

Abstract
In the present study, the neuroprotective effects of the adipokine leptin, and the molecular mechanism involved, have been studied in rat and mice cortical neurons exposed to N-methyl-D-Aspartate (NMDA) in vitro. In rat cortical neurons, leptin elicited neuroprotective effects against NMDA-induced cell death which were concentration-dependent (10–100 ng/ml) and largest when the adipokine was preincubated for 2 hours before the neurotoxic stimulus. In both rat and mouse cortical neurons, leptin-induced neuroprotection was fully antagonized by Paxilline (Pax, 0.01–1 μM) and Iberiotoxin (Ibtx, 1–100 nM), two blockers of Ca2+- and voltage-activated K+ channels (Slo1 BK channels), with EC50s (38±10 nM and 5±2 nM for Pax and Ibtx, respectively) close to those reported for Pax- and Ibtx-induced BK channel blockade; the BK channel opener NS1619 (1–30 μM) induced a concentration-dependent protection against NMDA-induced excitotoxicity. Moreover, cortical neurons from mice lacking one or both alleles coding for Slo1 BK channel pore-forming subunits were insensitive to leptin-induced neuroprotection. Finally, leptin exposure dose-dependently (10–100 ng/ml) increased intracellular Ca2+ levels in rat cortical neurons. In conclusion, our results suggest that Slo1 BK channel activation following increases in intracellular Ca2+ levels is a critical step for leptin-induced neuroprotection in NMDA-exposed cortical neurons in vitro, thus highlighting leptin-based intervention via BK channel activation as a potential strategy to counteract neurodegenerative diseases.
Keywords: voltage- and calcium-activated potassium channels (BK channels), leptin, N-methyl-D-Aspartate (NMDA), neuroprotection, paxilline, iberiotoxin, cortical neurons, intracellular Ca2+ concentration
1. Introduction
Leptin, a 16-kDa peptide hormone produced by white adipocytes, controls appetitive behaviors by acting on hypothalamic neurons involved in food intake and energy expenditure (Halaas et al., 1995). Leptin receptors are expressed in diverse brain regions such as the hippocampus, the cortex, and the cerebellum, being often located at axonal and synaptic subcellular sites (Harvey, 2007). Extrahypothalamic actions of leptin are increasingly being recognized; for example, leptin influences synaptic plasticity in hippocampal neurons (Shanley et al., 2001; Oomura et al., 2006) and inhibits epileptiform-like activity in hippocampal (Shanley et al., 2002a and 2002b; O’Malley et al., 2005) as well as in neocortical neurons (Xu et al., 2008). Several studies have also highlighted the ability of leptin to exert both in vitro and in vivo neuroprotective effects against oxygen-glucose deprivation, hypoxia, ischemia, neurotrophic factor withdrawal, and excitotoxic or oxidative stimuli in neuronal populations from distinct brain areas (Dicou et al., 2001; Zhang et al., 2007; Weng et al., 2007; Guo et al., 2008; Signore et al., 2008; Valerio et al., 2009; Gavello et al., 2012).
Among the molecular mechanisms responsible for the effects of leptin, neuronal silencing via activation of potassium (K+) channels appears to play a major role. Among leptin-sensitive K+ channels, activation of ATP-sensitive K+ channels (KATP) has been proposed to mediate leptin-induced suppression of excitability of hypothalamic neurons (Spanswick et al., 1997; Irani et al. 2008). Similarly, leptin-induced insulin release suppression from pancreatic β cells also depends on KATP activation (Kieffer et al., 1997). More recently, pharmacological evidence has suggested that large-conductance, Ca2+- and voltage-activated K+ channels (Slo1 BK channels), which are particularly abundant in axons and nerve terminals (Knaus et al., 1996; Misonou et al., 2006), where they stabilize the neuronal membrane potential and regulate excitatory neurotransmitter release (Brenner et al., 2005; Gu et al., 2007; Jin et al., 2000; Raffaelli et al., 2004; Dirnagl et al., 2003; Martire et al., 2010), mediate at least part of leptin’s effects of neuronal excitability. In fact, activation of BK channels mediates leptin-induced inhibition of gastric mucosal vagal afferents (Kentish et al., 2013), and hippocampal neuronal firing (Shanley et al., 2002a) and epileptiform-like events (Shanley et al., 2002b). Notably, BK channel activation exerts strong neuroprotective effects in animal models of cerebral ischemia (Gribkoff et al., 2001), and attenuated cerebral edema and neurologic motor impairment after traumatic brain injury (Cheney et al., 2001). Activation of BK channels also seems to mediate leptin effects on primary hippocampal neuronal excitability during hypoxia (Gavello et al., 2012).
Despite these results, direct evidence for BK channel activation by this adipokine during neuroprotection is lacking. Therefore, in the present study, the neuroprotective potential of BK channel activation by leptin and the underlying molecular mechanism(s), have been assessed in cortical neurons exposed to the ionotropic glutamate receptor agonist N-methyl-D-Aspartate (NMDA), a classical excitotoxic insult. The results obtained indicate that leptin is endowed with significant neuroprotective effects in both rat and mouse cortical neurons exposed to NMDA; the pharmacological blockade of BK channels, or the lack of one (Slo1+/− mice) or both (Slo1−/− mice) Slo1 alleles fully counteracted leptin-mediated neuroprotection. Furthermore, intracellular Ca2+ concentration ([Ca2+]i) monitoring in single mouse cortical neurons revealed that acute leptin (10–100 ng/ml) application prompted an oscillatory behavior in [Ca2+]i. These results reveal that the activation of BK channels is an obligatory step for leptin-induced neuroprotection, highlighting leptin-based intervention via BK channel activation as a potential strategy to treat neurodegenerative diseases.
2. Materials and Methods
2.1 Animals
Animals were kept under standard conditions of temperature, humidity and light, and were supplied with standard food and water ad libitum. Animals were handled in accordance with the recommendations of the National Institutes of Health Guide for the Care and Use of Laboratory Animals and in accordance with a protocol approved by the institutional animal care committees. All efforts were made to minimize animal suffering and to reduce the number of animals used. Pregnant Wistar rats were purchased from a commercial source (Charles River, Calco, Italy), while wild-type, heterozygous and Slo1 knockout FVB/NJ mice were obtained from Prof. R. Aldrich (University of Texas, Austin, TX, USA), and genotyped as previous described (Meredith et al., 2004). Briefly, genomic DNA from tail snips was amplified by PCR using primers Neo 5′ (5′-ATA GCC TGA AGA ACG AGA TCA GC-3′) and RA 14025 3′ (5′-CCT CAA GAA GGG GAC TCT AAA C-3′), amplifying the Slo1−/− allele product of 800 bp, and the exon 1 5′-3 (5′-TTC ATC ATC TTG CTC TGG CGG ACG-3′) and WT 3′-2 (5′-CCA TAG TCA CCA ATA GCC C-3′) amplifying the wild-type product of 332 bp.
2.2 Rat and mouse cortical cell cultures
Primary cultures of rat and mouse cortical neurons were prepared from embryos at 15–17 days of gestation; embryonic age (E) was calculated by considering E0.5 the day when a vaginal plug was detected. Briefly, pregnant animals were anesthetized with diethyl ether (Carl Roth GmbH & Co KG, Karlsruhe, Germany) and sacrificed by cervical dislocation. Cortical tissues from embryos were dissected in ice-cold medium (HBSS, Hank’s Balanced Salt Solution, supplemented with 27 mM glucose, 20 mM sucrose, 4 mM sodium bicarbonate), centrifuged, and the resulting pellet was mechanically dissociated with a glass pipette. Cells were resuspended in plating medium consisting in Eagle’s MEM (MEM, Earle’s salts, supplied bicarbonate-free) supplemented with 5% fetal bovine serum (FBS, Biochrom AG, Berlin, Germany), 5% horse serum (HS, Sigma Aldrich, Taufkirchen, Gemany), 2 mM L-glutamine, 20 mM glucose, 26 mM bicarbonate, and plated on 24-well plates (Thermo Fisher Scientific, Waltham, MA, USA) or on 18 mm glass coverslips (Glaswarenfabrik Karl Hecht KG, Sondheim, Germany) coated with 100 μg/ml poly(D)-lysine (Sigma Aldrich) at a density of four embryo cerebral hemispheres/10 ml; when single-embryo dissections were necessary, resuspension volumes were changed accordingly. Glial replication was inhibited by 24 hrs exposure to 10 μM cytosine arabinofuranoside (Sigma Aldrich) after 4 days in vitro (DIV). After this treatment, the medium was supplemented with 10% HS and partially substituted twice a week. All the experiments were performed at 12–16 DIV. HBSS, Eagle’s MEM, and glutamine were purchased from LifeTechnologies (Oslo, Norway). All other reagents were from Sigma Aldrich.
2.3 Cellular treatments and assessment of neuronal survival
Prior to drug exposure, cortical neurons were washed thoroughly to remove serum using HEPES control salt solution (HCSS, 120 mM NaCl, 5.4 mM KCl, 0.8 mM MgCl2, 20 mM HEPES, 15 mM glucose, 1.8 mM CaCl2, 10 mM NaOH, pH 7.4). NMDA (Sigma Aldrich) exposure was carried out in HCSS for 15 min at room temperature, followed by NMDA washout in media stock (MS, MEM supplemented with 20 mM glucose and 26 mM bicarbonate). Recombinant human leptin (R&D Systems Inc., Minneapolis, MN, USA) was used for mouse and rat cortical neurons, as similar affinities have been measured for human leptin at human, mouse, and rat leptin receptors (Mistrik et al., 2004). Leptin was added simultaneously to NMDA exposure, as well as 15 min, 2 hrs, or 6 hrs before NMDA exposure, according to the experimental protocol. Paxilline, iberiotoxin, or NS1619 were added at the desired concentrations from stock solutions in DMSO (maximal final DMSO concentration was <1%) 15 min before leptin application and kept throughout the experiment. After NMDA exposure, cultures were washed several times with HCSS buffer and maintained in MS for 24 hrs.
Neuronal mitochondrial function was assessed by the 3-[4,5-dimethylthiazol-2-yl]-2,5 diphenyl tetrazolium bromide (MTT) assay (Aras et al., 2008), a colorimetric assay which evaluates the ability of metabolically active cells to cleave a tetrazolium salt such as MTT into a colored formazan product; to this aim, neurons were incubated in the dark at 37 °C in a 5% CO2 atmosphere for 1 h with 0.5 mg/ml MTT (Sigma Aldrich) and solubilized with acidic isopropanol. The absorbance was measured at 570 nm (background subtraction at 650 nm). Data were expressed as percentage of the absorbance measured in untreated cells. Neuronal cell viability was evaluated by propidium iodide (PI) fluorescence (Boscia et al., 2006). PI is a very stable and highly polar compound that only enters cells with damaged or leaky plasma membranes, binds to DNA, and emits a bright red fluorescence when exposed to blue-green light. At the end of the experiment, cells were incubated with 5 μg/ml PI for 1 h. PI uptake was recorded by a digital camera (Media Cybernetics, Silver Springs, MD, USA) mounted on a Nikon Eclipse 400 fluorescence microscope (Nikon Instruments, Florence, Italy; excitation 510–560 nm, emission 590 nm). Three phase contrast digital images of randomly selected microscopic fields were used for data analysis using ImageJ software (NIH, Bethesda, MD, USA).
2.4 Ca2+ imaging
For Ca2+-imaging experiments, the cells grown on glass coverslips were loaded for 20–30 min with 5 μM 1-[2-(5-carboxyoxazol-2-yl)-6-aminobenzofuran-5-oxy]-2-(21-amino-51-methylphenoxy)-ethane-N,N,N1,N1-tetraacetic acid penta-acetoxymethyl ester (Fura-2 AM; LifeTechnologies) in control buffer medium containing (in mM): 140 NaCl, 3 KCl, 1 CaCl2, 1 MgCl2, 10 HEPES, 10 glucose, pH 7.4 with NaOH. At the end of the Fura-2 AM-loading period, the coverslip was introduced into a microscope chamber on the stage of an inverted microscope (Axiovert 100, Carl Zeiss, Jena, Germany) equipped with a 20x objective (Neofluar, Carl Zeiss). Cells were washed twice with the control buffer medium, and maintained in the same solution throughout the experiment. Fluorescence measurements were performed at room temperature with a combination of a monochromator (Polychrome II, TILL Photonics, Gräfelfing, Germany) and a cooled CCD camera (IMAGO, pco, Kelheim, Germany) operated with TILLvisION (TILL Photonics) software. Pairs of images (excitation at 340 and 380 nm) of Fura-2 fluorescence intensity were measured every 1 s; background fluorescence was subtracted in each experiment. Fluorescence values were converted to [Ca2+]i using a calibration curve, as described (Grynkiewicz et al., 1985).
2.5 Statistical analysis
All data are shown as mean ± S.E.M. Unless stated otherwise, comparison between groups was made using the Student’s paired or unpaired t-test, as necessary. The analysis of one-way variance (ANOVA) followed by the Newman-Keuls test was performed to assess significance among multiple experimental groups. A P value < 0.05 was considered as statistically significant.
3. Results
3.1 Leptin protects rat cortical neurons against NMDA-induced neurotoxicity
Rat primary cortical neurons exposed for 15 min to increasing concentrations of NMDA (30–300 μM; plus 10 μM glycine) showed, over the next 24 hrs, a concentration-dependent reduction in mitochondrial function, as measured by the MTT assay (Fig. 1A). This effect was entirely prevented upon co-exposure with the noncompetitive NMDA receptor antagonist MK-801 (10 μM) (P = 0.78 versus control). Exposure to 100 μM NMDA resulted in approximately 40% reduction in mitochondrial MTT-reducing potential; this concentration was used in all subsequent experiments. Leptin (10–100 ng/ml), when applied only during the 15 min of NMDA exposure, did not significantly enhance cell survival (Fig. 1B, white bars). By contrast, when leptin exposure was started before the NMDA insult and kept throughout, significant neuroprotective effects occurred; in particular, leptin pre-treatment for 2 hrs before NMDA application elicited robust (about 60%) and dose-dependent neuroprotection (Fig. 2B, red bars). Shorter (15 min, blue bars) or longer (6 hrs, green bars) pretreatment durations caused much less intense neuroprotective effects (Fig. 1B). In neurons not exposed to NMDA, leptin had no significant effect on MTT reduction (data not shown). Since the 30 ng/ml leptin concentration was the lowest exerting maximal cytoprotective effects when pre-incubated for 2 hrs, this adipokine concentration and this preincubation time were chosen to assess leptin-induced neuronal protection by an independent method, namely PI incorporation. As shown in Figs. 1C and 1D, the exposure of cortical neurons for 15 min to 100 μM NMDA resulted in about 50% of PI-positive neurons after 24 hrs; 2 hrs of leptin pre-treatment significantly reduced NMDA-induced neuronal death (P < 0.05 versus NMDA alone). As previously described for the MTT assay, leptin failed to modify PI incorporation in the absence of the neurotoxic stimulus (Fig. 1D).
Figure 1. Leptin reduces NMDA-induced neuronal death in rat cortical neurons in vitro.
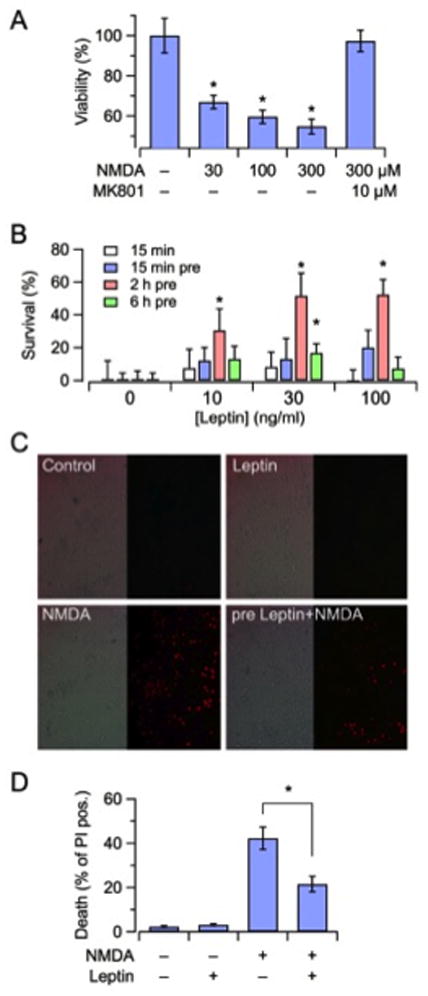
(A) Mitochondrial function, measured as MTT absorbance, was assessed 24 hrs after a 15-min exposure to the indicated concentrations of NMDA in the absence or presence of the NMDA-antagonist MK801. (B) Time course (15–360 min) and concentration dependence (10, 30, 100 ng/ml) of leptin effect on neurotoxicity, induced by 100 μM NMDA. Data are expressed as percentage of leptin-induced neuroprotection relative to controls (100 μM NMDA, no leptin treatment). (C) Photomicrographs of representative 12 DIV neuronal cultures after 24 hrs of exposure to the following experimental conditions; sham treatment (Control), leptin (2 hrs preincubation at 30 ng/ml), NMDA (15 min at 100 μM), leptin (30 ng/ml, 2 hrs preincubation) followed by NMDA (15 min at 100 μM). Left, bright field; Right, matched propidium iodide (PI) fluorescence images. All the photomicrographs were taken at 20x magnification. (D) Quantification of PI fluorescence data for the same experimental groups shown in panel C. Each data point is the mean ± S.E.M. of at least three separate experiments. The asterisk denotes statistical difference with P < 0.05.
Figure 2. Effect of BK channel modulators on leptin-induced neuroprotection in rat cortical neurons.
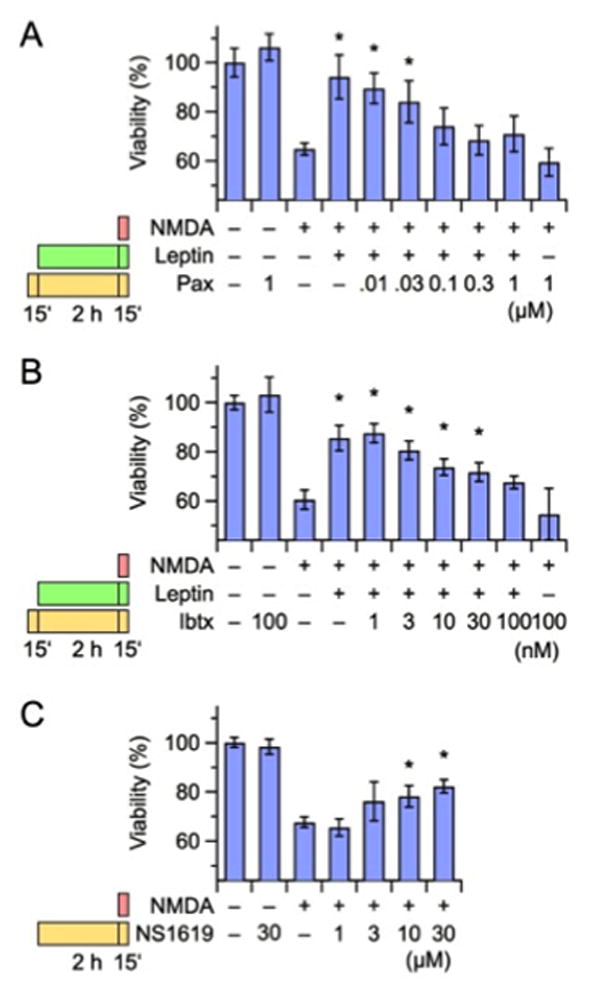
(A, B) Concentration-dependent effect of the BK blockers paxilline (A, Pax) and iberiotoxin (B, Ibtx) on leptin-induced neuroprotection of 100 μM NMDA-induced neuronal death. (C) Concentration-dependent effect of the BK channel activator NS1619 on NMDA-induced neuronal death. In each panel, the bars on the left indicate the drug exposure protocol. Neuronal viability was measured 24 hrs after the insult with the MTT assay. Data are expressed as percentage of neuronal viability in untreated, control neurons. Each data point is the mean ± S.E.M. of five separate experiments, each performed in triplicate. Asterisks denote values statistically different from respective controls (NMDA alone; P < 0.05).
3.2 The pharmacological modulation of BK channels interferes with leptin-induced neuroprotection
To investigate the role of BK channels in leptin-induced neuroprotection during NMDA exposure, the effect of BK channel modulators in this in vitro model were tested. Both natural BK channel blockers Paxilline (Pax; 0.01–1 μM; Fig. 2A) and Iberiotoxin (Ibtx; 1–100 nM; Fig. 2B) reversed leptin-induced neuroprotection in a dose-dependent manner; the EC50 values were 38 ± 10 nM and 5 ± 2 nM, respectively, for Pax and Ibtx. Both these values are rather close to the reported EC50 values for Pax- and Ibtx-induced BK channel blockade (about 10 nM for both; Nardi et al., 2003). A concentration of 100 nM of either drug completely prevented leptin neuroprotective effects in cortical neurons exposed to 100 μM NMDA (P = 0.25 for Pax; P = 0.06 for Ibtx). These results strongly indicated that the activation of BK channels mediates leptin-induced neuroprotective effects against NMDA-induced death of cortical neurons in vitro. Consistent with this, pre-treatment of the cells (2 hrs) with the BK channel opener NS1619 (1–30 μM) induced a concentration-dependent protection against NMDA-induced excitotoxicity, with 30 μM being the concentration associated with the highest level of neuroprotection (Fig. 2C); higher NS1619 concentrations could not be used, as they proved to be neurotoxic (data not shown).
3.3 Leptin fails to exert neuroprotective effects against NMDA-induced excitotoxicity in Slo1+/− and Slo1−/− mouse neuronal cultures
To provide further evidence for the role of BK channels in leptin-mediated neuroprotection, the effects of this adipokine were studied in primary cortical neurons derived from BL6 wild-type mice (Slo1+/+) and mice heterozygous (Slo1+/−) or homozygous (Slo1−/−) for a deletion of the Slo1 gene, encoding the pore-forming α subunit of BK channels (Meredith et al., 2004). Single-brain cultures from mouse embryos were exposed to NMDA in the presence or absence of leptin. NMDA (100 μM) exposure similarly impaired mitochondrial function in cortical neurons from Slo1+/+, Slo1+/−, and Slo1−/− mice (P = 0.12; Fig. 3). In NMDA-exposed cortical cultures from wild-type mice, leptin (30 ng/ml; 2 hrs preincubation) elicited neuroprotective effects whose extent (about 60%) was identical to that previously described in rat cortical neurons (see Fig. 1). In addition, as previously shown for rat neurons, leptin-induced neuroprotection in wild-type mouse cortical neurons was fully antagonized by Pax (1 μM) or Ibtx (100 nM). By contrast, leptin (30 ng/ml; 2 hrs preincubation) failed to display any significant neuroprotective effects in cortical neurons from Slo1+/− (P = 0.19 versus control, NMDA-only exposed neurons) or Slo1−/− mice (P = 0.70 versus control, NMDA-only exposed neurons).
Figure 3. Leptin-mediated neuroprotection from NMDA-induced injury is dependent on Slo1 gene expression.
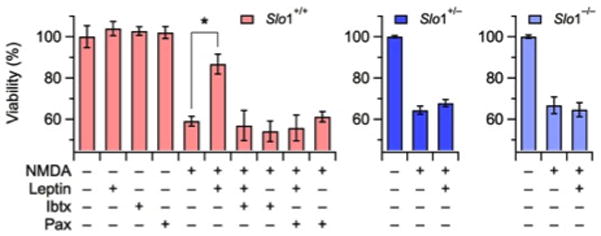
Viability of cortical neurons isolated from wild-type (Slo1+/+; red), heterozygous (Slo1+/−; blue) or homozygous (Slo1−/−; light blue) Slo1 knockout mouse embryos; neurons were pre-incubated with 30 ng/ml leptin for 2 hrs before 15-min treatment with 100 μM NMDA. Neuronal viability was measured with the MTT assay 24 hrs after the insult; neuronal viability is expressed as percent of the controls (corresponding to untreated neurons from each experimental group). The BK channel blockers paxilline (Pax, 1 μM) and iberiotoxin (Ibtx, 100 nM) were applied to wild-type neurons as described in Fig. 2. Data are mean ± S.E.M. of three independent experiments, each performed in triplicate. The asterisk denotes a value statistically different from the respective control (NMDA alone; P < 0.05).
3.4 Leptin-induced increase in [Ca2+]i in mice cortical neurons
To investigate the mechanism(s) by which leptin activated BK currents, [Ca2+]i were monitored in leptin-exposed mouse cortical neurons. As shown in Fig. 4A, leptin (10–100 o 160 ng/ml) induced transient increases in [Ca2+]i in about ?% of the cells (?/?; Agnese, puoi mettere i dati per tutte le concentrazioni e poi decidiamo!). The Ca2+i level increased from … ± … nM (basal) to … ± … nM (peak) (P < 0.05); in most cells, [Ca2+]i oscillations appeared soon after leptin application, although in few cells transient increases in [Ca2+]i were largely spread over the entire duration of leptin exposure (15 min). Fig. 4B shows a summary of the dose-dependent effects of leptin on [Ca2+]i.
Figure 4. Effect of leptin on [Ca2+]i in cortical neurons.
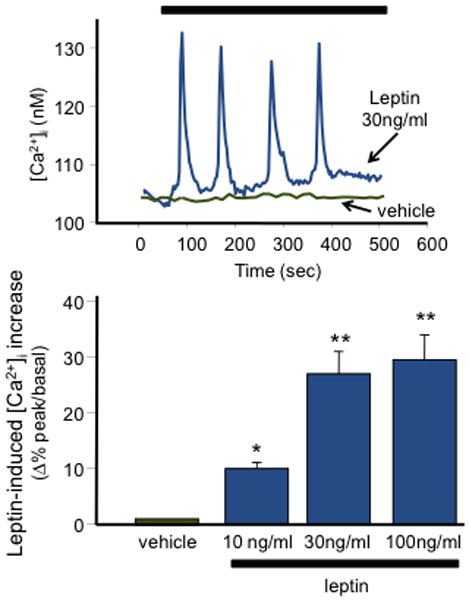
(A) Vehicle- (green trace) or leptin (30 ng/ml; blue trace)-induced changes in Fura-2 measured [Ca2+]i in 12 DIV mouse cortical neurons. The bar on top of the graph corresponds to the duration of vehicle or leptin exposure. (B) Concentration dependence of leptin-induced enhancement of peak [Ca2+]i. Each bar is the mean ± S.E.M. of the data from … cells recorded in … experimental sessions.
4. Discussion
The adipokine leptin has a firmly established role in promoting appetite suppression and energy expenditure. In addition to appetitive behaviors, leptin is also known to exert neuroprotective effects in several in vitro and in vivo models of neurotoxicity (Signore et al., 2008). In the present study we demonstrated that leptin exerts significant neuroprotection in an in vitro model of rat and mouse cortical neurons excitotoxicity triggered by exposure to NMDA. Leptin-induced neuroprotection appeared to be strongly dependent on the concentration and the timing of exposure: significant neuroprotective actions were only observed when the adipokine was applied 2 hours before the toxic insult, while leptin resulted largely ineffective after shorter or longer incubation times. By contrast, leptin achieved similar neuroprotective effects in cortical neurons exposed in vitro to oxygen and glucose deprivation (OGD) when applied 15 minutes before or 180 minutes after the neurotoxic stimulus (Valerio et al., 2009), suggesting that the neuroprotective mechanisms triggered by leptin during OGD and NMDA displayed a differential time-dependence. Interestingly, Pax and Ibtx, two well-known BK channel inhibitors, completely counteracted leptin-induced neuroprotection; both drugs displayed EC50s in the low nanomolar concentration range, consistent with their BK channel blocking actions (Nardi et al., 2003), and suggesting that BK channel opening is a crucial mechanism for leptin-induced neuroprotection during NMDA exposure. Consistent with this, the prototypical BK channel opener NS1619, similarly to leptin, also promoted neuroprotective effects; however, the selectivity of this compound for BK channels has been questioned (Gáspár et al., 2008), and additional molecular mechanisms such as the inhibition of voltage-dependent Ca2+ channels (Sheldon et al., 1997), voltage-dependent K+ channels, and KATP channels (Edwards et al., 1994) have been called into play to explain NS1619-induced neuroprotection. BMS-204352, an opener of BK and Kv7 potassium channels (Schrøder et al., 2001; Schrøder et al., 2003; Dupuis et al., 2002), also showed strong neuroprotective effects in animal models of cerebral ischemia (Gribkoff et al., 2001), and attenuated cerebral edema and neurologic motor impairment after traumatic brain injury (Cheney et al., 2001), although it failed to show superior efficacy compared to placebo in acute stroke patients (Jensen, 2002). In keeping with the neuroprotective role of BK channels, BK channel blockers aggravated neuronal damage in ischemic hippocampal organotypic cultures (Rundén-Pran et al., 2002).
BK channels are tetramers of pore-forming Slo1 α subunits encoded by the Kcnma1 gene. Slo1 subunits can assemble with distinct auxiliary β subunits (β1-β4), in a tissue-specific manner; such an association significantly modifies the biophysical and pharmacological properties of BK channels (Lu et al., 2006; King et al., 2006; Ghatta et al., 2006). To further substantiate the involvement of BK channels in leptin-mediated effects, we investigated the neuroprotective actions of this adipokine in neurons from mice carrying a deletion of exon 1 of the Kcnma1 locus (Meredith et al., 2004). The results obtained showed that leptin afforded strong neuroprotection only in cortical neurons from wild-type animals, whereas homozygous mice knockouts for the Slo1 gene were fully resistant to leptin effects; cortical neurons from heterozygous Slo1 knockout mice were also insensitive to leptin. These data confirm that BK channels are crucial mediators of leptin-induced neuroprotection; moreover, the fact that even a partial reduction in functional BK channels (such as that predicted to occur in Slo1+/− heterozygous animals) ablates leptin effects, suggests that a tight control of BK channel density is required for neuronal protection by this adipokine. Noteworthy, in these experiments, despite the dramatic difference in leptin sensitivity, NMDA treatment failed to induce greater toxicity in cortical neurons from Slo1−/− or Slo1+/− animals when compared to Slo1+/+ neurons, a result consistent with the lack of potentiation of NMDA-induced toxicity in Slo1+/+ neurons by BK blockers found in the present experiments. These results suggest that BK channels, though involved in leptin-induced protection, do not mediate NMDA-induced damage. By contrast, Liao et al. (2010) showed greater neurotoxicity by intra-cerebral injection of NMDA in Slo1−/− mice when compared to Slo1+/+ mice; however, it should be emphasized that, in our in vitro model, the extracellular concentrations of endogenous glutamate are unlikely to achieve the high neurotoxic levels observed in the intact tissue in vivo during NMDA-induced neurodegeneration.
In attempt to investigate the mechanism by which BK channels participated in leptin-induced neuroprotection, we studied by single-cell [Ca2+]i imaging whether leptin induced changes in [Ca2+]i availability. As a matter of fact, when used at concentrations similar to those triggering neuroprotection (30–100 ng/ml), leptin was found to trigger transient increases in [Ca2+]i in cortical neurons, in most cells appearing shortly after leptin exposure. Although this result clearly suggests that acute leptin application enhances [Ca2+]i, the molecular mechanism by which an increased [Ca2+]i leads to the activation of BK currents is yet unknown. Direct activation seems unlikely, given that [Ca2+]i changes larger than those herein observed are required to directly enhance BK currents (reference!). In support of this view, patch-clamp experiments performed in both mouse cortical neurons and in HEK293-T cells constitutively expressing leptin receptors (Kellerer et al., 1997) and transfected with Slo1 channels failed to demonstrate a direct current enhancement after few (10–15) min of leptin (30 ng/ml) application (data not shown). Thus, also considering the 2 hour time lag between leptin exposure and neuroprotection, it seems likely that leptin-induced sub-threshold [Ca2+]i challenge may upregulate BK current activity via an indirect mechanism.
Leptin-induced BK channel activation in neurons has been first proposed by Shanley et al. (2002a; b) to explain the ability of this hormone to inhibit hippocampal neurons excitability. However, in these studies, despite similar slow kinetics and apparent increase in [Ca2+]i sensitivity of BK channels, leptin failed to increase [Ca2+]i; rather, it was subsequently proposed that phosphatidylinositol 3-kinase (PI3K) was involved in BK currents activation by leptin, an effect consequent to the ability of this enzyme to alter actin dynamics, and to cluster and activate BK channels in the neuronal membrane (O’Malley et al., 2005). However, leptin-induced changes in [Ca2+]i have been described in porcine somatotropes (Glavaski-Joksimovic et al., 2004) and in mouse hypothalamic astrocytes (Hsuchou et al., 2009). Moreover, both stimulatory and inhibitory effects of leptin on [Ca2+]i have been reported in hippocampal neurons (Shanley et al., 2001) and in cerebellar granular cells; while stimulatory effects have been related to the ability of leptin to facilitate NMDA receptor-mediated Ca2+ influx (Irving et al., 2006), inhibitory actions have been largely ascribed to the inhibition of voltage-gated Ca2+ channels (Jo et al., 2005). Leptin-induced PI3K activation and [Ca2+]i changes do not appear as mutually-exclusive phenomena; in mouse macrophages, leptin triggers migration by promoting [Ca2+]i mobilization and by activating janus kinase/signal transducers and activators of transduction (JAK/STAT), mitogen-activated protein kinase (MAPK), and PI3K pathways (Gruen et al., 2007); by contrast, in HEK293 cells stably expressing full-length leptin receptor (OB-Rb), leptin inhibits lysophosphatidic acid-induced intracellular Ca2+ mobilization, an effect abrogated by PI3K inhibitors (Eiras et al., 2004).
In conclusion, our results suggest that leptin-dependent BK channel activation is a fundamental step in the neuroprotective effects triggered by this adipokine against NMDA-induced cortical neuronal degeneration. This cellular mechanism seems to render cortical neurons more “tolerant” to a neurotoxic insult. Thus, despite a fairly rigid time frame for leptin-induced neuroprotection, one can speculate that leptin-induced cellular effects, such as an enhanced [Ca2+]i availability, could act as a long-lasting preconditioning event. Further work will be needed to decipher the molecular pathways by which the observed oscillatory [Ca2+]i behavior triggered by leptin renders neurons resistant to the subsequent neurotoxic stimulus, and to set on firmer grounds the potential utility of leptin in the prevention and/or treatment of neurodegenerative disorders.
Acknowledgments
The present study was supported by grants from Telethon (GGP07125), the Fondazione San Paolo–IMI (Project Neuroscience), Regione Molise (Convenzione AIFA/Regione Molise), and PRIN 2009 (MT), and by the German Research Foundation, DFG FOR 1738 (to SHH, TH, MB).
Abbreviations
- NMDA
N-methyl-D-Aspartate
- BK channels
large conductance Ca2+- and voltage-activated K+ channels
- Pax
Paxilline
- Ibtx
Iberiotoxin
- OGD
oxygen-glucose deprivation
- KATP
ATP-sensitive K+ channels
- E
embryonic age
- HBSS
Hank’s Balanced Salt Solution
- MEM
Minimum Essential Media
- FBS
fetal bovine serum
- HS
horse serum
- DIV
days in vitro
- HCSS
Hepes control salt solution
- MS
media stock
- HEK293T cells
human embryonic kidney 293T cells
- MTT
3-[4,5-dimethylthiazol-2-yl]-2,5 diphenyl tetrazolium bromide
- DMEM
Dulbecco’s modified Eagle’s Medium
- Fura2-AM
1-[2-(5-carboxyoxazol-2-yl)-6-aminobenzofuran-5-oxy]-2-(21-amino-51-methylphenoxy)-ethane-N,N,N1,N1-tetraacetic acid penta-acetoxymethyl ester
- EC50
effective concentration 50
- [Ca2+]i
intracellular calcium concentration
- RCK domain
regulator of conductance of K domain
- PI3K
phosphatidylinositol 3-kinase
Footnotes
Conflict of interest
The authors declare that they have no conflict of interest.
References
- 1.Aras MA, Harnett KA, Aizenman E. Assessment of cell viability in primary neuronal cultures. Curr Protoc Neurosci. 2008;Chapter 7(Unit 7.18) doi: 10.1002/0471142301.ns0718s44. [DOI] [PubMed] [Google Scholar]
- 2.Boscia F, Annunziato L, Taglialatela M. Retigabine and flupirtine exert neuroprotective actions in organotypic hippocampal cultures. Neuropharmacology. 2006;51:283–294. doi: 10.1016/j.neuropharm.2006.03.024. [DOI] [PubMed] [Google Scholar]
- 3.Brenner R, Chen QH, Vilaythong A, Toney GM, Noebels JL, Aldrich RW. BK channel beta4 subunit reduces dentate gyrus excitability and protects against temporal lobe seizures. Nat Neurosci. 2005;8:1752–1759. doi: 10.1038/nn1573. [DOI] [PubMed] [Google Scholar]
- 4.Cheney JA, Weisser JD, Bareyre FM, Laurer HL, Saatman KE, Raghupathi R, Gribkoff V, Starrett JE, Jr, McIntosh TK. The maxi-K channel opener BMS-204352 attenuates regional cerebral edema and neurologic motor impairment after experimental brain injury. J Cereb Blood Flow Metab. 2001;21:396–403. doi: 10.1097/00004647-200104000-00008. [DOI] [PubMed] [Google Scholar]
- 5.Dicou E, Attoub S, Gressens P. Neuroprotective effects of leptin in vivo and in vitro. Neuroreport. 2001;12:3947–3951. doi: 10.1097/00001756-200112210-00019. [DOI] [PubMed] [Google Scholar]
- 6.Dirnagl U, Simon RP, Hallenbeck JM. Ischemic tolerance and endogenous neuroprotection. Trends Neurosci. 2003;26:248–254. doi: 10.1016/S0166-2236(03)00071-7. [DOI] [PubMed] [Google Scholar]
- 7.Dupuis DS, Schrøder RL, Jespersen T, Christensen JK, Christophersen P, Jensen BS, Olesen SP. Activation of KCNQ5 channels stably expressed in HEK293 cells by BMS-204352. Eur J Pharmacol. 2002;437:129–137. doi: 10.1016/s0014-2999(02)01287-6. [DOI] [PubMed] [Google Scholar]
- 8.Edwards G, Niederste-Hollenberg A, Schneider J, Noack T, Weston AH. Ion channel modulation by NS 1619, the putative BKCa channel opener, in vascular smooth muscle. Br J Pharmacol. 1994;113:1538–1547. doi: 10.1111/j.1476-5381.1994.tb17171.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Eiras S, Camiña JP, Diaz-Rodriguez E, Gualillo O, Casanueva FF. Leptin inhibits lysophosphatidic acid-induced intracellular calcium rise by a protein kinase C-dependent mechanism. J Cell Physiol. 2004;201:214–226. doi: 10.1002/jcp.20046. [DOI] [PubMed] [Google Scholar]
- 10.Gáspár T, Katakam P, Snipes JA, Kis B, Domoki F, Bari F, Busija DW. Delayed neuronal preconditioning by NS1619 is independent of calcium activated potassium channels. J Neurochem. 2008;105:1115–1128. doi: 10.1111/j.1471-4159.2007.05210.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gavello D, Rojo-Ruiz J, Marcantoni A, Franchino C, Carbone E, Carabelli V. Leptin counteracts the hypoxia-induced inhibition of spontaneously firing hippocampal neurons: a microelectrode array study. PLoS One. 2012;7:e41530. doi: 10.1371/journal.pone.0041530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ghatta S, Nimmagadda D, Xu X, O’Rourke ST. Large-conductance, calcium-activated potassium channels: structural and functional implications. Pharmacol Ther. 2006;110:103–116. doi: 10.1016/j.pharmthera.2005.10.007. [DOI] [PubMed] [Google Scholar]
- 13.Glavaski-Joksimovic A, Rowe EW, Jeftinija K, Scanes CG, Anderson LL, Jeftinija S. Effects of leptin on intracellular calcium concentrations in isolated porcine somatotropes. Neuroendocrinology. 2004;80:73–82. doi: 10.1159/000081843. [DOI] [PubMed] [Google Scholar]
- 14.Gribkoff VK, Starrett JE, Jr, Dworetzky SI, Hewawasam P, Boissard CG, Cook DA, Frantz SW, Heman K, Hibbard JR, Huston K, Johnson G, Krishnan BS, Kinney GG, Lombardo LA, Meanwell NA, Molinoff PB, Myers RA, Moon SL, Ortiz A, Pajor L, Pieschl RL, Post-Munson DJ, Signor LJ, Srinivas N, Taber MT, Thalody G, Tronjnacki JT, Wiener H, Yeleswaram K, Yeola SW. Targeting acute ischemia stroke with calcium-sensitive opener of maxi-K potassium channels. Nat Med. 2001;7:471–477. doi: 10.1038/86546. [DOI] [PubMed] [Google Scholar]
- 15.Gruen ML, Hao M, Piston DW, Hasty AH. Leptin requires canonical migratory signaling pathways for induction of monocyte and macrophage chemotaxis. Am J Physiol Cell Physiol. 2007;293:C1481–1488. doi: 10.1152/ajpcell.00062.2007. [DOI] [PubMed] [Google Scholar]
- 16.Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- 17.Gu N, Vervaeke K, Storm JF. BK potassium channels facilitate high-frequency firing and cause early spike frequency adaptation in rat CA1 hippocampal pyramidal cells. J Physiol. 2007;580:859–882. doi: 10.1113/jphysiol.2006.126367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guo Z, Jiang H, Xu X, Duan M, Mattson MP. Leptin-mediated cell survival signaling in hippocampal neurons mediated by JAK STAT3 and mitochondrial stabilization. J Biol Chem. 2008;283:1754–1763. doi: 10.1074/jbc.M703753200. [DOI] [PubMed] [Google Scholar]
- 19.Halaas JL, Gajiwala KS, Maffei M, Cohen SL, Chait BT, Rabinowitz D, Lallone RL, Burley SK, Friedman JM. Weight-reducing effects of the plasma protein encoded by the obese gene. Science. 1995;269:543–546. doi: 10.1126/science.7624777. [DOI] [PubMed] [Google Scholar]
- 20.Harvey J. Leptin regulation of neuronal excitability and cognitive function. Curr Opin Pharmacol. 2007;7:643–647. doi: 10.1016/j.coph.2007.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Horrigan FT, Aldrich RW. Coupling between voltage sensor activation, Ca2+ binding and channel opening in large conductance (BK) potassium channels. J Gen Physiol. 2002;120:267–305. doi: 10.1085/jgp.20028605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hsuchou H, He Y, Kastin AJ, Tu H, Markadakis EN, Rogers RC, Fossier PB, Pan W. Obesity induces functional astrocytic leptin receptors in hypothalamus. Brain. 2009;132:889–902. doi: 10.1093/brain/awp029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Irani BG, Le Foll C, Dunn-Meynell A, Levin BE. Effects of leptin on rat ventromedial hypothalamic neurons. Endocrinology. 2008;149:5146–5154. doi: 10.1210/en.2008-0357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Irving AJ, Wallace L, Durakoglugil D, Harvey J. Leptin enhances NR2B-mediated N-methyl-D-aspartate responses via a mitogen-activated protein kinase-dependent process in cerebellar granule cells. Neuroscience. 2006;138:1137–1148. doi: 10.1016/j.neuroscience.2005.11.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jensen BS. BMS-204352: a potassium channel opener developed for the treatment of stroke. CNS Drug Rev. 2002;8:353–360. doi: 10.1111/j.1527-3458.2002.tb00233.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jin W, Sugaya A, Tsuda T, Ohguchi H, Sugaya E. Relationship between large conductance calcium-activated potassium channel and bursting activity. Brain Res. 2000;860:21–28. doi: 10.1016/s0006-8993(00)01943-0. [DOI] [PubMed] [Google Scholar]
- 27.Jo YH, Chen YJ, Chua SC, Jr, Talmage DA, Role LW. Integration of endocannabinoid and leptin signaling in an appetite-related neural circuit. Neuron. 2005;48:1055–1066. doi: 10.1016/j.neuron.2005.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kellerer M, Koch M, Metzinger E, Mushack J, Capp E, Häring HU. Leptin activates PI-3 kinase in C2C12 myotubes via janus kinase-2 (JAK-2) and insulin receptor substrate-2 (IRS-2) dependent pathways. Diabetologia. 1997;40:1358–1362. doi: 10.1007/s001250050832. [DOI] [PubMed] [Google Scholar]
- 29.Kentish SJ, O’Donnell TA, Isaacs NJ, Young RL, Li H, Harrington AM, Brierley SM, Wittert GA, Blackshaw LA, Page AJ. Gastric vagal afferent modulation by leptin is influenced by food intake status. J Physiol. 2013;591:1921–1934. doi: 10.1113/jphysiol.2012.247577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kieffer TJ, Heller RS, Leech CA, Holz GG, Habener JF. Leptin suppression of insulin secretion by the activation of ATP-sensitive K+ channels in pancreatic beta-cells. Diabetes. 1997;46:1087–1093. doi: 10.2337/diab.46.6.1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.King JT, Lovell PV, Rishniw M, Kotlikoff MI, Zeeman ML, McCobb DP. Beta2 and beta4 subunits of BK channels confer differential sensitivity to acute modulation by steroid hormones. J Neurophysiol. 2006;95:2878–2888. doi: 10.1152/jn.01352.2005. [DOI] [PubMed] [Google Scholar]
- 32.Knaus HG, Schwarzer C, Koch RO, Ederhart A, Kaczorowski GJ, Glossmann H, Wunder F, Pongs O, Garcia ML, Sperk G. Distribution of high-conductance Ca(2+)-activated K+ channels in rat brain: targeting to axons and nerve terminals. J Neurosci. 1996;16:955–963. doi: 10.1523/JNEUROSCI.16-03-00955.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liao Y, Kristiansen AM, Oksvold CP, Tuvnes FA, Gu N, Rundén-Pran E, Ruth P, Sausbier M, Storm JF. Neuronal Ca2+-activated K+ channels limit brain infarction and promote survival. PLoS One. 2010;5:e15601. doi: 10.1371/journal.pone.0015601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lu R, Alioua A, Kumar Y, Eghbali M, Stefani E, Toro L. MaxiK channel partners: physiological impact. J Physiol. 2006;570:65–72. doi: 10.1113/jphysiol.2005.098913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Martire M, Barrese V, D’Amico M, Iannotti FA, Pizzarelli R, Samengo I, Viggiano D, Ruth P, Cherubini E, Taglialatela M. Pre-synaptic BK channels selectively control glutamate versus GABA release from cortical and hippocampal nerve terminals. J Neurochem. 2010;115:411–422. doi: 10.1111/j.1471-4159.2010.06938.x. [DOI] [PubMed] [Google Scholar]
- 36.Meredith AL, Thorneloe KS, Werner ME, Nelson MT, Aldrich RW. Overactive bladder and incontinence in the absence of the BK large conductance Ca2+-activated K+ channel. J Biol Chem. 2004;279:36746–36752. doi: 10.1074/jbc.M405621200. [DOI] [PubMed] [Google Scholar]
- 37.Misonou H, Menegola M, Buchwalder L, Park EW, Meredith A, Rhodes KJ, Aldrich RW, Trimmer SJ. Immunolocalization of the Ca2+-activated K+ channel Slo1 in axons and nerve terminals of mammalian brain and cultured neurons. J Comp Neurol. 2006;496:289–302. doi: 10.1002/cne.20931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mistrík P, Moreau F, Allen JM. BiaCore analysis of leptin-leptin receptor interaction: evidence for 1:1 stoichiometry. Anal Biochem. 2004;327:271–277. doi: 10.1016/j.ab.2004.01.022. [DOI] [PubMed] [Google Scholar]
- 39.Nardi A, Calderone V, Chericoni S, Morelli I. Natural modulators of large-conductance calcium-activated potassium channels. Planta Med. 2003;69:885–892. doi: 10.1055/s-2003-45095. [DOI] [PubMed] [Google Scholar]
- 40.Oberhauser A, Alvarez O, Latorre R. Activation by divalent cations of a Ca2+-activated K+ channel from skeletal muscle membrane. J Gen Physiol. 1988;92:67–86. doi: 10.1085/jgp.92.1.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.O’Malley D, Irving AJ, Harvey J. Leptin-induced dynamic alterations in the actin cytoskeleton mediate the activation and synaptic clustering of BK channels. FASEB J. 2005;19:1917–1919. doi: 10.1096/fj.05-4166fje. [DOI] [PubMed] [Google Scholar]
- 42.Oomura Y, Hori N, Shiraishi T, Fukunaga K, Takeda H, Tsuji M, Matsumiya T, Ishibashi M, Aou S, Li XL, Kohno D, Uramura K, Sougawa H, Yada T, Wayner MJ, Sasaki K. Leptin facilitates learning and memory performance and enhances hippocampal CA1 long-term potentiation and CaMK II phosphorylation in rats. Peptides. 2006;27:2738–2749. doi: 10.1016/j.peptides.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 43.Raffaelli G, Saviane C, Mohajerani MH, Pedarzani P, Cherubini E. BK potassium channels control transmitter release at CA3-CA3 synapses in the rat hippocampus. J Physiol. 2004;557:147–157. doi: 10.1113/jphysiol.2004.062661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rundén-Pran E, Haug FM, Storm JF, Ottersen OP. BK channel activity determines the extent of cell degeneration after oxygen and glucose deprivation: a study in organotypical hippocampal slice cultures. Neuroscience. 2002;112:277–288. doi: 10.1016/s0306-4522(02)00092-1. [DOI] [PubMed] [Google Scholar]
- 45.Schrøder RL, Jespersen T, Christophersen P, Strøbaek D, Jensen BS, Olesen SP. KCNQ4 channel activation by BMS-204352 and retigabine. Neuropharmacology. 2003;44:553. doi: 10.1016/s0028-3908(01)00029-6. [DOI] [PubMed] [Google Scholar]
- 46.Schrøder RL, Strøbaek D, Olesen SP, Christophersen P. Voltage-independent KCNQ4 currents induced by (+/−)BMS-204352. Pflügers Arch. 2003;446:607–616. doi: 10.1007/s00424-003-1116-x. [DOI] [PubMed] [Google Scholar]
- 47.Signore AP, Zhang F, Weng Z, Gao Y, Chen J. Leptin neuroprotection in the CNS: mechanisms and therapeutic potentials. J Neurochem. 2008;106:1977–1990. doi: 10.1111/j.1471-4159.2008.05457.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shanley LJ, Irving AJ, Harvey J. Leptin enhances NMDA receptor function and modulates hippocampal synaptic plasticity. J Neurosci. 2001;21:RC186. doi: 10.1523/JNEUROSCI.21-24-j0001.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shanley LJ, Irving AJ, Rae MG, Ashford ML, Harvey J. Leptin inhibits rat hippocampal neurons via activation of large conductance calcium-activated K+ channels. Nat Neurosci. 2002a;5:299–300. doi: 10.1038/nn824. [DOI] [PubMed] [Google Scholar]
- 50.Shanley LJ, O’Malley D, Irving AJ, Ashford ML, Harvey J. Leptin inhibits epileptiform-like activity in rat hippocampal neurons via PI 3-kinase-driven activation of BK channels. J Physiol. 2002b;545:933–944. doi: 10.1113/jphysiol.2002.029488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sheldon JH, Norton NW, Argentieri TM. Inhibition of guinea pig detrusor contraction by NS-1619 is associated with activation of BKCa and inhibition of calcium currents. J Pharmacol Exp Ther. 1997;283:1193–1200. [PubMed] [Google Scholar]
- 52.Spanswick D, Smith MA, Groppi VE, Logan SD, Ashford ML. Leptin inhibits hypothalamic neurons by activation of ATP-sensitive potassium channels. Nature. 1997;390:521–525. doi: 10.1038/37379. [DOI] [PubMed] [Google Scholar]
- 53.Valerio A, Dossena M, Bertolotti P, Boroni F, Sarnico I, Faraco G, Chiarugi A, Frontini A, Giordano A, Liou HC, De Simoni MG, Spano P, Carruba MO, Pizzi M, Nisoli E. Leptin is induced in the ischemic cerebral cortex and exerts neuroprotection through NF-kappaB/c-Rel-dependent transcription. Stroke. 2009;40:610–617. doi: 10.1161/STROKEAHA.108.528588. [DOI] [PubMed] [Google Scholar]
- 54.Weng Z, Signore AP, Gao Y, Wang S, Zhang F, Hastings T, Yin XM, Chen J. Leptin protects against 6-hydroxydopamine-induced dopaminergic cell death via mitogen-activated protein kinase signaling. J Biol Chem. 2007;282:34479–34491. doi: 10.1074/jbc.M705426200. [DOI] [PubMed] [Google Scholar]
- 55.Xu L, Rensing N, Yang XF, Zhang HX, Thio LL, Rothman SM, Weisenfeld AE, Wong M, Yamada KA. Leptin inhibits 4-aminopyridine- and pentylenetetrazole-induced seizures and AMPAR-mediated synaptic transmission in rodents. J Clin Invest. 2008;118:272–280. doi: 10.1172/JCI33009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhang F, Wang S, Signore AP, Chen J. Neuroprotective effects of leptin against ischemic injury induced by oxygen-glucose deprivation and transient cerebral ischemia. Stroke. 2007;38:2329–2336. doi: 10.1161/STROKEAHA.107.482786. [DOI] [PubMed] [Google Scholar]