **<0.01, ***<0.001. Representative results from 3 independent MEF isolates are shown.
RNA virus infection also induces inflammatory cytokine expression in an RLR/RIG-I-dependent manner (20). Next, we determined the role of IKKβ in RLR-induced expression of TNFα and IL-6. LPS-induced TNFα expression was abolished in IKKβ−/− MEFs, while TNFα was still induced after virus infection of IKKβ−/− MEFs but at a reduced level (Fig. 4B). Therefore, IKKβ presence is more crucial for TLR versus RLR-induced TNFα expression. Furthermore, LPS-induced IL-6 was substantially reduced in IKKβ−/− MEFs but not in virus-infected MEFs, which showed enhanced IL-6 expression in IKKβ−/− MEFs (Fig. 4C). These results therefore indicate that IKKβ is essential for TLR-induced but not RLR-induced inflammatory cytokine gene expression.
Differential IKKβ/NF-κB and IRF-3 activation by TLR versus RLR engagement in cDCs
Overexpression of components of both TLR and RLR pathways have been shown to strongly activate NF-κB (49, 50). However, it is possible that natural triggering of these pathways does not equivalently impact NF-κB activation. We hypothesized that the difference in IKKβ/NF-κB requirement between cDCs and pDCs could be linked to a differential activation of this signaling module in TLR versus RLR pathways. To test this, we first determined the level of IKKβ activation after engagement of the TLR and RLR pathway in cDCs. LPS induced robust activation-specific phosphorylation of IKKβ (Fig. 5A), consistent with strong activation of NF-κB in cDCs (Fig. S4). In striking contrast, RLR engagement in cDCs by SeV infection showed little or no activation of IKKβ (Fig. 5A), which corresponded to weaker and less sustained NF-κB activation (Fig. S4). However, both SeV and NDV infection led to substantial increase in IRF-3 activation, while LPS induced significantly less IRF-3 activation (Fig. 5B). Thus, TLR engagement specifically leads to high IKKβ activation in cDCs. We next determined whether a similar response is triggered in pDCs following TLR activation by virus infection. Importantly, a substantial increase in phosphorylated IKKβ was observed in response to SeV infection of pDCs (Fig. 5C). Thus, strong IKKβ activation specifically occurs in response to TLR engagement by distinct agonists in both cDCs (i.e., LPS) and pDCs (i.e., virus infection).
Fig. 5. Differential IKKβ/NF-κB and IRF-3 activation by TLR versus RLR engagement in cDCs.

(A) Phosphorylation of IKKβ induced by LPS and SeV in cDCs determined by Western Blotting. (B) Activation of IRF-3 induced by LPS and SeV in cDCs determined by Native Gel Western Blotting. (C) Phosphorylation of IKKβ induced by SeV infection in splenic pDCs determined by Western Blotting. (D) Effect of PS-1145 on synergistic induction of IFN-β by LPS plus SeV infection in cDCs determined by real-time PCR. P-values are specified as *<0.05, **<0.01, ***<0.001. Representative results from 2 or 3 independent experiments are shown.
As RLR engagement results in weak, likely suboptimal activation of NF-κB in cDCs, we asked if TLR stimulation by virtue of its capacity to robustly activate NF-κB could enhance virus-induced IFN expression in cDCs. Indeed, LPS treatment of cDCs together with SeV infection synergistically increased levels of IFN-β mRNA compared to LPS or SeV alone (Fig. 5D) or LPS and NDV (data not shown). Furthermore, PS-1145 decreased SeV+LPS-induced IFN-β expression to levels comparable to SeV alone (Fig. 5D). Therefore, strong TLR-induced IKKβ/NF-κB activation can enhance RLR-induced IFN-β expression in cDCs, indicating that lack of a requirement for IKKβ/NF-κB signaling in type 1 IFN gene expression downstream of RLRs is likely due to weak activation of this pathway in cDCs.
TLR and RLR pathways induce distinct gene expression patterns in cDCs
We hypothesized that differential IKKβ/NF-κB activation by TLR versus RLR pathways may also impact gene expression patterns induced by these PRR. Both NF-κB target inflammatory cytokines (IL-1α, IL-1β, TNF-α and IL-6) and Th1 cytokines (IL-12 p40, IL-12 p35 and IL-18) (44) were strongly induced by LPS in cDCs (Fig. 6). In contrast, mRNA induction of these genes following SeV or NDV infection was substantially lower in comparison with LPS (Fig. 6). On the other hand, IFN-β was strongly induced by virus/RLR but only weakly by LPS/TLR (Fig. 6). These results therefore indicate that differential activation of IKKβ/NF-κB by TLR versus RLR pathways in cDCs can also lead to induction of distinct gene expression responses.
Fig. 6. TLR and RLR pathways induce distinct gene expression patterns in cDCs.
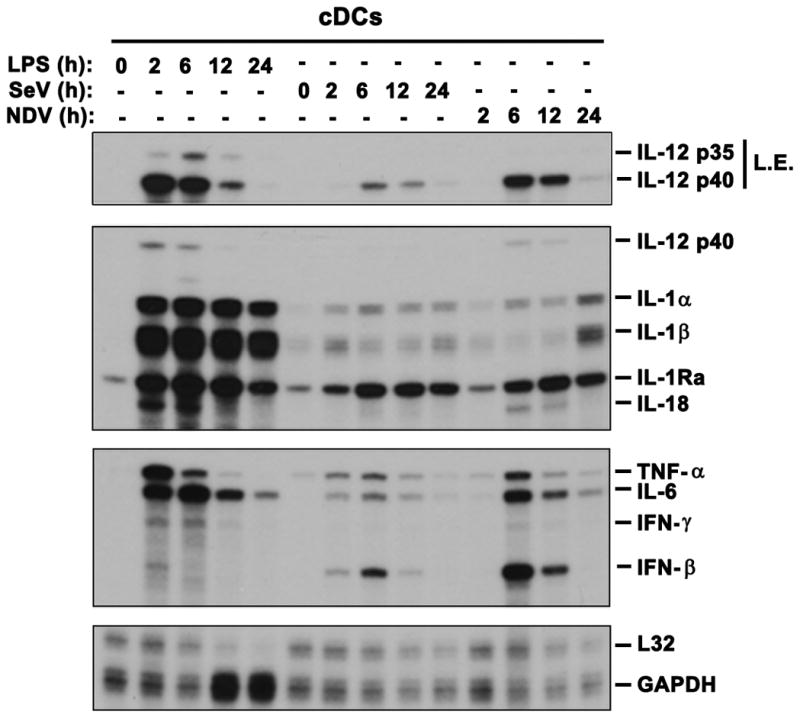
Total RNA was prepared by TRIzol reagent for Ribonuclease Protection Assay for determining expression of indicated genes. LPS: 100ng/uL, SeV: 200HAU/mL, NDV: MOI 2. L.E.: long exposure to allow visualization of IL-12 p35.
Discussion
Although IKKβ/NF-κB is activated by all known microbial PRRs (3), the role of this signaling module in mediating transcriptional responses to different classes of PRR is not well understood. We demonstrate here that IKKβ/NF-κB is selectively required for TLR - but not RLR - induced expression of type 1 IFN and inflammatory cytokines. Consequently, cell-types (such as cDCs and MEFs) that rely on RLR signaling for RNA virus-triggered induction of type 1 IFNs do not require NF-κB signaling, while cell-types (e.g., pDCs) that respond to RNA viruses via TLR-mediated signaling pathways are dependent on IKKβ/NF-κB signaling for optimal induction of type 1 IFNs. Mechanistically, we find that difference in NF-κB ‘signal strength’ may underlie this differential requirement for the IKKβ/NF-κB module in transcriptional responses downstream of RLRs versus TLRs in DC subsets: RLR stimulation in cDCs results in weaker activation of IKKβ/NF-κB than does TLR stimulation in pDCs by the same RNA virus trigger.
This selective utilization of IKKβ/NF-κB for induction of type 1 IFNs by the TLR pathway may explain, at least in part, why pDCs produce such copious amounts of type I IFNs. In pDCs, endosomal vesicle TLR signaling leads to high and sustained IRF7 activation via assembly of a unique MyD88-IRF7 transcriptional-transductional signaling complex (35, 51). The additional activation of NF-κB by TLR signaling in pDCs may then allow synergistic interactions between NF-κB and IRF7 leading to the characteristic high-level IFN expression that is a defining feature of this cell-type. The requirement for NF-κB in high-level IFN production by pDCs may result from CBP/p300 histone acetyl transferase transcriptional co-activator recruitment to the ifnβ locus, and perhaps to other type 1 IFN gene loci.
In contrast to NF-κB, IRF-3 is activated significantly more strongly by RLRs than by the TLRs. High-level IRF-3 activation in cell-types (e.g., cDCs, MEFs) where RLRs are the primary sensors and producers of type 1 IFNs after RNA virus infection may therefore minimize a requirement for strong IKKβ-induced NF-κB activation for type 1 IFN gene expression (52). Nonetheless, our earlier work has demonstrated an important role for NF-κB very early in IFN-β induction by RLRs (37), consistent with the idea that an initial, relatively-weak NF-κB signal serves to recruit essential coactivators to type I IFN gene loci immediately after RLR activation, after which strong IRF-3 activity can drive IFN-β gene expression (52). While both TLR or RLR pathway components may potently activate NF-κB in overexpression studies (49, 50), our results indicate that the natural activation of these two pathways leads to significantly different activation levels of IKKβ/NF-κB, with consequently distinct effects on the induction of type 1 IFN genes.
Genome-wide gene expression studies have previously revealed that transcriptional mechanisms play a key role in determining specificity of host responses against different infectious agents (1, 2). For example, studies in DCs have shown that pro-inflammatory cytokines (e.g., TNFα, IL-1α/β, and IL-6) are strongly induced by bacteria but weakly by viruses while, conversely, type 1 IFN genes are strongly induced by viruses but only weakly by bacteria (1). As the molecular basis for this specificity is unclear, we explored whether differential activation of NF-κB versus IRF3 pathways by TLRs and RLRs leads to the induction of distinct gene expression patterns, besides type 1 IFNs. A side-by-side comparison of LPS stimulation versus SeV and NDV infection of cDCs showed a dramatic difference in induction of inflammatory cytokines versus IFN-β expression by these stimuli. These results therefore indicate that (a) strong activation and critical requirement for IKKβ/NF-κB by TLR signaling is crucial for high-level expression of inflammatory cytokines (e.g., TNFα, IL-1α/β, and IL-6), while conversely, (b) low activation and lack of requirement of IKKβ/NF-κB by RLR is likely responsible for low expression of inflammatory cytokines following RNA virus infection. More generally, differential activation of IKKβ/NF-κB and IRF3 axes may allow induction of distinct gene expression responses by infectious agents activating different PRRs in distinct cell-types. These findings as they relate to our discoveries in cDCs versus pDCs are schematically summarized in Fig. 7.
Fig. 7. Regulation of RLR and TLR-induced transcriptional responses in cDCs and pDCs.

RLR engagement in cDCs leads to strong IRF-3 but weak NF-κB activation resulting in high type 1 IFN but low inflammatory cytokine expression. In contrast, TLR-induced combined IRF-7 and NF-κB activation in pDCs leads to very high type 1 IFN expression, while impact on inflammatory cytokines remains to be defined.
Supplementary Material
Acknowledgments
We acknowledge help provided by Flow Cytometry and Molecular Genomics core facilities at the Moffitt Cancer Center (MCC). IKKβ+/− mice were kindly provided by Dr. Zhi-Wei Li (MCC).
Footnotes
This work was supported by NIH R01 AI059715, DOD BC011057 and institutional funds from Moffitt Cancer Center to A.A.B. This work was also partially supported by an NIAID grants U19AI083025 and U01AI095611 (to A.G-S.) and by an NIH R21 AI104212 (to S.B.).
References
- 1.Huang Q, Liu D, Majewski P, Schulte LC, Korn JM, Young RA, Lander ES, Hacohen N. The plasticity of dendritic cell responses to pathogens and their components. Science. 2001;294:870–875. doi: 10.1126/science.294.5543.870. [DOI] [PubMed] [Google Scholar]
- 2.Jenner RG, Young RA. Insights into host responses against pathogens from transcriptional profiling. Nat Rev Microbiol. 2005;3:281–294. doi: 10.1038/nrmicro1126. [DOI] [PubMed] [Google Scholar]
- 3.Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 4.Medzhitov R. Recognition of microorganisms and activation of the immune response. Nature. 2007;449:819–826. doi: 10.1038/nature06246. [DOI] [PubMed] [Google Scholar]
- 5.Ishii KJ, Koyama S, Nakagawa A, Coban C, Akira S. Host innate immune receptors and beyond: making sense of microbial infections. Cell Host Microbe. 2008;3:352–363. doi: 10.1016/j.chom.2008.05.003. [DOI] [PubMed] [Google Scholar]
- 6.Barber GN. STING-dependent cytosolic DNA sensing pathways. Trends Immunol. 2013 doi: 10.1016/j.it.2013.10.010. [DOI] [PubMed] [Google Scholar]
- 7.Iwasaki A, Medzhitov R. Regulation of adaptive immunity by the innate immune system. Science. 2010;327:291–295. doi: 10.1126/science.1183021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Blasius AL, Beutler B. Intracellular toll-like receptors. Immunity. 2010;32:305–315. doi: 10.1016/j.immuni.2010.03.012. [DOI] [PubMed] [Google Scholar]
- 9.Gautier EL, Shay T, Miller J, Greter M, Jakubzick C, Ivanov S, Helft J, Chow A, Elpek KG, Gordonov S, Mazloom AR, Ma’ayan A, Chua WJ, Hansen TH, Turley SJ, Merad M, Randolph GJ C. Immunological Genome. Gene-expression profiles and transcriptional regulatory pathways that underlie the identity and diversity of mouse tissue macrophages. Nat Immunol. 2012;13:1118–1128. doi: 10.1038/ni.2419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Greter M, Helft J, Chow A, Hashimoto D, Mortha A, Agudo-Cantero J, Bogunovic M, Gautier EL, Miller J, Leboeuf M, Lu G, Aloman C, Brown BD, Pollard JW, Xiong H, Randolph GJ, Chipuk JE, Frenette PS, Merad M. GM-CSF controls nonlymphoid tissue dendritic cell homeostasis but is dispensable for the differentiation of inflammatory dendritic cells. Immunity. 2012;36:1031–1046. doi: 10.1016/j.immuni.2012.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu K, Victora GD, Schwickert TA, Guermonprez P, Meredith MM, Yao K, Chu FF, Randolph GJ, Rudensky AY, Nussenzweig M. In vivo analysis of dendritic cell development and homeostasis. Science. 2009;324:392–397. doi: 10.1126/science.1170540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Randolph GJ, Ochando J, Partida-Sanchez S. Migration of dendritic cell subsets and their precursors. Annual review of immunology. 2008;26:293–316. doi: 10.1146/annurev.immunol.26.021607.090254. [DOI] [PubMed] [Google Scholar]
- 13.Steinman RM, Hemmi H. Dendritic cells: translating innate to adaptive immunity. Curr Top Microbiol Immunol. 2006;311:17–58. doi: 10.1007/3-540-32636-7_2. [DOI] [PubMed] [Google Scholar]
- 14.Reis e Sousa C. Dendritic cells in a mature age. Nature reviews Immunology. 2006;6:476–483. doi: 10.1038/nri1845. [DOI] [PubMed] [Google Scholar]
- 15.Liu YJ. IPC: professional type 1 interferon-producing cells and plasmacytoid dendritic cell precursors. Annual review of immunology. 2005;23:275–306. doi: 10.1146/annurev.immunol.23.021704.115633. [DOI] [PubMed] [Google Scholar]
- 16.Lund JM, Linehan MM, Iijima N, Iwasaki A. Cutting Edge: Plasmacytoid dendritic cells provide innate immune protection against mucosal viral infection in situ. J Immunol. 2006;177:7510–7514. doi: 10.4049/jimmunol.177.11.7510. [DOI] [PubMed] [Google Scholar]
- 17.Prakash A, Smith E, Lee CK, Levy DE. Tissue-specific positive feedback requirements for production of type I interferon following virus infection. J Biol Chem. 2005;280:18651–18657. doi: 10.1074/jbc.M501289200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dalod M, Hamilton T, Salomon R, Salazar-Mather TP, Henry SC, Hamilton JD, Biron CA. Dendritic cell responses to early murine cytomegalovirus infection: subset functional specialization and differential regulation by interferon alpha/beta. J Exp Med. 2003;197:885–898. doi: 10.1084/jem.20021522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Garcia-Sastre A, Biron CA. Type 1 interferons and the virus-host relationship: a lesson in detente. Science. 2006;312:879–882. doi: 10.1126/science.1125676. [DOI] [PubMed] [Google Scholar]
- 20.Kato H, Sato S, Yoneyama M, Yamamoto M, Uematsu S, Matsui K, Tsujimura T, Takeda K, Fujita T, Takeuchi O, Akira S. Cell type-specific involvement of RIG-I in antiviral response. Immunity. 2005;23:19–28. doi: 10.1016/j.immuni.2005.04.010. [DOI] [PubMed] [Google Scholar]
- 21.Kumagai Y, Takeuchi O, Kato H, Kumar H, Matsui K, Morii E, Aozasa K, Kawai T, Akira S. Alveolar macrophages are the primary interferon-alpha producer in pulmonary infection with RNA viruses. Immunity. 2007;27:240–252. doi: 10.1016/j.immuni.2007.07.013. [DOI] [PubMed] [Google Scholar]
- 22.Loo YM, Fornek J, Crochet N, Bajwa G, Perwitasari O, Martinez-Sobrido L, Akira S, Gill MA, Garcia-Sastre A, Katze MG, Gale M., Jr Distinct RIG-I and MDA5 signaling by RNA viruses in innate immunity. J Virol. 2008;82:335–345. doi: 10.1128/JVI.01080-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Akira S, Takeda K. Toll-like receptor signalling. Nature reviews Immunology. 2004;4:499–511. doi: 10.1038/nri1391. [DOI] [PubMed] [Google Scholar]
- 24.Honda K, Takaoka A, Taniguchi T. Type I inteferon gene induction by the interferon regulatory factor family of transcription factors. Immunity. 2006;25:349–360. doi: 10.1016/j.immuni.2006.08.009. [DOI] [PubMed] [Google Scholar]
- 25.Saccani S, Pantano S, Natoli G. Two waves of nuclear factor kappaB recruitment to target promoters. J Exp Med. 2001;193:1351–1359. doi: 10.1084/jem.193.12.1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Saccani S, Pantano S, Natoli G. Modulation of NF-kappaB activity by exchange of dimers. Mol Cell. 2003;11:1563–1574. doi: 10.1016/s1097-2765(03)00227-2. [DOI] [PubMed] [Google Scholar]
- 27.Werner SL, Barken D, Hoffmann A. Stimulus specificity of gene expression programs determined by temporal control of IKK activity. Science. 2005;309:1857–1861. doi: 10.1126/science.1113319. [DOI] [PubMed] [Google Scholar]
- 28.Covert MW, Leung TH, Gaston JE, Baltimore D. Achieving stability of lipopolysaccharide-induced NF-kappaB activation. Science. 2005;309:1854–1857. doi: 10.1126/science.1112304. [DOI] [PubMed] [Google Scholar]
- 29.Hayden MS, West AP, Ghosh S. NF-kappaB and the immune response. Oncogene. 2006;25:6758–6780. doi: 10.1038/sj.onc.1209943. [DOI] [PubMed] [Google Scholar]
- 30.Hiscott J. Convergence of the NF-kappaB and IRF pathways in the regulation of the innate antiviral response. Cytokine Growth Factor Rev. 2007;18:483–490. doi: 10.1016/j.cytogfr.2007.06.002. [DOI] [PubMed] [Google Scholar]
- 31.Li Q, I, Verma M. NF-kappaB regulation in the immune system. Nature reviews Immunology. 2002;2:725–734. doi: 10.1038/nri910. [DOI] [PubMed] [Google Scholar]
- 32.Sato M, Suemori H, Hata N, Asagiri M, Ogasawara K, Nakao K, Nakaya T, Katsuki M, Noguchi S, Tanaka N, Taniguchi T. Distinct and essential roles of transcription factors IRF-3 and IRF-7 in response to viruses for IFN-alpha/beta gene induction. Immunity. 2000;13:539–548. doi: 10.1016/s1074-7613(00)00053-4. [DOI] [PubMed] [Google Scholar]
- 33.Sharma S, tenOever BR, Grandvaux N, Zhou GP, Lin R, Hiscott J. Triggering the interferon antiviral response through an IKK-related pathway. Science. 2003;300:1148–1151. doi: 10.1126/science.1081315. [DOI] [PubMed] [Google Scholar]
- 34.Honda K, Yanai H, Negishi H, Asagiri M, Sato M, Mizutani T, Shimada N, Ohba Y, Takaoka A, Yoshida N, Taniguchi T. IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature. 2005;434:772–777. doi: 10.1038/nature03464. [DOI] [PubMed] [Google Scholar]
- 35.Honda K, Ohba Y, Yanai H, Negishi H, Mizutani T, Takaoka A, Taya C, Taniguchi T. Spatiotemporal regulation of MyD88-IRF-7 signalling for robust type-I interferon induction. Nature. 2005;434:1035–1040. doi: 10.1038/nature03547. [DOI] [PubMed] [Google Scholar]
- 36.Hayden MS, Ghosh S. Shared principles in NF-kappaB signaling. Cell. 2008;132:344–362. doi: 10.1016/j.cell.2008.01.020. [DOI] [PubMed] [Google Scholar]
- 37.Wang J, Basagoudanavar SH, Wang X, Hopewell E, Albrecht R, Garcia-Sastre A, Balachandran S, Beg AA. NF-{kappa}B RelA Subunit Is Crucial for Early IFN-{beta} Expression and Resistance to RNA Virus Replication. J Immunol. 2010;185:1720–1729. doi: 10.4049/jimmunol.1000114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang X, Hussain S, Wang EJ, Li MO, Garcia-Sastre A, Beg AA. Lack of essential role of NF-kappa B p50, RelA, and cRel subunits in virus-induced type 1 IFN expression. J Immunol. 2007;178:6770–6776. doi: 10.4049/jimmunol.178.11.6770. [DOI] [PubMed] [Google Scholar]
- 39.Ouaaz F, Arron J, Zheng Y, Choi Y, Beg AA. Dendritic cell development and survival require distinct NF-kappaB subunits. Immunity. 2002;16:257–270. doi: 10.1016/s1074-7613(02)00272-8. [DOI] [PubMed] [Google Scholar]
- 40.Li ZW, Chu W, Hu Y, Delhase M, Deerinck T, Ellisman M, Johnson R, Karin M. The IKKbeta subunit of IkappaB kinase (IKK) is essential for nuclear factor kappaB activation and prevention of apoptosis. J Exp Med. 1999;189:1839–1845. doi: 10.1084/jem.189.11.1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hideshima T, Chauhan D, Richardson P, Mitsiades C, Mitsiades N, Hayashi T, Munshi N, Dang L, Castro A, Palombella V, Adams J, Anderson KC. NF-kappa B as a therapeutic target in multiple myeloma. J Biol Chem. 2002;277:16639–16647. doi: 10.1074/jbc.M200360200. [DOI] [PubMed] [Google Scholar]
- 42.Wang X, Hussain S, Wang EJ, Wang X, Li MO, Garcia-Sastre A, Beg AA. Lack of essential role of NF-kappa B p50, RelA, and cRel subunits in virus-induced type 1 IFN expression. J Immunol. 2007;178:6770–6776. doi: 10.4049/jimmunol.178.11.6770. [DOI] [PubMed] [Google Scholar]
- 43.Iwamura T, Yoneyama M, Yamaguchi K, Suhara W, Mori W, Shiota K, Okabe Y, Namiki H, Fujita T. Induction of IRF-3/-7 kinase and NF-kappaB in response to double-stranded RNA and virus infection: common and unique pathways. Genes Cells. 2001;6:375–388. doi: 10.1046/j.1365-2443.2001.00426.x. [DOI] [PubMed] [Google Scholar]
- 44.Wang J, Wang X, Hussain S, Zheng Y, Sanjabi S, Ouaaz F, Beg AA. Distinct roles of different NF-kappa B subunits in regulating inflammatory and T cell stimulatory gene expression in dendritic cells. J Immunol. 2007;178:6777–6788. doi: 10.4049/jimmunol.178.11.6777. [DOI] [PubMed] [Google Scholar]
- 45.Diebold SS, Montoya M, Unger H, Alexopoulou L, Roy P, Haswell LE, Al-Shamkhani A, Flavell R, Borrow P, Reis e Sousa C. Viral infection switches non-plasmacytoid dendritic cells into high interferon producers. Nature. 2003;424:324–328. doi: 10.1038/nature01783. [DOI] [PubMed] [Google Scholar]
- 46.Beg AA, Finco TS, Nantermet PV, Baldwin AS. Tumor necrosis factor and interleukin-1 lead to phosphorylation and loss of IκB: a mechanism for NF-κB activation. Mol Cell Biol. 1993;13:3301–3310. doi: 10.1128/mcb.13.6.3301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Basagoudanavar SH, Thapa RJ, Nogusa S, Wang J, Beg AA, Balachandran S. Distinct roles for the NF-kappa B RelA subunit during antiviral innate immune responses. J Virol. 2011;85:2599–2610. doi: 10.1128/JVI.02213-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yemelyanov A, Gasparian A, Lindholm P, Dang L, Pierce JW, Kisseljov F, Karseladze A, Budunova I. Effects of IKK inhibitor PS1145 on NF-kappaB function, proliferation, apoptosis and invasion activity in prostate carcinoma cells. Oncogene. 2006;25:387–398. doi: 10.1038/sj.onc.1209066. [DOI] [PubMed] [Google Scholar]
- 49.Kawai T, Takahashi K, Sato S, Coban C, Kumar H, Kato H, Ishii KJ, Takeuchi O, Akira S. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat Immunol. 2005;6:981–988. doi: 10.1038/ni1243. [DOI] [PubMed] [Google Scholar]
- 50.Yoneyama M, Kikuchi M, Natsukawa T, Shinobu N, Imaizumi T, Miyagishi M, Taira K, Akira S, Fujita T. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat Immunol. 2004;5:730–737. doi: 10.1038/ni1087. [DOI] [PubMed] [Google Scholar]
- 51.Honda K, Yanai H, Mizutani T, Negishi H, Shimada N, Suzuki N, Ohba Y, Takaoka A, Yeh WC, Taniguchi T. Role of a transductional-transcriptional processor complex involving MyD88 and IRF-7 in Toll-like receptor signaling. Proc Natl Acad Sci U S A. 2004;101:15416–15421. doi: 10.1073/pnas.0406933101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Balachandran S, Beg AA. Defining emerging roles for NF-kappaB in antivirus responses: revisiting the interferon-beta enhanceosome paradigm. PLoS Pathog. 2011;7:e1002165. doi: 10.1371/journal.ppat.1002165. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.