

Abstract
Previous studies using mannose-binding lectin (MBL) and complement C4 deficient mice have suggested that the lectin pathway (LP) is not required for the development of inflammatory arthritis in the collagen antibody-induced arthritis (CAIA) model. MBL, ficolins and collectin-11 are key LP pattern recognition molecules that associate with three serine proteases, MASP-1, MASP-2 and MASP-3, and also with two MBL-associated proteins designated sMAP and MAp44. Recent studies have shown that MAp44, an alternatively spliced product of the MASP-1/3 gene, is a competitive inhibitor of the binding of the recognition molecules to all three MASPs. In these studies we examined the effect of treatment of mice with adenovirus (Ad) programmed to express human MAp44 (AdhMAp44) on the development of CAIA. AdhMAp44 and Ad programming Green fluorescent protein (AdGFP) expression were injected intraperitoneally in C57BL/6 wild-type mice prior to the induction of CAIA. AdhMAp44 significantly reduced the clinical disease activity score (CDA) by 81% compared to mice injected with AdGFP. Similarly, histopathologic injury scores for inflammation, pannus, cartilage and bone damage, as well as C3 deposition in the cartilage and synovium, were significantly reduced by AdhMAp44 pretreatment. Mice treated with AdmMAp44, programming expression of mouse MAp44, also showed significantly decreased CDA and histopathologic injury scores. Additionally, administration of AdhMAp44 significantly diminished the severity of Ross River Virus-induced arthritis, a LP-dependent model. Our study provides conclusive evidence that an intact complement LP is essential to initiate CAIA, and that MAp44 may be an appropriate treatment for inflammatory arthritis.
Keywords: complement, arthritis, immune complex, inflammation
INTRODUCTION
Rheumatoid arthritis (RA) is a complex autoimmune disease with genetic and environmental components, affecting approximately 1% of the population worldwide (1). Autoantibodies, especially as constituents of immune complexes (ICs), play a central role in triggering inflammation in this disease (2, 3). In the present report we studied the pathophysiology of RA employing a mouse model, collagen antibody-induced arthritis (CAIA), where ICs are formed in the joint between anti-collagen type II (CII) monoclonal antibodies (mAbs) and CII, one of the major matrix proteins of the articular cartilage.
The complement system is a central part of the innate immune system and has been found to play a major role in the development of inflammation and tissue damage in CAIA and other animal models of inflammatory arthritis (4-7). Activation of the complement system is thought to contribute to inflammation and tissue damage in human RA, especially in very early disease (8-10). ICs containing Abs of the IgG isotype are found in the cartilage and synovium of the joints of patients with RA and have been implicated in induction of local tissue damage through activation of the complement system (11-13).
The complement system can be activated by three pathways: the classical pathway (CP), the lectin pathway (LP), and the alternative pathway (AP). IgG Abs in arthritis-related IC in human RA have previously been shown to activate both the CP and AP of the complement system (14-16). In CAIA, using pathway-specific functional deficiencies of the complement system developed through gene targeting and inactivation of specific pathway components, it was previously concluded that the AP alone is both necessary and sufficient for the development of CAIA through its role in both the initiation process and amplification loop (4). The lack of a role for the LP was inferred using MBL-A/C and C4 deficient mice (4, 6, 17), and for the CP using C4 and C1q deficient mice (4, 17), where disease was largely unchanged.
The CP is initiated by C1q binding to Ab in an IC, leading to activation of C1r, C1s and to the subsequent formation of the CP C3 convertase, C4b2b. The LP is initiated when members of a family of pattern recognition molecules designated the collectins, whose members are mannose-binding lectin (MBL), ficolins (the three in humans are designated H, L, and M), and Collectin-11 (also termed CL-K1) bind along, with MBL-associated serine proteases (MASP-1, MASP-2, and MASP-3), to arrays of specific monosaccharides or modified carbohydrates present on the surface of microorganisms and other target surfaces and molecules. Notably, in humans one MBL is found whereas in mice two, MBL-A and MBL-C, are present; in addition, two ficolins (Ficolin-A and Ficolin-B) are found in mice along with Collectin-11 (18-21). This process leads to the formation of the shared CP/LP C3 convertase, C4b2b, through the activities of MASP-2 (22) in a manner that requires initial engagement of MASP-1 (23, 24). The AP is initiated by spontaneous turnover of C3 with transient formation of hydrolyzed C3 (H2O), followed by binding of factor B (FB) with cleavage by factor D (FD) and generation of the AP initiation C3 convertase C3(H2O)Bb (25). Cleavage of C3 also exposes a short-lived thioester in C3b that covalently attaches to amine and carboxyl groups on target surfaces (25). Following formation of C3b through any of the three pathways, the amplification loop is initiated through the binding of FB and cleavage by FD to form the C3bBb C3 convertase (26).
The AP may also be initiated by properdin bound to target-containing molecular patterns (27) or by adherent IgG or IgA (16, 28). In addition, it was reported that the AP in mice is dependent on MASP-1/3 cleavage of pro-FD to form mature FD in the circulation (29) and that mice lacking MASP-1 and MASP-3 have a defective AP and LP (30). Recently, it has been shown that MASP-1/3−/−/fH−/− mice have pro-FD in their circulation and the AP was present but defective (31). In addition, a functional AP was present in the serum of a patient reported to lack MASP-1 and MASP-3, although it could not be determined whether a partial defect was present (32).
MBL, ficolins and Collectin-11 circulate in complex with MASP-1, - 2 and -3 and two additional proteins (MAp19 and MAp44, also known as sMAP and MAP1 or MASP-1 isoform 3, respectively) (32-34). MASPs are present as pro-enzymes and become activated once MBL, ficolins or Collectin-11 bind to ligands. Three proteins, MASP-1, MASP-3 and MAp44 are translated from mRNAs formed by alternative splicing of RNA encoded by the MASP-1 gene (32). MASP-1 and MASP-3 are two proteases which share their first five domains (CUB1-EGFCUB2-CCP1-CCP2) but have different serine protease domains encoded by distinct exons (35). MAp44 shares the first four domains with MASP-1 and MASP-3, followed by 17 unique C-terminal amino acid residues encoded by a separate exon (33). Since the first three domains mediate binding to MBL, MAp44, MASP-1 and MASP-3 bind to the same site on MBL. MAp44, which lacks a serine protease domain, can thus compete with MASPs for binding to MBL and other collectins, and through this mechanism regulate activity of the LP (36). MASP-2 activation strictly depends on an initiating activation of MASP-1 because inhibition of MASP-1 prevents autoactivation of MASP-2 (24), and no LP is present in mice lacking MASP-1 (30). MAp44 may also displace MASP-1 and MASP-2 from MBL or ficolins, further inhibiting the activation of MASP-2 and the subsequent cleavage of C4 and C2 (36). Through these activities, MAp44 is considered to be a natural endogenous inhibitor of the LP (37).
In the present report we utilize CAIA to evaluate the role of the LP in inflammatory arthritis. Previously, studies in mice deficient in different components of the complement system have shown that the AP is both necessary and sufficient to mediate CAIA as neither the LP nor the CP appear to be required (4, 17). Additionally, mice lacking MASP-1, MASP-3 and MAp44 (MASP-1/3−/−) were shown to be resistant to CAIA (38), which was considered to be likely because they lacked mature FD and a functional AP (30). Although the AP may initiate and amplify CAIA, the LP (and CP) may also function to initiate the disease process. Since the LP has not previously been shown to play an essential role in CAIA, we hypothesized that ficolin-A or -B or Collectin-11 may mediate recognition of ligands independently of MBL-A/C. In addition, direct MASP-mediated cleavage of C3 (39) could have allowed C3 activation and engagement of the amplification loop even in the absence of MBL-A/C or C4. The use of MAp44 as an endogenous inhibitor of all of the MASPs, due to its interference with the interactions between the recognition molecules and MASPs, should allow a more complete impairment of the LP and better assessment of its role in the development of CAIA.
Herein we examined the effect of replication defective adenovirus (Ad) serotype 5 vectors containing the human (AdhMAp44) or mouse (AdmMAp44) genes on the initiation of CAIA. Using this approach our study provides strong support for a key role of the LP in the initiation and pathogenesis of CAIA.
MATERIALS AND METHODS
Mice
Eight to ten week-old WT C57BL/6 male mice (n = 73) were used for this study. We obtained C4−/− mice originally from Dr. Michael Carroll and C1q/MBL−/− mice from Dr. Gregory Stahl. Our laboratory has now maintained colonies of both C4−/− and C1q/MBL−/− C57BL/6 homozygous mice; sera from these mice were used for various ELISAs. WT C57BL/6 mice were obtained from Jackson Laboratories. All mice were weighed prior to use and were kept in a barrier animal facility with a climate-controlled environment with 12 h light/dark cycles. Filter top cages were used with three mice in each cage. During the course of this study, all experimental mice were fed breeder's chow provided by the Center for Laboratory Animal Care, University of Colorado School of Medicine.
Construction of AdMAp44 vectors
Human AdMAp44 (AdhMAp44) construct was generated by Welgen, Inc (Worcester, MA) using the human MAp44 cDNA purchased from Thermo Fisher (Waltham, MA). The HA-Tag (Human influenza hemagglutinin, sequence “YPYDVPDYA”) was added to the C-terminus of the MAp44 molecule to facilitate the detection of recombinant MAp44 in the circulation of mice generated by the administration of AdhMap44 or AdmMAp44. To detect the presence of HA in the sera of mice with and without CAIA, anti-HA tag antibodies were used. Information about the vectors and specific elements described herein is found in Supplemental Figure 1. Additional RGD sequences were added to the construct for AdhMAp44, but not AdmMAp44, to stimulate receptors for adenoviral entry in synovial cells other than the CAR (40). Briefly, pBSK-MAp-1-HA was cleaved with Xho1/Xba1 and the MAp44-HA fragment was ligated to the pEntCMV shuttle vector digested with the same enzymes. Positive clones were screened and sequenced for confirmation. pEntCMV-MAp44-HA was treated with LR Clonase II (Invitrogen) and ligated with the plasmid pAd5. The recombination products were used to transform E. coli. After incubation overnight, clones were selected and grown, and cosmid DNA was purified. The purified cosmid DNA (2 mg) was digested with Pac1 and then transfected into 293 cells with Lipofectamine 2000 (Life Technologies) according to the manufacturer's instructions. The 293 cells were grown at 37°C with 5% CO2. Ad plaque growth was apparent by 7 days after transfection. The titer of virus particles (vp) was further amplified to 1012 vp/ml. The amplified Ad was purified on 2 sequential cesium chloride gradients and then dialyzed against PBS, pH 7.4, containing 10% glycerol. The titer of the purified virus was estimated from the absorption at 260 nm. The final titer of AdhMAp44 was 1.0x1012 particles per ml. Ad with cytomegalovirus (CMV) sequences and programming expression of green fluorescent protein (GFP) (AdGFP) was used as a negative control for all CAIA studies.
Human recombinant MAp44
Human recombinant MAp44 (hrMAp44) was produced as described in detail elsewhere (36). In brief mammalian cells, HEK293F cells, were transfected with a vector encoding human Map44 using PEI as transfection reagent. Map44 expressed in the supernatants was purified by affinity chromatography on MBL coated beads.
Induction of collagen antibody-induced arthritis
CAIA was induced in WT mice using a cocktail of 5 mAbs to bovine CII (Arthritomab-CIA, Chondrex) suspended in sterile PBS as previously described (4, 17, 38). WT mice were injected i.p. with 4 mg/mouse of Arthritomab on day 0 and 50 ug/mouse of LPS from E. coli strain 0111B4 (Chondrex) on day 3 to synchronize the development of arthritis according to the standard protocol suggested by the supplier of Arthritomab and LPS. Mice started to develop arthritis at day 4 and were sacrificed at day 10. Mice develop very mild transient arthritis with no histological damage (data not shown) for a few days when anti-CII mAbs or LPS alone were injected i.p., using 6 mg/mouse Arthritomab or 50 ug/mouse LPS (Supplemental Fig. S2A). Furthermore, mice injected with anti-CII mAbs or LPS alone developed inconsistent disease as evident from disease prevalence, making it difficult to assess the cause and effect relationship of complement inhibitors (Supplemental Fig. S2B). Therefore, an injection of anti-CII mAbs followed by an injection of LPS is required for a sustained production of disease for a minimum of 10 days with a clinical course and histological damage sufficient to observe the effects of complement inhibitors (Supplemental Fig. S2A). Clinical disease activity (CDA) was examined daily until day 10 by observers blinded to the treatment as per our previously published studies (4, 17, 38).
In Ad studies, WT mice were injected i.p. at day -5, day 0, and day 3 with AdhMAp44 (at either a higher dose (1×1011) or a lower dose (5.0 ×1010)), AdGFP (1×1011 particles), or PBS alone (n = 5 for each treatment). Arthritomab was injected as usual at day 0. For the local joint injection experiment, 50 ul containing 5.0 ×1010 particles of AdmMAp44 or of AdGFP were injected in the right knee joint at day -5, day 0 (after the anti-CII mAb injection), and at day 3 (after LPS injection).
Ross River Virus-induced mouse model of inflammatory arthritis
Three-to-four week old WT C57BL/6 mice were inoculated in the left rear footpad with 103 PFU of Ross River virus (RRV) in a volume of 10 μl as previously described (41-43). Disease scores were determined by assessing grip strength, hind limb weakness, and altered gait as previously described (41-43). In RRV-induced arthritis, AdhMAp44 and AdGFP were injected with doses of viral particles identical to those used in the CAIA studies for knee injection, i.e., 5×1010 at days -3, 0, and 3 in the right rear footpad.
Histopathology and immunohistochemistry of all joints
Knee joints from both fore limbs, and the right hind limb knee joint, ankle and paw from WT mice with CAIA at day 10 were fixed in 4% paraformaldehyde and examined by immunohistochemical staining (IHS) for Toluidine-blue (T-blue) and C3 deposition according to our published methods (4, 17, 44). Additionally, IHS was used to detect integrin αvβ5 (antibody dilution 1:500) in the synovium and knee joint sections from mice transduced with AdhMAp44 and AdGFP. Hemotoxylin (VWR) staining was used to show the presence of the synovium. Toluidine-blue stain was used to assess histopathology for determination of inflammation, pannus formation, and cartilage and bone damage according to published criteria (4, 17, 44). Seven μm sections were cut for histology and processed for T-blue and C3 IHS. All slides for histopathology and C3 deposition were observed under light microscopy at a magnification of 20X or 10X in a blinded fashion and scored according to published criteria (4, 17, 44). The knee joints from untreated C3−/− mice on C57BL/6 background were used as negative controls.
Immunohistochemistry to detect GFP and the HA tag in organs
Knee joints from the right hind limb, liver, spleen and kidney from WT mice with and without CAIA at day 10 were fixed in 4% paraformaldehyde and examined by IHS for GFP. Anti-GFP polyclonal rabbit (dilution 1:200) and the secondary antibody (goat anti-rabbit, Alexa Flour 488 (1:200 dilution) (Invitrogen)) were used to detect GFP. Sections were visualized under UV-light using an Olympus (Model - BX51) microscope. The presence of green fluorescence under UV-light indicated the presence of GFP expression in tissues.
Additionally, we examined for the presence of HA in the synovium from the knee joints of mice injected i.p. with AdhMAp44 and AdmMAp44 at day 10. At the time of sacrifice liver, spleen, kidney and knee joints were collected and fixed in 10% neutral-buffered formalin, processed and sections were cut. After staining with anti-HA antibody (dilution 1:1000) (Cell Signal), and the development of color using anti-Rabbit En Vision plus Polymer HRP-conjugated followed by DAB plus Chromogen (Dako), the sections were visualized by light microscopy and photographed.
Examination of in vivo transduction efficiency and Western blot analysis
Since both AdhMAp44 and AdmMAp44 constructs utilized a HA tag, we analyzed sera at days -5 or -2, 0, 3 and 10 after the i.p. injections for the presence of HA using Western blot analysis. Similarly, we analyzed the sera from mice injected in the knee joints with AdmMAp44 at days -5, 0, 3 and 10 for the presence of HA using Western blot analysis.
Western blot analysis
A 10% Bis-Tris reducing SDS gel was used for separation of proteins in mouse serum. After transfer, the blots were incubated overnight at 4° C with rabbit Ab specific for HA (dilution 1:1000) (Cell Signal). Anti-rabbit HRP-conjugated Ab was used as the secondary Ab (dilution 1:2000) (Hycult Biotech). The blots were washed 3×10 min in 1xPBS 0.5% Tween 20 and developed for 3 min using a 1:1 mixture of SuperSignal West Pico chemiluminescent substrate (Thermo Scientific). The presence of HA and MAp44 bands at ~43-50 kDa in serum identified the presence in circulation of AdhMAp44 or AdmMAp4, indicating successful synthesis and secretion. We also evaluated whether the expressed hMAp44 protein was functionally active, i.e. did it bind to MBL. This was examined by incubation of sera with mannose-agarose beads, which bind MBL and, thus, should indirectly bind MAp44 through its interaction with MBL. The material eluted from the beads was analyzed by Western blot analysis and probed with anti-HA Ab as noted above.
Quantitative RT- PCR for mRNA expression levels
Knee joints were harvested from mice with CAIA at day 10 after induction. Total RNA was extracted using an RNAeasy mini kit (Qiagen) from the left knee joints of all experimental mice injected i.p. with PBS, AdhMAp44 LD, AdhMap44 HD, or AdGFP at days -5, 0, and 3. The presence of mouse MBL-A, MBL-C, Ficolin-A (FCN-A), MASP-1, MASP-2, MASP-3, FD, TNF-α, IL-1α and IL-1β were analyzed in the samples by RT-PCR using 40 cycles according to published methods (45). All RT-PCR data were analyzed using a cDNA based standard curve. The standard curves for mRNA encoding FD, MASP-1, MASP-2, and MASP-3 were constructed by using mRNA from mouse adipose tissue for FD and liver for MBL-A, MBL-C, FCN-A, TNF-α, IL-1α and IL-1β, and the MASPs, respectively. In parallel, the baseline mRNA levels for various targets from knee joint of age-matched WT mice without CAIA and without any treatment were also determined. Primer sequences used to determine mRNA concentrations are available upon request from the corresponding authors.
Human MAp44 assay
A sandwich type immunoassay method was used to determine the absolute levels of human MAp44 present in the circulation of WT mice injected with PBS, AdGFP or AdhMAp44 (low dose or high dose) according to recently published studies (33). This assay is highly specific and sensitive for human MAp44 and can be used to determine the levels of human MAp44 in mouse serum. To measure the levels of MAp44, serum from each mouse obtained at day -5, day 0, day 3 and at day 10 was diluted 1:15 in a binding buffer. A standard human plasma pool with a known level of human MAp44 was used to establish a standard curve. All samples were tested in duplicate, and three quality controls were included in each assay as described (33).
Measurements of absolute levels of C5a in serum
Serum levels of C5a before (day -5) and after (day 10) the development of disease in WT mice injected with AdhMAp44 or AdGFP or PBS were measured using standard ELISA protocols according to our published methods (46).
Measurements of LP induced C3 in serum
LP induced C3 activation using sera from CAIA mice, at day 10, treated with PBS, AdGFP, AdhMAp44 LD, or AdhMAp44 HD was determined using ELISA plates pre-coated with mannan particles, according to previously described methods (47). Sera from WT mice with no CAIA were used as a positive control. Sera from C3−/− and MBL/Df−/− mice were used as a negative controls.
Analysis of recombinant human MAp44 on LPS induced C3 activation
The effect of recombinant human MAp44 (rhMAp44) on LPS-induced C3 activation via the LP and AP was determined using ELISA. Ninety-six well ELISA plates were pre-coated with 5 ug/well LPS from E. coli strain 0111B4. Serum from a WT mouse with no disease was diluted (1:10) and pre-treated for 30 min with rhMAp44 (10 ug/10 ul of serum) in GBV+ buffer (Ca2+ -sufficient buffer) or in Mg2+ EGTA buffer (Ca2+ - deficient buffer). LPS induced C3 activation was measured according to described methods (48). In parallel, serum from a WT mouse was also pre-treated with an inhibitory anti-factor B antibody (4 ug/10 ul of serum) as a positive control to inhibit C3 activation specifically from the AP. Serum from C3−/− mice was used as a negative control and there was no C3 activation as expected.
Statistical analyses
Student's t test was used to calculate p-values using the GraphPad Prism® 4 statistical program. The data in all graphs and histograms are shown as the mean + SEM with p < 0.05 considered significant. Pearson correlation was used to calculate r2 value among histological parameters, C3 deposition and CDA. Preliminary analyses using a null hypothesis for w-statistics indicated that the data were normally distributed.
RESULTS
Human AdMAp44 prevents initiation of disease in mice with CAIA
The generation of AdGFP, AdhMap44, and AdmMAp44 is described in detail in Materials and Methods and illustrated in Supplemental Fig. 1. To determine the effect of human MAp44 expression, we examined the development of CAIA in WT mice treated with a higher and lower dose of AdhMAp44 as compared to AdGFP and PBS buffer alone. The mice were treated with AdhMAp44, AdGFP or PBS alone on days -5, 0, and 3. Anti-CII mAb was injected i.p. on day 0. The prevalence of disease in each condition was 100% at day 10 (Fig. 1A). The CDA in WT mice injected with a higher (HD) or lower dose (LD) of AdhMAp44 was significantly reduced by 81% and 75%, respectively, as compared to WT mice injected with an equivalent higher dose of AdGFP (Fig. 1B). Specifically, the CDA at day 10 in mice treated with the HD and LD of AdhMAp44 was 2.0 ± 0.4 and 2.6 ± 0.4, respectively. In contrast, in mice treated with PBS and AdGFP, the CDA at day 10 was 10.6 ± 1.8 and 8.8 ± 2.0, respectively. These results demonstrate that while pretreatment with either dose of AdhMAp44 did not prevent CAIA, it significantly reduced the severity.
FIGURE 1.
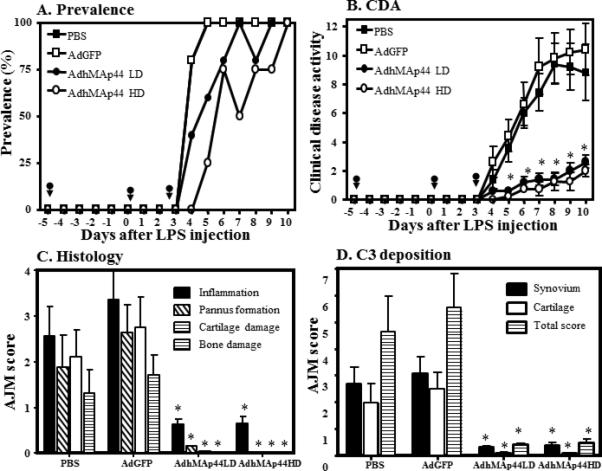
Substantial decrease in the CDA of anti-CII mAb-induced arthritis by pretreatment with AdhMAp44. Higher (HD) and lower (LD) doses of AdhMAp44 particles were used. A. Prevalence of arthritis (%) over the duration of the experiment. B. CDA over the duration of the experiment. Black arrows show the injection time of AdhMAp44 or AdGFP at days -5, 0 and 3. *p < 0.05 in comparison with AdGFP treatment for data in B. C. All joint mean (AJM) histopathologic scores for inflammation (black solid bar), pannus formation (white hatched bar), cartilage damage (white empty bar) and bone damage (white line bar) from the five joints (2 forepaws, right hind knee, right hind ankle and right hind paw) was performed following tissue processing and toluidine-blue staining of sections. D. AJM for C3 deposition in the five joints examined within the synovium (black solid bar), on the surface of cartilage (white empty bar), and total score (white line bar) is presented. The data are expressed as mean of disease ± SEM (n = 5 each time point). *p values for specific histologic scoring results in C and D are included in the Results.
A significant decrease (p < 0.034) was seen in the total histopathology score as well as in the individual scores for inflammation (p < 0.0080), pannus (p < 0.006), cartilage damage (p < 0.007), and bone damage (p < 0.009) in mice treated with a HD of AdhMAp44 in comparison to mice treated with AdGFP (Fig. 1C). Almost identical results were observed in mice treated with a LD of AdhMAp44 (Fig. 1C). The correlation coefficient (r2) between CDA and histology scores was highly positive (0.99). Representative tissue sections of histology are shown in knee joints (Fig. 2A, C) and ankle (Fig. 2B, D) of mice injected i.p. with AdGFP or AdhMAp44 (HD), respectively.
FIGURE 2.
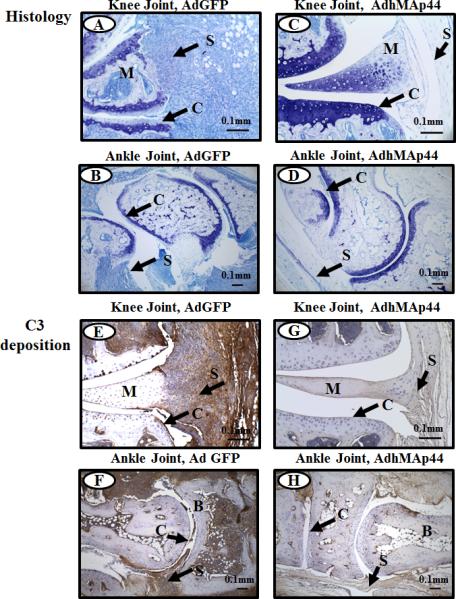
Representative histopathology and C3 deposition images from the knee joints of WT mice treated with AdGFP or AdhMAp44 (HD). The top two panels from left to right (A & C) show staining with toluidine-blue (blue color) from the knee joints of WT mice treated with AdGFP (left panel) and AdhMAp44 (right panel). The second two panels from left to right (B & D) show staining with Toluidine-blue (blue color) from the ankle joints of WT mice treated with AdGFP (left panel) and AdhMAp44 (right panel). The third set of two panels from left to right (E & G) show staining with anti-C3 Ab (brown color) from the knee joints of WT mice treated with AdGFP (left panel) and AdhMAp44 (right panel). The fourth set of two panels from left to right (F & H) show staining with anti-C3 Ab (brown color) from the ankle joints of WT mice treated with AdGFP (left panel) and AdhMAp44 (right panel). Areas of synovium (S-black arrow), cartilage (C-black arrow), bone (B) and meniscus (M) are identified. Data from using a LD or a HD of AdhMAp44 in the study were indistinguishable; therefore, we show representative pictures from only the HD of AdhMAp44. Magnification for all knee joint images shown in Fig. 2 is 20X, and magnification for all ankle joints shown in Fig. 2 is 10X. Scale bar is 0.1mm (100μm).
The level of C3 deposition in the synovium and cartilage was also significantly decreased in mice injected with either dose of AdhMAp44 in comparison to the mice injected with AdGFP or PBS (Fig. 1D). A significant decrease was seen in the total C3 deposition score as well as in the individual scores for synovium (p < 0.001) and cartilage (p < 0.003) in mice treated with a LD of AdhMAp44 in comparison to mice treated with AdGFP (Fig. 1D). Almost identical results were seen in mice treated with a HD of AdhMAp44 (Fig. 1D). Overall there was more than a 90% decrease in C3 deposition in the knee joint of mice either treated with a LD or with a HD of AdhMAp44. Representative tissue sections of C3 deposition are shown in knee joints (Fig. 2E, G) and ankle joints (Fig. 2F, H) of mice injected i.p. with AdGFP or AdhMAp44 (HD), respectively. There was no effect on the weight of mice before, during and after the development of disease in mice treated with a LD or HD of AdhMAp44 as compared to AdGFP or PBS (Supplemental Fig. 2C).
Effect of AdhMAp44 on C5a levels in serum
A 22% (p < 0.045) and 45% (p <0.001) decrease in the level of C5a at day 10 was noted using sera from mice treated with AdhMAp44 using a LD or a HD, respectively, as compared to AdGFP-transduced mice (Fig. 3A). At day -5, i.e. prior to treatment, the absolute levels of C5a were identical and there was no significant difference as expected in all treatment groups. The decrease in the absolute levels of C5a in serum of CAIA were consistent with the anticipated effect in the model of AdMAp44 treatment on complement activation.
FIGURE 3.
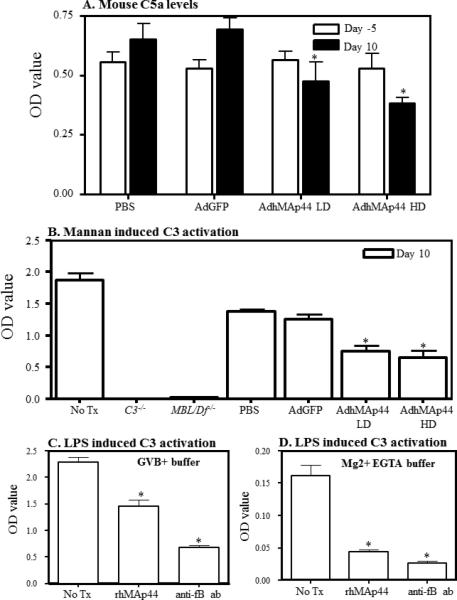
Effects of AdhMAp44 on serum C5a levels and on mannan-induced C3 activation. A. Decrease in the absolute levels of C5a in the circulation of AdhMAp44 treated mice. Mice were injected i.p. with PBS, AdGFP or AdhMAp44. The absolute levels of C5a were measured using sera from mice before (day -5) after the induction of CAIA (day 10). The data in A represent the mean ± SEM based on n = 5 for each group. LD = low dose AdhMAp44. HD = high dose AdhMAp44. *p < 0.05 in comparison with AdGFP treatment. B. ELISA showing a decrease in mannan-induced (LP) C3 activation, in vitro, using sera from AdhMAp44 treated mice. Sera obtained from mice injected i.p. with PBS, AdGFP or AdhMAp44, at day 10, were analyzed for C3 activation in vitro. Sera from WT mice (No Tx), without CAIA, were used as a positive control. Sera from C3−/− and MBL/Df−/− mice were used as negative controls. The data in B represent mean ± SEM based on n =4 mice in all groups. *p < 0.05 in comparison with AdGFP treatment. C. ELISA showing an overall decrease in LPS-induced C3 activation in vitro in GVB buffer with Ca2+ (all complement pathways are active) by recombinant human MAp44 in vitro. WT sera from mice without any CAIA were pre-treated with rhMAp44 or anti-fB inhibitory antibody. D. ELISA showing a decrease in LPS-induced C3 activation in Ca2+-deficient buffer with Mg2+ EGTA (AP only is active) by recombinant human MAp44 in vitro. WT sera from mice without any CAIA were pre-treated with rhMAp44 or anti-fB inhibitory antibody. The data in C & D represent the mean ± SEM based on n = 4 for each treatment group. p < 0.05 in comparison with no Tx sera from WT mice.
Effect of AdhMAp44 on mannan-induced C3 activation
Mannan particles specifically activate C3 through the LP. There was a 40% and 49% decrease at day 10 in the C3 activation induced by sera from mice treated with AdhMap44 LD and AdhMAp44 HD vs. AdGFP, respectively (Fig. 3B). The O.D. values for WT serum were 1.187 ± 0.108, PBS 1.383 ± 0.035, AdGFP 1.258 ± 0.078, AdhMAp44 LD 0.750 ± 0.086 (p < 0.002 vs. AdGFP), and AdhMAp44 HD 0.646 ± 0.122 (p < 0.003 vs. Ad GFP). In contrast, no decrease in C3 activation was seen in the sera from mice treated with PBS or AdGFP alone. There was no C3 activation using sera from C3−/− and MBL/Df−/− mice, as expected. Sera from C3−/− and MBL/Df−/− mice were used as negative controls for ELISA. These results show that recombinant human MAp44 present in the circulation of CAIA mice affected activation of the LP.
Effect of recombinant human MAp44 on LPS-induced C3 deposition
LPS is known to activate complement through both the LP and AP. LPS is used in our disease model of CAIA after the passive infusion of anti-CII mAbs, although the mechanism whereby LPS enhances disease is unknown. The possibility exists that rhMAp44 may inhibit the enhancing effects of LPS on the initiation of CAIA through the LP or AP of the complement system. We examined this question through induction of C3 deposition in vitro using LPS-coated plates and serum in the presence of Ca2+ - sufficient buffer, enabling all 3 complement pathways, or Ca2+ - deficient buffer with Mg2+ EGTA where only the AP is active. LPS-induced C3 deposition in the presence of Ca2+ was decreased from 2.28 ± 0.10 O.D. to 1.45 ± 0.13 O.D. in the absence or presence of rhMAp44, a 36% reduction (p < 0.002), and decreased to 0.679 ± 0.042 in the presence of anti-FB mAb, a 70% reduction (p < 0.001) (Fig. 3C). More marked results were observed in Ca2+ - deficient buffer where the O.D. values for LPS-induced C3 deposition using no treatment were 0.161 ± 0.017, with rhMAp44 were 0.044 ± 0.003 (p < 0.0004), and with anti-FB mAb were 0.027 ± 0.003 (p < 0.0002) (Fig. 3D); these decreases were 72% and 83%, respectively. These results suggest that rhMAp44 may inhibit the induction of complement in part through its effects on the AP.
Human MAp44 is present in the circulation of mice after injection of AdhMAp44
Using an ELISA human MAp44 was found to be present in the sera at day -5, day 0, day 3 and at day 10 from mice with CAIA treated with a LD or HD of AdhMAp44, but not in the sera from mice injected with PBS or AdGFP alone (Fig. 4A-D). At day 0, a large and highly significant increase (P <0.001) in the levels of human MAp44 was seen in the circulation of mice treated with a LD or HD dose of AdhMAp44 (Fig. 4). At day 3, the levels of human MAp44 were 808.6 ± 170.54 (ng/ml) and 1841.0 ± 1173.9 (ng/ml) in the circulation of mice treated with a LD or HD of AdhMAp44, respectively (Fig. 4C). At day 10, the levels of human MAp44 were 238.39 ± 70.66 (ng/ml) and 127.25 ± 56.08 (ng/ml) in the circulation of mice treated with a LD or HD of AdhMAp44, respectively (Fig. 4D). The differences in the level of human MAp44 between a LD and HD of AdhMAp44, at day 10, were not statistically significant (p > 0.5). As expected no human MAp44 was detected using sera from WT mice without any treatment (data not shown). The presence of human MAp44 in the circulation at day 0, at day 3 and at day 10 of mice demonstrates that Ad-targeted cells were effectively transduced with AdhMAp44 to produce human MAp44.
FIGURE 4.
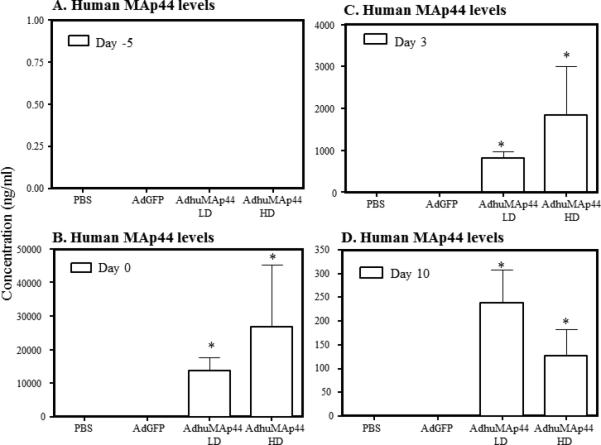
Levels of human MAp44 in the circulation of CAIA mice treated with or without AdMAp44. A. Levels of human MAp44 in the circulation of mice at day -5 prior to injecting i.p. with PBS or AdGFP or of LD and HD AdhMAp44. B. Levels of human MAp44, at day 0, in the circulation of mice injected with PBS, AdGFP, or AdhMAp44. C. Levels human MAp44 at day 3 injected with PBS, AdGFP, or AdhMAp44. D. Levels at day 10 of human MAp44 in the circulation of mice with CAIA injected with PBS, AdGFP, or AdhMAp44. ELISAs were performed for human MAp44. Sera from mice with CAIA injected with LD or HD dose of AdhMAp44 exhibit somewhat similar levels of human MAp44. In contrast, mice injected with PBS or AdGFP have no detectable levels of human MAp44. The data represent the mean ± SEM (ng/ml) based on n = 5 for each group, except for mice injected with a HD of AdhMAp44 (n = 4). *p < 0.01 in comparison with PBS or AdGFP treatment.
AdmMAp44 treatment also substantially prevents clinical disease activity in mice with CAIA
To confirm that the effect of AdhMAp44 was not due to non-physiologic effects of the human protein in mouse, the effects of expression of mouse MAp44 were also examined. To evaluate this question, mice were injected i.p. with AdmMAp44 and control AdGFP as described in Materials and Methods. At day 10 the CDA in WT mice injected with AdGFP and AdmMAp44 was 9.0 ± 1.65 and 3.4 ± 1.60, respectively (Fig. 5B). Thus, at day 10 CDA was reduced by 60% (p < 0.026) in mice pre-treated with AdmMAp44 as compared to mice pre-treated identically with AdGFP. The prevalence of disease at day 10 in WT mice injected with AdGFP or AdmMAp44 was 100% and 60%, respectively (Fig. 5A). There was no significant effect on the weight of mice treated with AdGFP as compared to AdmMAp44 (Supplemental Fig. 2D). These data demonstrate that, similarly to AdhMAp44, treatment with AdmMAp44 prevents the development of CAIA.
FIGURE 5.
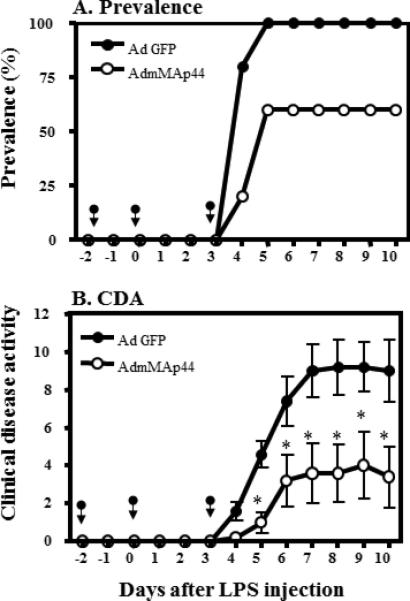
Effects of AdmMAp44 on CAIA. The data are derived from the indicated days after anti-CII mAb and LPS injections. The arrows in panels A and B indicate the days of injection of AdGFP or AdmMAp44. A. Prevalence of arthritis (%) over the duration of the experiment. B. CDA over the duration of the experiment. The data represent the mean ± SEM for each group (n = 5). *p < 0.05 in comparison to AdGFP treatment.
Exogenous mouse MAp44 expression prevents histological changes and C3 deposition in the joints in CAIA
To further examine the treatment effect of AdmMAp44, histopathologic analyses were performed in fixed joints from mice injected i.p. with AdGFP or with AdmMAp44. A significant decrease (p < 0.023) was seen in the total histopathology score as well as in the individual scores for inflammation (p < 0.040), pannus (p < 0.015), cartilage damage (p < 0.021), and bone damage (p < 0.026) in mice treated with AdmMAp44 as compared to AdGFP (Fig. 6A). The correlation coefficient (r2) between CDA and histology scores was 0.93 (Fig. 6B). The levels of C3 deposition in the synovium and cartilage were also significantly decreased in mice transduced with AdmMAp44 in comparison to transduction with AdGFP (p < 0.02) (Fig. 6C). The correlation coefficient (r2) between CDA and C3 deposition scores was highly positive (0.95) (Fig. 6D). Representative tissue sections are shown in knee joints (Supplemental Fig. 3A, C) and ankle (Supplemental Fig. 3B, D) of mice injected i.p. with AdGFP or AdmMAp44. Representative tissue sections of C3 deposition are shown in knee joints (Supplemental Fig. 3E, G) and ankle (Supplemental Fig. 3F, H) of mice injected with AdGFP or AdmMAp44.
FIGURE 6.
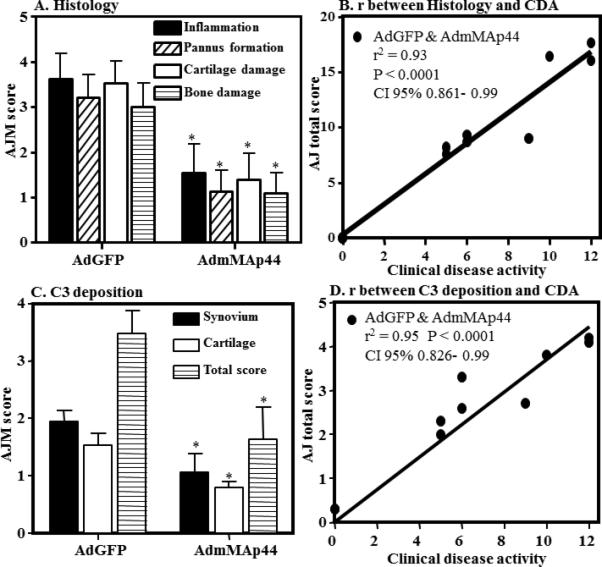
Decrease in scoring for inflammation, pannus, cartilage damage, and bone damage as well as staining for C3 deposition in knee joints of mice with CAIA treated with AdmMAp44 compared to AdGFP. A. AJM of histopathologic score for inflammation (black solid bar), pannus formation (white hatched bar), cartilage damage (white empty bar) and bone damage (white line bar) from the five joints (2 forepaws, right hind knee, right hind ankle and right hind paw) was performed following tissue processing and toluidine-blue staining of sections. B. Pearson correlation (r) between histology scores (total scores) and CDA. C. AJM for C3 deposition score from the five joints in the synovium (black solid bar), on the surface of cartilage (white empty bar), and total score (white line bar). D. Pearson correlation (r) between C3 deposition total scores (synovium and cartilage) and CDA. The data are expressed as mean of disease ± SEM (n = 5). *p values for histologic scoring data in A and C are included in the Results.
Systemic effects of AdmMAp44 injected into the right knee joint
AdmMAp44 or AdGFP were injected three times in the right knee during the development of CAIA at days -5, 0, and 3 (with anti-CII mAb injected at day 0); the un-injected left knee served as a control. The overall CDA in all of the indicated joints in mice with CAIA pretreated with AdmMAp44 was significantly reduced by 55% as compared to mice injected with AdGFP; the CDA values were AdGFP 10.1 ± 1.26 and AdmMAp44 4.5 ± 1.40, respectively (p < 0.008) (Fig. 7A). The prevalence of disease at day 10 in CAIA mice pretreated with AdGFP or AdmMAp44 was 100% and 70%, respectively (Fig. 7B). Injection in the right knee joint led to a decreased CDA in this joint (Fig. 7C). Following injection in the right knee joint, decreases in CDA were also observed in the left hind limb (Fig. 7D), right forepaw (Fig. 7E), and left forepaw (Fig. 7F). This experiment was repeated two times with identical injection schedules in the right knee, and the data were pooled. Thus, in addition to demonstrating that AdmMAp44 prevents CAIA in mice within the locally injected right knee joint, the effect was systemic because the CDA in all joints was substantially diminished.
FIGURE 7.
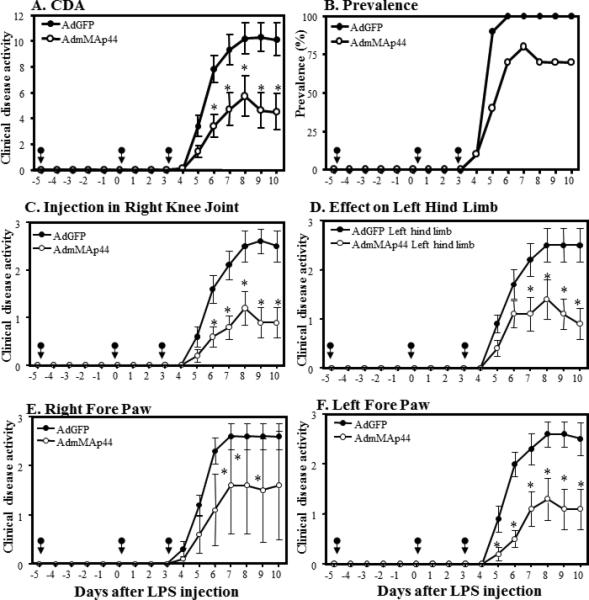
Decrease in the overall CDA by local right knee joint injection of AdmMAp44 or AdGFP on arthritis induced by anti-CII mAb. WT mice were injected three times locally in the right knee joint at days -5, 0, and 3. The effects were examined on both forepaws and the left hind limb. The data shown are derived from the indicated days after the mAb and LPS injection. A. CDA in all joints over the duration of the experiment. B. Prevalence of arthritis (%) in all joints over the duration of the experiment. C. CDA in the right knee joint over the duration of the experiment. D. CDA in the left hind limb over the duration of the experiment. E. CDA in the right fore paw over the duration of the experiment in. F. CDA in the left fore paw over the duration of the experiment. The data represent the mean ± SEM for each group (n = 5). *p < 0.05 for AdmMAp44 in comparison to treatment with AdGFP. Black arrows show the injection time of AdmMAp44 and AdGFP.
AdhMAp44 treatment decreases severity of Ross River Virus-induced arthritis and myositis
To assess the role of AdhMAp44 in another model of LP-dependent musculoskeletal inflammatory disease, we used a mouse model of Ross River virus (RRV)-induced arthritis and myositis (43) (Supplemental Fig. 4). Previous studies have implicated a role for the LP of the complement system in the development of RRV-induced disease, as both C3- and MBL-deficient mice exhibit reduced RRV-induced disease severity and tissue destruction compared to WT mice (41, 43). To evaluate whether human MAp44 could mitigate RRV-induced disease, AdhMAp44 or AdGFP was administered in the right rear footpad to mice at days -3, 0, and 3 with inoculation with RRV in the left rear footpad at day 0. As shown, pretreatment with AdhMAp44 significantly (p < 0.05) diminished the severity of RRV-induced disease signs (Supplemental Fig. 4A). There was no effect on weight (Supplemental Fig. 4B).
Presence of HA-tagged mouse MAp44 in circulation after AdmMAp44 or AdhMAp44 treatment
The presence of Ad-derived mouse or human MAp44 in the serum was examined, using an ELISA for the HA tag, in mice injected with AdmMAp44 or AdhMAp44 before and after the induction of CAIA (Fig. 8). Sera from mice injected i.p. with AdmMAp44 were examined at days -2, 0, 3, and 10 using Western blot analysis for the HA tag (Fig. 8A). Likewise, sera from mice injected with AdmMAp44 in the right knee joint were examined at days -5, 0, 3, and 10 using Western blot analysis for the HA tag (Fig. 8B). A band of ~43-50 kDa was present in mice injected i.p. with AdmMAp44 but was missing at day -2 prior to injection as expected (Fig. 8A lane 2). Similarly, a band of ~43-50 kDa was present in the sera of mice injected in the knee joint with AdmMAp44 but it was not present at day -5 prior to injection (Fig. 7B lane 2). HA was not detected using serum from untreated mice with no disease (Fig. 8A, B lane 1). The presence of HA in sera at days 0, 3 and 10 clearly indicates that mouse recombinant MAp44 was present in the circulation. In addition, human MAp44 generated after i.p.administration of AdhMap44 was apparently functional because it bound to MBL, as confirmed using mannanagarose beads to pull down the HA-tagged protein from serum (Fig. 8C). These results also show that AdmMAp44 or AdhMap44 effectively transduced cells and led to recombinant protein expression in vivo regardless of the injection route. We used Western blot analysis to detect the HA tag since an ELISA to measure mouse MAp44 is not available.
FIGURE 8.
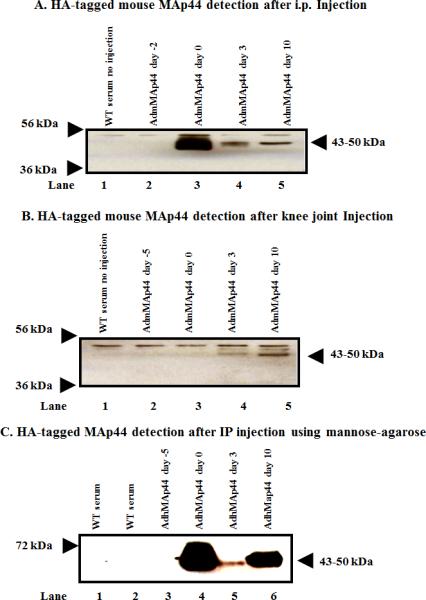
In vivo transduction and expression efficiency of AdmMAp44 or AdhMAp44 assessed by using Western blot analysis for the HA tag on mouse MAp44 in the sera of WT mice before and after the induction of CAIA. Mice were injected in separate studies with AdmMAp44 or AdhMap44 i.p. and also locally in the right knee joint. After SDS-PAGE and transfer to nitrocellulose, the blots were probed with anti-HA rabbit antibody. The presence of a HA band (~43-50 kDa) in serum indicates the successful transduction of cells and protein expression in mice treated with AdmMAp44. A. Presence of HA band in serum at day 0 (lane 3), at day 3 (lane 4) and at day 10 (lane 5) after mice were injected i.p. with AdmMAp44 at day -2 (lane 2). B. Presence of HA band in serum at day 3 (lane 4) and at day 10 (lane 5) after mice were injected in the right knee joint with AdmMAp44 at day -5 (lane 2). Serum from a WT mouse with no injection of adenoviral vectors was used as a negative control (lane 1). C. MAp44 bound to MBL in serum after i.p. injection of AdhMAp44 at day -5. MBL-MAp44 complexes were pulled down using mannose-agarose beads, and the presence of the HA tag on human MAp44 in serum at day 0 (lane 4), day 3 (lane 5), and day 10 (lane 6) was detected using rabbit anti-HA antibody. Serum from a WT mouse without mannose-agarose preparation (lane 1) as well as serum from a WT mouse with no injections with Ad vectors (lane 2) were also examined as controls.
In vivo transduction efficiency in the knee joint synovium through detection of GFP and HA
To specifically determine whether the synovium in the knee joint of mice with CAIA was transduced with AdmMAp44, we used IHS. After mice were injected i.p. with AdGFP or AdmMAp44, the presence of GFP and the HA tag was assessed in the synovium of the knee joint (Fig. 9). We found that GFP was clearly visible by IHS in the knee joints of mice with CAIA at day 10 after injection of AdGFP (Fig. 9C). In contrast, no green fluorescence was visible in mice injected with PBS (Fig. 9A) or AdmMAp44 (Fig. 9E). However, we found that the HA tag was detectable as sand grain particles in cells in mice injected i.p. with AdmMAp44 (Fig. 9F) but not in mice injected with PBS (Fig. 9B) or AdGFP (Fig. 9D). These data show that AdmMAp44 transduced a subset of cells in the synovium. We do not know the exact identity of the cells but they could be synovioctyes, lymphocytes, neutrophils and/or macrophages because of the presence in CAIA of these cell populations (46).
FIGURE 9.
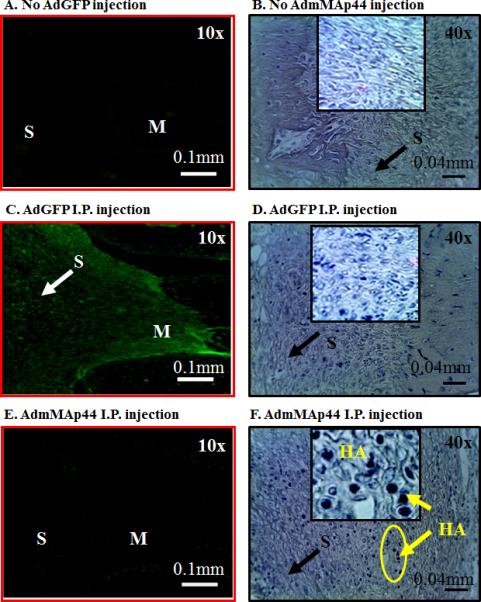
Representative IHS in mice with CAIA comparing the in vivo transduction of AdGFP and AdmMAp44 injected i.p. at day -2 and day 0. All mice were sacrificed at day 10 to assay by IHS the presence of GFP by immunofluorescence or the HA tag (sand grain blue particles) using anti-HA tag antibodies. A. Mice were not injected with AdGFP or AdmMAp44 but only injected two times with PBS. No green fluorescence was seen in the synovium and in the meniscus. C. Mice were injected two times with AdGFP, and green fluorescence protein is clearly visible in the synovium as marked by a white arrow. E. Mice were injected two times with AdmMAp44 and there was no green fluorescence in the synovium. B. Mice were neither injected with AdGFP nor AdmMAp44 but injected two times with PBS. No sand grain blue particles were seen in the synovium or in the meniniscus. D. Mice were injected two times with AdGFP and a no sand grain blue particles are visible in the synovium marked by a black arrow. F. Mice were injected two times with AdmMAp44 and there were very distinct sand grain blue particles (HA) present throughout the synovium. The circular area of the synovium has been enhanced in an insert by 4X to more clearly demonstrate sand grain blue particles of HA stain. Areas of synovium (S) and meniscus (M) are identified. Magnification for all images in left panels is 10X. Scale bar is 0.1mm (100μm). Magnification for all images in right panels is 40X. Scale bar is 0.04 mm (40μm).
Effect of AdMAp44 on lectin, MASPs, FD and cytokine mRNA levels in the knee joints with CAIA
To determine the effect of human MAp44 expression on expression of MBL-A, MBL-C, FCNA, MASP-1, MASP-2, MASP-3 and FD, as well as on pro-inflammatory cytokines, we measured the mRNA levels at day 10 from the knee joints of mice with CAIA injected i.p. three times with PBS, AdGFP, AdhMAp44 LD or AdhMap44 HD (Table 1). Minimum baseline mRNA levels for all targets were present in the knee joints of mice without CAIA and without any treatment (Table 1). Liver was used as a positive control to examine the mRNA levels of all ten targets. There was a 42% and 60% decrease in the mRNA levels of FCN-A in the knee joints of mice treated with a LD or with a HD of AdhMAp44 respectively compared with AdGFP-treated mice; this decrease was significant (P < 0.0017 for LD, P < 0.08 for HD). Minimal levels of mRNA for MBL-A or MBL-C were present below 40 cycles of PCR in the knee joints of mice treated with PBS, AdGFP, AdhMAp44 LD, or AdhMAp44 HD. There was a statistically significant (p < 0.05) decrease in the mRNA levels in the knee joints of MASP-1, MASP-2, MASP-3 and FD in mice injected with either dose of AdhMAp44 compared to mice injected with AdGFP or PBS. There was also a significant decrease (p < 0.02 or greater) of 66%, 87% and 85% in the mRNA levels of TNF-α, IL-1α and IL-1β respectively in the knee joints of mice treated with either the LD or HD of AdhMAp44 (Table 1). These mRNA data show that treatment of CAIA mice with AdhMap44 decreased the mRNA levels in the joints of FCN-A, FD, and MASPs as well as of pro-inflammatory cytokines.
Table 1.
mRNA for factor D, MASPs and cytokines in the knee joints of WT mice with and without CAIA1
| mRNA | No CAIA2 | PBS | AdGFP | AdhMAp44 LD3 | AdhMAp44 HD4 |
|---|---|---|---|---|---|
| Factor D | 28.27 ± 10.7 | 1523.6 ± 523.8 | 709.8 ± 73.9 | 295.31 ± 52.2 | 324.0 ± 120.5 |
| p | 0.0017 | 0.018 | |||
| Decrease | 58% | 54% | |||
| MASP-1 | 6.83 ± 0.9 | 40.6 ± 523.8 | 51.3 ± 3.7 | 31.8 ± 1.6 | 22.7 ± 3.2 |
| p | 0.001 | 0.001 | |||
| Decrease | 38% | 56% | |||
| MASP-3 | 1.38 ± 0.13 | 16.6 ± 2.7 | 14.2 ± 0.13 | 8.9 ± 0.76 | 5.2 ± 0.55 |
| p | 0.025 | 0.0035 | |||
| Decrease | 38% | 56% | |||
| MASP-2 | 0.39 ± 0.09 | 1.3 ± 0.15 | 1.2 ± 0.13 | 0.73 ± 0.1 | 0.53 ± 0.2 |
| p | 0.044 | 0.010 | |||
| Decrease | 39% | 56% | |||
| MBL-A5 | 0.29 ± 0.09 | 0.05 ± 0.01 | 0.04 ± 0.01 | 0.01 ± 0.004 | 0.01 ± 0.004 |
| MBL-C5 | 1.13 ± 0.27 | 0.002 ± 0.002 | 0.016 ± 0.006 | 0.006 ± 0.002 | 0.003 ± 0.002 |
| FCN-A | 145.1 ± 34.2 | 366.3 ± 55.3 | 295.6 ± 34.1 | 170.0 ± 32.6 | 117.3 ± 30.1 |
| p | 0.028 | 0.069 | |||
| Decrease | 42% | 60% | |||
| TNF-α | 1.06 ± 0.134 | 15.1 ± 2.1 | 12.8 ± 2.1 | 3.09 ± 0.6 | 3.9 ± 0.37 |
| p | 0.002 | 0.007 | |||
| Decrease | 76% | 66% | |||
| IL-1α | 1.36 ± 0.19 | 4.7 ± 1.2 | 4.0 ± 0.75 | 0.99 ± 0.23 | 0.53 ± 0.14 |
| p | 0.005 | 0.005 | |||
| Decrease | 75% | 87% | |||
| IL-1β | 2.91 ± 0.50 | 41.3 ± 6.1 | 29.8 ± 8.5 | 5.2 ± 0.50 | 4.40 ± 0.2 |
| p | 0.02 | 0.03 | |||
| Decrease | 83% | 85% |
Data are expressed in pg/ng 18s rRNA with mean ± SEM based on the indicated number of mice (n). CAIA mice treated with PBS (n=5), AdGFP (n = 5), AdhMAp44 LD (n = 5) and AdhMAp44 HD (n =5).
Baseline levels for various mRNA targets from the knee joint of WT mice without CAIA and without any treatment (n = 4).
LD = Low dose
HD = high dose.
The mRNA levels of MBL-A and MBL-C were very low.
Liver from WT mice used as a positive control to measuring the mRNA levels (data not shown). All p-values for different mRNAs in mice treated either with AdhMAp44 LD or with AdhMAp44 HD were compared with the corresponding values of WT mice treated with AdGFP. The percent (%) decrease in mRNA levels in mice treated with AdhMAp44 in each column has been shown compared with mice treated with AdGFP. p values were compared between mice treated either with AdGFP and AdhMAp44 LD or AdhMAp44 HD. p < 0.05 were considered statistically significant.
DISCUSSION
Through the use of Ad-programmed expression of both human and mouse MAp44, these studies have revealed an unexpected but essential role for the LP of the complement system in the development of CAIA. Prevention of clinical disease with adenovirus expressed MAp44 was associated with a decrease in cartilage and synovial C3 deposition, marked improvement in histologic injury scores, and decreases in local mRNA levels of LP and AP components as well pro-inflammatory cytokines. Both human and mouse MAp44 appeared to be effective in ameliorating disease, and the molecules were functional as assessed by binding to MBL in vivo and inhibition of the LP and AP ex vivo. Treatment with Ad was effective using both systemic and local articular injection, with a systemic amelioration found in the latter situation likely due to the resulting effects of circulating recombinant MAp44. In sum, our studies have revealed a central role for the LP in the initiation of complement activation, and that an understanding of the molecular pathogenesis of inflammatory arthritis must expand to incorporate a role for the LP.
Prior studies using mice deficient in MBL-A/C or C4 had demonstrated no effect on the development of joint damage in CAIA. MBL is a major pattern recognition molecule within the LP, and the major means by which the LP activates C3 is through MASP-1- and MASP-2-mediated cleavage of C4 and C2 to generate the shared CP/LP C3 convertase C4b2b. The absence of an apparent effect of deletion of MBL-A/C or C4 on the evolution of tissue injury in CAIA had suggested that no major role existed for the LP. However, recent findings indicate the presence of additional pattern recognition molecules that could be important in the initiation of the LP (19). In addition, the suggestion that MASP-1 could directly activate C3 (39), and the possible engagement of the extrinsic pathway of complement activation through engagement by MASPs of the coagulation pathway proteases (49), strongly suggested a re-examination of the role of the LP in CAIA. In addition, the recent understanding that MAp44 acts as a competitive inhibitor of all of the MASPs provided a means to more broadly impact the activation of the LP (36).
MAp44 is a product of alternative splicing within the MASP-1/3 gene (33). The heart has the highest mRNA expression of MAp44 mRNA in humans followed by weaker expression in the liver, brain and cervix (36); MAp44 is also produced in skeletal muscle (34) and is absent in adipose tissue and lungs. The role of MAp44 in the human complement system has been well documented, and the recent demonstration that MAp44 preserves cardiac function by decreasing levels of MBL and local C3 deposition and preventing thrombogenesis is consistent with a similar role in the mouse (37).
MAp44 interacts with MBL and ficolins with nM affinities and forms a Ca2+ dependent homo-dimer (34). MAp44 blocks interactions between MBL and ficolins with the MASPs by competitive inhibition or displacement, disrupting the activation complexes and thus impairing LP-mediated complement activation (50). The results from in vitro structural and functional studies have been corroborated by in vivo studies showing that MAp44 attenuates myocardial injury and arterial thrombogenesis in MBL-A/C-dependent models (37). Therefore, MAp44 was suggested as a natural in vivo endogenous inhibitor of the LP (37). Although the CAIA model does not require MBL-A/C engagement, we hypothesized that if MAp44 inhibits other collectin-MASP interactions, it could affect the development of CAIA in mice. Since large amounts of purified MAp44 protein were not available for experiments, we used AdhMAp44 and AdmMAp44 to generate sustained in vivo production of human and mouse HA-tagged MAp44 for CAIA studies. We found that there was more than a 40% decrease in C3 activation via LP when mice were treated with AdhMAp44 vs. AdGFP. Furthermore, at day 10, mice treated with AdhMAp44 (LD) and AdhMAp44 (HD) demonstrated a decrease of 22% and 45%, respectively, in the absolute levels of C5a. Although this decrease was significant and consistent with the intended effect of MASP inhibition, the lack of a decrease of C5a in a similar magnitude as the C3 deposition in the joint (~90% decrease) indicates that changes in systemic levels of C5a do not fully reflect the local complement activation that occurs during the development of injury.
The concentration of MAp44 in human serum is 1.7ug/ml (33, 34). Although the levels of mouse MAp44 are not known, the demonstration of a marked clinical effect showed that we had achieved a therapeutically effective dose. This is consistent with the high levels of human MAp44 at day 0 detected using both ELISA and Western blot analysis, following just a single injection of AdhMAp44 at day -5. The measured levels of human MAp44 were in the 100-300 ng/ml range using serum obtained at 10 days, which was a point of lower levels as assessed by pull-down experiments.
Inhibition of MAp44 likely has a direct relevance to the human disease(s) because there is now agreement on the similar roles played by MASP-1 in the cleavage pro-FD into active FD in both mice and humans (29, 51, 52), although it appears that pro-FD cleavage is not essential to develop AP-dependent disease in every experimental model (31). Thus, inhibition of the LP by AdMAp44 might affect the AP indirectly because MASP-1 activates MASP-2, leading to engagement of the amplification loop (32), or directly because pro-FD is cleaved to allow AP activation, or because C3 and/or Factor B cleavage is mediated by MASPs. Nevertheless, although there are undoubtedly some species-specific and disease-specific differences in the necessity of the LP pathway for AP activation between mouse and human, there appears to be substantially shared activities across the two species.
Adenovirus type 5 was chosen as a delivery vehicle for the current studies because it uses the Coxsackie-Adenovirus Receptor (CAR, a cell adhesion molecule) to bind to cells (40), and subsequent internalization takes place by binding of an Arg-Gly-Asp (RGD) sequence to the integrins αvβ5 and αvβ3 (53-55). Synoviocytes express CAR- or RGD-binding integrin on their surface, which are used by adenoviruses to enter into cells. We found by using FACS analysis that a fibroblast-like synoviocyte (FLS) cell line derived from the synovium of a mouse with CIA expressed substantial levels (~60%) of the integrin α5 on their surface (data not shown). IHS data also showed that RGD binding integrins are highly expressed in the mouse synovium with and without arthritis (data not shown) (56-58). The inclusion of the RGD sequence in the AdhMAp44 construct markedly improved delivery into synovial cells (24); administration of AdmMAp44 without RGD only partially attenuated CAIA. Therefore, RGD sequences in AdhMAp44 served as an effective delivery vehicle for homing AdhMAp44 in the synovium of knee joints of mice without affecting the disease itself; the control AdGFP also contained RGD but did not inhibit synovitis. For example, adenovirus containing the mouse interleukin 1 receptor antagonist (mIL-1Ra) inhibited collagen-induced arthritis (CIA) (40), and it has been shown that Ad vectors carrying the human adiponectin APN (Ad-APN) gene significantly reduced CIA and C3 deposition in the knee joints (59).
CAIA is an appropriate model in which to study the role of the LP. The complement system, a part of innate immunity, protects from invading pathogens but also plays a central role in the pathological process of an autoimmune and inflammatory disease such as RA. Our studies have previously shown that in CAIA, the AP is the main contributor to the development of tissue injury (4). Additionally, MASP-1 and MASP-3 cleave a zymogen of FD called pro-FD (29). It was shown that mice lacking MASP-1 and MASP-3 lack both the LP and have reduced AP activity (29, 30). Similarly, it has been reported that patients deficient in MASP-1 and MASP-3 have reduced but detectable AP activity (32). No cleavage of pro-FD was observed in the circulation of fH−/−/MASP-1/3−/− mice; AP activity was reduced in these mice and activation was possible only after injecting Cobra Venom Factor (31). Overall, MASP-1/3−/− mice exhibit defective AP activation because there is only pro-FD and not mature FD in circulation, even in the presence of plasmin, thrombin and kallikreins (29, 30, 38). Consistent with these observations, we have shown that MASP-1/3−/− mice not only have defective AP of the complement system but are markedly resistant to CAIA (38). Thus, for the development of CAIA, the AP of the complement system is necessary and mice lacking any component of the AP are resistant to arthritis (4, 27, 44).
In the current CAIA studies we have made several new observations. First, AdhMAp44 dramatically attenuates CAIA in mice. Recombinant hMAp44 was detectable on day 0 through day 10 in the circulation of CAIA mice treated with AdhMAp44. In addition, administration of AdhMAp44 diminished the severity of RRV-induced arthritis in mice. Second, AdmMAp44 also attenuated CAIA in mice using either a systemic or local injection as the delivery route. Third, there was a decrease in the levels of C5a in the circulation of WT mice treated with AdhMAp44 compared with AdGFP-treated mice, which is consistent with the intended effect of MAp44 expression (Figure 3A). Fourth, MAp44 not only inhibited interactions between MASPs and its ligands but also resulted in reduced levels of MASP-1, MASP-2, MASP-3 and pro-FD mRNA expression, as well as pro-inflammatory cytokine expression, in the knee joints of mice treated with AdhMAp44. And fifth, the ameliorative effects of MAp44 are not specific to one model of arthritis, as when we used another mouse model, RRV-induced arthritis that is not dependent on the AP of complement but it is partially dependent on the LP ligand MBL (41-43), we again found that AdhMAp44 significantly decreased arthritis.
Furthermore, our results suggest that the mechanisms by which the LP affects the initiation and development of CAIA deserve further attention. What is not understood is why, if the LP is essential for the development of CAIA, there is no effect of C4 deficiency on the disease when the major means by which C3 is activated is through the shared CP/LP C4b2b C3 convertase. This result suggests the presence of a bypass mechanism, perhaps related to direct MASP-mediated C3 activation or use of the extrinsic (clotting) pathway to cleave C3 and/or C5. Both possibilities should be explored. Also uncertain is which of the pattern recognition molecules are engaged during CAIA, and what injury-associated ligand(s) are elaborated during the evolution of CAIA.
Our in vitro results demonstrating only a modest effect of MAp44 on in vitro C3 deposition as well as C5a generation induced through either the LP or AP by adherent anti-CII mAbs (data not shown) suggests that it is not the anti-CII mAbs themselves which engage pattern recognition molecules. In contrast, more than a 40% decrease in the C3 activation induced by mannan in the sera from mice treated with AdhMAp44 indicated that human MAp44 specifically inhibited the LP of the complement. MAp44 might be inhibiting interactions between ficolins or Collectins bound to MASPs. We also can't rule out the possibility that AdhMAp44 may have affected C3 activation induced by LPS because all of the mice with CAIA have been injected with LPS in order to induce severe disease. This observation is based on the fact that rhMAp44, in vitro, inhibited 36% and 72% of LPS-induced C3 activation using sera from WT mice in either a GVB+ (Ca2+-sufficient) or a Mg2+ EGTA (Ca2+-deficient) buffer, respectively. It has been shown that sera from mice deficient in all MASPs showed lower C3 deposition activity on bacteria compared with sera from WT mice; addition of rMASP-3 to serum deficient in all MASPs restored C3 deposition (60). Furthermore, rMASP-3 enzyme was activated by incubation with bacteria in the presence of MBL-A but not MBL-C (60). Thus, complexes of MASP-3-lectins-bacteria can trigger the AP and MAp44 can inhibit it.
Finally, we conclude that Ad-mediated gene transfer of MAp44 can be used as a potential tool for the treatment of arthritis. Human MAp44 was present in the circulation and MAp44 delivered by this approach can have long-term inhibitory effects on inflammatory arthritis even with reduced transduction efficiency. Furthermore, an Ad gene delivery system is highly efficient at transferring genes to a variety of proliferating and quiescent cells both in vitro and in vivo (61). It has also been shown that genetically modified Ad5 vectors with short-shafted fibers are highly efficient in transduction of RA fibroblast-like synoviocytes (FLS) and of human and murine synovium (62). The marked prevention of disease using AdhMAp44 sequences provides evidence that Ad vectors containing complement inhibitor genes and/or use of recombinant MAp44 alone or along with nanoparticles or fused to targeting molecules, should be evaluated as a treatment for inflammatory arthritis in humans.
Supplementary Material
Acknowledgements
The authors thank Ms. Umarani Pugazhenthi, University of Colorado Anschutz Medical Campus PCR core for performing quantitative RT-PCR from the knee joints of mice with CAIA.
AdhMap44 and AdmMAp44 constructs were custom prepared by Welgen, Inc (Worcester, MA) and the company's website is www.welgenic.com.
This work was supported by NIH grant AR051749 to VMH and a grant from the Lundbeck Foundation for TRK and ST.
Abbreviations used in this paper
- AJM
all joint mean
- AP
alternative pathway
- CAIA
collagen antibody-induced arthritis
- CIA
collagen-induced arthritis
- RRV
Ross River Virus
- CP
classical pathway
- CII
type II collagen
- CDA
clinical disease activity
- LP
lectin pathway
- MBL
Mannose-binding lectin
- MAC
membrane attack complex
- Map44
MBL-associated protein of 44 kDA
- rMAp44
recombinant Map44
- MAp19
MBL-associated protein of 19kDa
- MASP-1
MBL-associated serine protease-1
- MASP-2
MBL-associated serine protease-2
- MASP-3
MBL-associated serine protease-3
- WT
wild type
References
- 1.Helmick CG, Felson DT, Lawrence RC, Gabriel S, Hirsch R, Kwoh CK, Liang MH, Kremers HM, Mayes MD, Merkel PA, Pillemer SR, Reveille JD, Stone JH. Estimates of the prevalence of arthritis and other rheumatic conditions in the United States. Part I. Arthritis and rheumatism. 2008;58:15–25. doi: 10.1002/art.23177. [DOI] [PubMed] [Google Scholar]
- 2.Arend WP, Firestein GS. Pre-rheumatoid arthritis: predisposition and transition to clinical synovitis. Nature reviews. Rheumatology. 2012;8:573–586. doi: 10.1038/nrrheum.2012.134. [DOI] [PubMed] [Google Scholar]
- 3.Klareskog L, Ronnelid J, Lundberg K, Padyukov L, Alfredsson L. Immunity to citrullinated proteins in rheumatoid arthritis. Annual review of immunology. 2008;26:651–675. doi: 10.1146/annurev.immunol.26.021607.090244. [DOI] [PubMed] [Google Scholar]
- 4.Banda NK, Thurman JM, Kraus D, Wood A, Carroll MC, Arend WP, Holers VM. Alternative complement pathway activation is essential for inflammation and joint destruction in the passive transfer model of collagen-induced arthritis. J. Immunol. 2006;177:1904–1912. doi: 10.4049/jimmunol.177.3.1904. [DOI] [PubMed] [Google Scholar]
- 5.Hietala MA, Jonsson IM, Tarkowski A, Kleinau S, Pekna M. Complement deficiency ameliorates collagen-induced arthritis in mice. Journal of Immunology. 2002;169:454–459. doi: 10.4049/jimmunol.169.1.454. [DOI] [PubMed] [Google Scholar]
- 6.Ji H, Ohmura K, Mahmood U, Lee DM, Hofhuis FM, Boackle SA, Takahashi K, Holers VM, Walport M, Gerard C, Ezekowitz A, Carroll MC, Brenner M, Weissleder R, Verbeek JS, Duchatelle V, Degott C, Benoist C, Mathis D. Arthritis critically dependent on innate immune system players. Immunity. 2002;16:157–168. doi: 10.1016/s1074-7613(02)00275-3. [DOI] [PubMed] [Google Scholar]
- 7.Wang Y, Kristan J, Hao L, Lenkoski CS, Shen Y, Matis LA. A role for complement in antibody-mediated inflammation: C5-deficient DBA/1 mice are resistant to collagen-induced arthritis. Journal of Immunology. 2000;164:4340–4347. doi: 10.4049/jimmunol.164.8.4340. [DOI] [PubMed] [Google Scholar]
- 8.Okroj M, Heinegard D, Holmdahl R, Blom AM. Rheumatoid arthritis and the complement system. Ann. Med. 2007;39:517–530. doi: 10.1080/07853890701477546. [DOI] [PubMed] [Google Scholar]
- 9.Sturfelt G, Truedsson L. Complement in the immunopathogenesis of rheumatic disease. Nat Rev Rheumatol. 2012;8:458–468. doi: 10.1038/nrrheum.2012.75. [DOI] [PubMed] [Google Scholar]
- 10.Zvaifler NJ. Rheumatoid synovitis. An extravascular immune complex disease. Arthritis Rheum. 1974;17:297–305. doi: 10.1002/art.1780170315. [DOI] [PubMed] [Google Scholar]
- 11.Cooke TD, Hurd ER, Jasin HE, Bienenstock J, Ziff M. Identification of immunoglobulins and complement in rheumatoid articular collagenous tissues. Arthritis Rheum. 1975;18:541–551. doi: 10.1002/art.1780180603. [DOI] [PubMed] [Google Scholar]
- 12.Ghose T, Woodbury JF, Ahmad S, Stevenson B. Immunopathological changes in rheumatoid arthritis and other joint diseases. J. Clin. Pathol. 1975;28:109–117. doi: 10.1136/jcp.28.2.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ohno O, Cooke TD. Electron microscopic morphology of immunoglobulin aggregates and their interactions in rheumatoid articular collagenous tissues. Arthritis Rheum. 1978;21:516–527. doi: 10.1002/art.1780210505. [DOI] [PubMed] [Google Scholar]
- 14.Banda NK, Wood AK, Takahashi K, Levitt B, Rudd PM, Royle L, Abrahams JL, Stahl GL, Holers VM, Arend WP. Initiation of the alternative pathway of murine complement by immune complexes is dependent on N-glycans in IgG antibodies. Arthritis Rheum. 2008;58:3081–3089. doi: 10.1002/art.23865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ratnoff WD, Fearon DT, Austen KF. The role of antibody in the activation of the alternative complement pathway. Springer Semin Immunopathol. 1983;6:361–371. doi: 10.1007/BF02116280. [DOI] [PubMed] [Google Scholar]
- 16.Wouters D, Voskuyl AE, Molenaar ET, Dijkmans BA, Hack CE. Evaluation of classical complement pathway activation in rheumatoid arthritis: measurement of C1q-C4 complexes as novel activation products. Arthritis Rheum. 2006;54:1143–1150. doi: 10.1002/art.21729. [DOI] [PubMed] [Google Scholar]
- 17.Banda NK, Takahashi K, Wood AK, Holers VM, Arend WP. Pathogenic complement activation in collagen antibody-induced arthritis in mice requires amplification by the alternative pathway. J. Immunol. 2007;179:4101–4109. doi: 10.4049/jimmunol.179.6.4101. [DOI] [PubMed] [Google Scholar]
- 18.Hansen S, Thiel S, Willis A, Holmskov U, Jensenius JC. Purification and characterization of two mannan-binding lectins from mouse serum. Journal of Immunology. 2000;164:2610–2618. doi: 10.4049/jimmunol.164.5.2610. [DOI] [PubMed] [Google Scholar]
- 19.Kawai T, Suzuki Y, Eda S, Kase T, Ohtani K, Sakai Y, Keshi H, Fukuoh A, Sakamoto T, Nozaki M, Copeland NG, Jenkins NA, Wakamiya N. Molecular cloning of mouse collectin liver 1. Bioscience, biotechnology, and biochemistry. 2002;66:2134–2145. doi: 10.1271/bbb.66.2134. [DOI] [PubMed] [Google Scholar]
- 20.Ohashi T, Erickson HP. Oligomeric structure and tissue distribution of ficolins from mouse, pig and human. Archives of biochemistry and biophysics. 1998;360:223–232. doi: 10.1006/abbi.1998.0957. [DOI] [PubMed] [Google Scholar]
- 21.Ohtani K, Suzuki Y, Eda S, Kawai T, Kase T, Yamazaki H, Shimada T, Keshi H, Sakai Y, Fukuoh A, Sakamoto T, Wakamiya N. Molecular cloning of a novel human collectin from liver (CL-L1). The Journal of biological chemistry. 1999;274:13681–13689. doi: 10.1074/jbc.274.19.13681. [DOI] [PubMed] [Google Scholar]
- 22.Thiel S, Vorup-Jensen T, Stover CM, Schwaeble W, Laursen SB, Poulsen K, Willis AC, Eggleton P, Hansen S, Holmskov U, Reid KB, Jensenius JC. A second serine protease associated with mannan-binding lectin that activates complement. Nature. 1997;386:506–510. doi: 10.1038/386506a0. [DOI] [PubMed] [Google Scholar]
- 23.Moller-Kristensen M, Thiel S, Sjoholm A, Matsushita M, Jensenius JC. Cooperation between MASP-1 and MASP-2 in the generation of C3 convertase through the MBL pathway. Int Immunol. 2007;19:141–149. doi: 10.1093/intimm/dxl131. [DOI] [PubMed] [Google Scholar]
- 24.Heja D, Kocsis A, Dobo J, Szilagyi K, Szasz R, Zavodszky P, Pal G, Gal P. Revised mechanism of complement lectin-pathway activation revealing the role of serine protease MASP-1 as the exclusive activator of MASP-2. Proc Natl Acad Sci U S A. 2012;109:10498–10503. doi: 10.1073/pnas.1202588109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pangburn MK, Schreiber RD, Muller-Eberhard HJ. C3b deposition during activation of the alternative complement pathway and the effect of deposition on the activating surface. Journal of Immunology. 1983;131:1930–1935. [PubMed] [Google Scholar]
- 26.Rosen BS, Cook KS, Yaglom J, Groves DL, Volanakis JE, Damm D, White T, Spiegelman BM. Adipsin and complement factor D activity: an immune-related defect in obesity. Science. 1989;244:1483–1487. doi: 10.1126/science.2734615. [DOI] [PubMed] [Google Scholar]
- 27.Kemper C, Atkinson JP, Hourcade DE. Properdin: emerging roles of a pattern-recognition molecule. Annu. Rev. Immunol. 2010;28:131–155. doi: 10.1146/annurev-immunol-030409-101250. [DOI] [PubMed] [Google Scholar]
- 28.Hiemstra PS, Biewenga J, Gorter A, Stuurman ME, Faber A, van Es LA, Daha MR. Activation of complement by human serum IgA, secretory IgA and IgA1 fragments. Mol. Immunol. 1988;25:527–533. doi: 10.1016/0161-5890(88)90074-0. [DOI] [PubMed] [Google Scholar]
- 29.Takahashi M, Ishida Y, Iwaki D, Kanno K, Suzuki T, Endo Y, Homma Y, Fujita T. Essential role of mannose-binding lectin-associated serine protease-1 in activation of the complement factor D. J Exp Med. 2010;207:29–37. doi: 10.1084/jem.20090633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Takahashi M, Iwaki D, Kanno K, Ishida Y, Xiong J, Matsushita M, Endo Y, Miura S, Ishii N, Sugamura K, Fujita T. Mannose-binding lectin (MBL)-associated serine protease (MASP)-1 contributes to activation of the lectin complement pathway. J Immunol. 2008;180:6132–6138. doi: 10.4049/jimmunol.180.9.6132. [DOI] [PubMed] [Google Scholar]
- 31.Ruseva MM, Takahashi M, Fujita T, Pickering MC. C3 dysregulation due to factor H deficiency is MASP-1 and MASP-3 independent in vivo. Clinical and experimental immunology. 2013 doi: 10.1111/cei.12244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Degn SE, Jensen L, Hansen AG, Duman D, Tekin M, Jensenius JC, Thiel S. Mannan-binding lectin-associated serine protease (MASP)-1 is crucial for lectin pathway activation in human serum, whereas neither MASP-1 nor MASP-3 is required for alternative pathway function. Journal of Immunology. 2012;189:3957–3969. doi: 10.4049/jimmunol.1201736. [DOI] [PubMed] [Google Scholar]
- 33.Degn SE, Jensen L, Gal P, Dobo J, Holmvad SH, Jensenius JC, Thiel S. Biological variations of MASP-3 and MAp44, two splice products of the MASP1 gene involved in regulation of the complement system. J Immunol Methods. 2010;361:37–50. doi: 10.1016/j.jim.2010.07.006. [DOI] [PubMed] [Google Scholar]
- 34.Skjoedt MO, Hummelshoj T, Palarasah Y, Honore C, Koch C, Skjodt K, Garred P. A novel mannose-binding lectin/ficolin-associated protein is highly expressed in heart and skeletal muscle tissues and inhibits complement activation. J Biol Chem. 2010;285:8234–8243. doi: 10.1074/jbc.M109.065805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dahl MR, Thiel S, Matsushita M, Fujita T, Willis AC, Christensen T, Vorup-Jensen T, Jensenius JC. MASP-3 and its association with distinct complexes of the mannan-binding lectin complement activation pathway. Immunity. 2001;15:127–135. doi: 10.1016/s1074-7613(01)00161-3. [DOI] [PubMed] [Google Scholar]
- 36.Degn SE, Hansen AG, Steffensen R, Jacobsen C, Jensenius JC, Thiel S. MAp44, a human protein associated with pattern recognition molecules of the complement system and regulating the lectin pathway of complement activation. J Immunol. 2009;183:7371–7378. doi: 10.4049/jimmunol.0902388. [DOI] [PubMed] [Google Scholar]
- 37.Pavlov VI, Skjoedt MO, Siow Tan Y, Rosbjerg A, Garred P, Stahl GL. Endogenous and natural complement inhibitor attenuates myocardial injury and arterial thrombogenesis. Circulation. 2012;126:2227–2235. doi: 10.1161/CIRCULATIONAHA.112.123968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Banda NK, Takahashi M, Levitt B, Glogowska M, Nicholas J, Takahashi K, Stahl GL, Fujita T, Arend WP, Holers VM. Essential role of complement mannose-binding lectin-associated serine proteases-1/3 in the murine collagen antibody-induced model of inflammatory arthritis. J. Immunol. 2010;185:5598–5606. doi: 10.4049/jimmunol.1001564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Matsushita M, Fujita T. Cleavage of the third component of complement (C3) by mannose-binding protein-associated serine protease (MASP) with subsequent complement activation. Immunobiology. 1995;194:443–448. doi: 10.1016/S0171-2985(11)80110-5. [DOI] [PubMed] [Google Scholar]
- 40.Bakker AC, Van de Loo FA, Joosten LA, Bennink MB, Arntz OJ, Dmitriev IP, Kashentsera EA, Curiel DT, van den Berg WB. A tropism-modified adenoviral vector increased the effectiveness of gene therapy for arthritis. Gene Ther. 2001;8:1785–1793. doi: 10.1038/sj.gt.3301612. [DOI] [PubMed] [Google Scholar]
- 41.Morrison TE, Fraser RJ, Smith PN, Mahalingam S, Heise MT. Complement contributes to inflammatory tissue destruction in a mouse model of Ross River virus-induced disease. J Virol. 2007;81:5132–5143. doi: 10.1128/JVI.02799-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Morrison TE, Simmons JD, Heise MT. Complement receptor 3 promotes severe ross river virus-induced disease. J Virol. 2008;82:11263–11272. doi: 10.1128/JVI.01352-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Morrison TE, Whitmore AC, Shabman RS, Lidbury BA, Mahalingam S, Heise MT. Characterization of Ross River virus tropism and virus-induced inflammation in a mouse model of viral arthritis and myositis. J Virol. 2006;80:737–749. doi: 10.1128/JVI.80.2.737-749.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Banda NK, Levitt B, Wood AK, Takahashi K, Stahl GL, Holers VM, Arend WP. Complement activation pathways in murine immune complex-induced arthritis and in C3a and C5a generation in vitro. Clin Exp Immunol. 2010;159:100–108. doi: 10.1111/j.1365-2249.2009.04035.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc. 2008;3:1101–1108. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- 46.Banda NK, Hyatt S, Antonioli AH, White JT, Glogowska M, Takahashi K, Merkel TJ, Stahl GL, Mueller-Ortiz S, Wetsel R, Arend WP, Holers VM. Role of C3a receptors, C5a receptors, and complement protein C6 deficiency in collagen antibody-induced arthritis in mice. Journal of Immunology. 2012;188:1469–1478. doi: 10.4049/jimmunol.1102310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Banda NK, Takahashi K. Analysis of the complement activation in mice. Methods in molecular biology. 2014;1100:365–371. doi: 10.1007/978-1-62703-724-2_31. [DOI] [PubMed] [Google Scholar]
- 48.Kimura Y, Miwa T, Zhou L, Song WC. Activator-specific requirement of properdin in the initiation and amplification of the alternative pathway complement. Blood. 2008;111:732–740. doi: 10.1182/blood-2007-05-089821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Presanis JS, Hajela K, Ambrus G, Gal P, Sim RB. Differential substrate and inhibitor profiles for human MASP-1 and MASP-2. Molecular Immunology. 2004;40:921–929. doi: 10.1016/j.molimm.2003.10.013. [DOI] [PubMed] [Google Scholar]
- 50.Degn SE, Jensen L, Olszowski T, Jensenius JC, Thiel S. Co-complexes of MASP-1 and MASP-2 associated with the soluble pattern-recognition molecules drive lectin pathway activation in a manner inhibitable by MAp44. J Immunol. 2013;191:1334–1345. doi: 10.4049/jimmunol.1300780. [DOI] [PubMed] [Google Scholar]
- 51.Takahashi M, Sekine H, Endo Y, Fujita T. Comment on “Mannan-binding lectin-associated serine protease (MASP)-1 is crucial for lectin pathway activation in human serum, whereas neither MASP-1 nor MASP-3 is required for alternative pathway function”. Journal of Immunology. 2013;190:2477. doi: 10.4049/jimmunol.1390002. [DOI] [PubMed] [Google Scholar]
- 52.Takahashi M, Sekine H, Fujita T. Comment on “The Pro-Factor D Cleaving Activity of MASP-1/-3 Is Not Required for Alternative Pathway Function”. Journal of Immunology. 2014;192:5448–5449. doi: 10.4049/jimmunol.1400766. [DOI] [PubMed] [Google Scholar]
- 53.Bai M, Harfe B, Freimuth P. Mutations that alter an Arg-Gly-Asp (RGD) sequence in the adenovirus type 2 penton base protein abolish its cell-rounding activity and delay virus reproduction in flat cells. J Virol. 1993;67:5198–5205. doi: 10.1128/jvi.67.9.5198-5205.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mathias P, Wickham T, Moore M, Nemerow G. Multiple adenovirus serotypes use alpha v integrins for infection. J Virol. 1994;68:6811–6814. doi: 10.1128/jvi.68.10.6811-6814.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wickham TJ, Mathias P, Cheresh DA, Nemerow GR. Integrins alpha v beta 3 and alpha v beta 5 promote adenovirus internalization but not virus attachment. Cell. 1993;73:309–319. doi: 10.1016/0092-8674(93)90231-e. [DOI] [PubMed] [Google Scholar]
- 56.Nikkari L, Haapasalmi K, Aho H, Torvinen A, Sheppard D, Larjava H, Heino J. Localization of the alpha v subfamily of integrins and their putative ligands in synovial lining cell layer. J Rheumatol. 1995;22:16–23. [PubMed] [Google Scholar]
- 57.Pirila L, Heino J. Altered integrin expression in rheumatoid synovial lining type B cells: in vitro cytokine regulation of alpha 1 beta 1, alpha 6 beta 1, and alpha v beta 5 integrins. J Rheumatol. 1996;23:1691–1698. [PubMed] [Google Scholar]
- 58.Rinaldi N, Weis D, Brado B, Schwarz-Eywill M, Lukoschek M, Pezzutto A, Keilholz U, Barth TF. Differential expression and functional behaviour of the alpha v and beta 3 integrin subunits in cytokine stimulated fibroblast-like cells derived from synovial tissue of rheumatoid arthritis and osteoarthritis in vitro. Ann Rheum Dis. 1997;56:729–736. doi: 10.1136/ard.56.12.729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ebina K, Oshima K, Matsuda M, Fukuhara A, Maeda K, Kihara S, Hashimoto J, Ochi T, Banda NK, Yoshikawa H, Shimomura I. Adenovirus-mediated gene transfer of adiponectin reduces the severity of collagen-induced arthritis in mice. Biochem Biophys Res Commun. 2009;378:186–191. doi: 10.1016/j.bbrc.2008.11.005. [DOI] [PubMed] [Google Scholar]
- 60.Iwaki D, Kanno K, Takahashi M, Endo Y, Matsushita M, Fujita T. The role of mannose-binding lectin-associated serine protease-3 in activation of the alternative complement pathway. J Immunol. 2011;187:3751–3758. doi: 10.4049/jimmunol.1100280. [DOI] [PubMed] [Google Scholar]
- 61.Wilson JM. Adenoviruses as gene-delivery vehicles. N Engl J Med. 1996;334:1185–1187. doi: 10.1056/NEJM199605023341809. [DOI] [PubMed] [Google Scholar]
- 62.Toh ML, Hong SS, van de Loo F, Franqueville L, Lindholm L, van den Berg W, Boulanger P, Miossec P. Enhancement of adenovirus-mediated gene delivery to rheumatoid arthritis synoviocytes and synovium by fiber modifications: role of arginine-glycine-aspartic acid (RGD)- and non-RGD-binding integrins. J Immunol. 2005;175:7687–7698. doi: 10.4049/jimmunol.175.11.7687. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.